

1. DISEASE CHARACTERISTICS
1.1 Name of the disease (synonyms)
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is an inheritable disease characterized by structural and functional abnormalities of the right ventricle (RV), with or without concomitant left ventricular (LV) disease. The diagnosis ARVC is made when a patient fulfils the recently revised criteria.1 Criteria encompass global and/or regional dysfunction and structural changes; repolarization abnormalities; depolarization and conduction abnormalities; arrhythmias; family history/the results of genetic testing; and tissue characterization by endomyocardial biopsy (EMB). Either localized or diffuse atrophy, with subsequent replacement by fibrous and fatty tissue mainly of the RV outflow tract, RV inflow tract and RV apex (‘triangle of dysplasia') represent the histopathological characteristics of ARVC. Synonyms: arrhythmogenic right ventricular dysplasia (ARVD); ARVC/dysplasia (ARVC/D); arrhythmogenic cardiomyopathy (ACM); arrhythmogenic left-dominant cardiomyopathy; and arrhythmogenic LV cardiomyopathy (ALVC).
1.2 OMIM# of the disease
107970 (ARVD1); 600996 (ARVD2); 602086 (ARVD3); 602087 (ARVD4); 604400 (ARVD5); 604401 (ARVD6); 607450 (ARVD8); 609040 (ARVD9); 610193 (ARVD10); 610476 (ARVD11); 611528 (ARVD12).
The symbol ARVD7 was previously used for a disorder later shown to be the same as desmin-related myopathy (601419 Myopathy, myofibrillar, 1; MFM1).
1.3 Name of the analyzed genes or DNA/chromosome segments
(1) Cytoskeletal protein genes: (a) Desmin gene (DES), locus 2q35; (b) Titin gene (TTN), locus 2q31.2#. (2) Nuclear envelope protein genes: (a) Lamin A/C gene (LMNA), locus 1q22#. (3) Desmosomal protein genes: (a) Desmocollin 2 gene (DSC2), locus 18q12.1; (b) Desmoglein 2 gene (DSG2), locus 18q12.1; (c) Desmoplakin gene (DSP), locus 6p24.3; (d) Junction plakoglobin gene (JUP), locus 17q21.2; (e) Plakophilin 2 gene (PKP2), locus 12p11.21. (4) Calcium/sodium-handling genes: (a) Phospholamban gene (PLN), locus 6q22.31#; (b) Ryanodine receptor 2 gene (RYR2), locus 1q43*. (5) Other genes: (a) Alpha-T-catenin (CTNNA3), locus 10q21.3#; (b) Transforming growth factor-β3 (TGFβ3), locus 14q24.3* (c) Transmembrane protein 43 (TMEM43), locus 3p25.1.
#Indicates gene not yet annotated as ARVC related in the OMIM database; *indicates the involvement of these genes is based on single publications2, 3 and therefore controversial.
1.4 OMIM# of the gene(s)
(1) Cytoskeletal protein genes: (a) 125660 and (b) 188840#. (2) Nuclear envelope protein genes: (a) 150330#. (3) Desmosomal protein genes: (a) 125645; (b) 125671; (c) 125647; (d) 173325; and (e) 602861. (4) Calcium/sodium-handling genes: (a) 172405# and (b) 180902*. (5) Other genes: (a) 607667#; (b) 190230* and (c) 612048.
#Indicates gene not yet annotated as ARVC related in the OMIM database; *indicates the involvement of these genes is based on single publications and therefore controversial.
1.5 Mutational spectrum
Until now, 13 ARVC genes have been identified. The majority of mutations in these genes are missense mutations.4 Mutations in PKP2, however, are often nonsense- and frameshift mutations.4 Less frequently, other types of mutations including large deletions/duplications, are detected.5, 6
1.6 Analytical methods
Sanger sequencing of coding regions and their flanking intronic sequences is used in combination with pre-screening methods, such as DGGE, DHPLC, CSCE and so on. The multiplex ligation-dependent probe amplification (MLPA) technique is being used to identify large deletions/duplications. Very recently, next generation sequencing (NGS) methods have been implemented.7
1.7 Analytical validation
Sequencing of both strands: When a mutation is found, it is usually validated in an independent experiment by direct sequencing or MLPA using a freshly prepared DNA solution.
1.8 Estimated frequency of the disease(incidence at birth (‘birth prevalence') or population prevalence)
If known to be variable between ethnic groups, please report):
The prevalence is estimated to be approximately 1 per 5000 individuals in the general population worldwide.8, 9 Higher numbers, however, are found in specific regions, for example, the Veneto region of Italy.10
1.9 Diagnostic Setting

Comment:
The most frequently mutated ARVC genes are the desmosomal genes PKP2, DSG2, DSC2, JUP and DSP. Among these, PKP2 is the most prevalent mutated gene responsible for 11–51% of ARVC patients.11, 12, 13 In certain populations, specific mutations can be found often due to a founder effect, such as the PLN p.Arg14del and PKP2 p.Arg79* mutations in the Netherlands and the TMEM43 p.Ser358Leu mutation in Newfoundland, Canada.14, 15, 16
ARVC patients carrying more than one disease-associated mutation often show a more severe phenotype, characterized by a younger age of onset and worse prognosis, suggesting a gene-dosage effect.5, 13, 17, 18 It is to be expected that the use of NGS techniques will result in the identification of an increasing number of patients with such complex genotypes.
When interpreting the pathogenicity of genetic variants in ARVC-related genes, one has to be aware that putative mutations including radical mutations (eg, truncating and nonsense mutations) can also be present in healthy controls, although less frequent.19
2. TEST CHARACTERISTICS
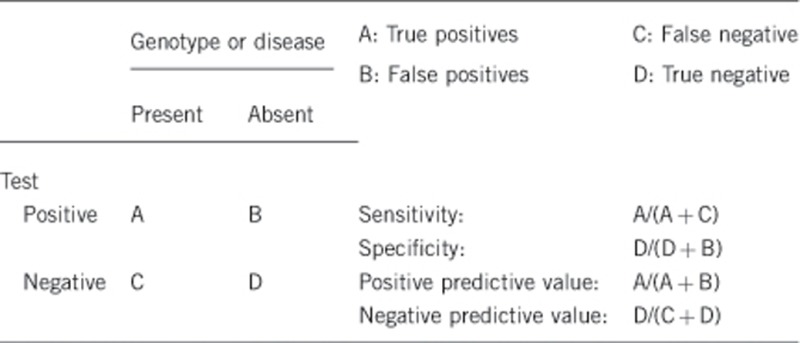
2.1 Analytical sensitivity
(proportion of positive tests if the genotype is present)
Almost 100%, also depending on the analytical method used. Preferential amplification of one allele can happen when one of the primers is located on a SNP. To minimize this risk, primers have to be verified using the SNPCheck software (https://ngrl.manchester.ac.uk/SNPCheckV3/snpcheck.htm) yearly.
2.2 Analytical specificity
(proportion of negative tests if the genotype is not present)
Almost 100%, depending on the analytical method used.
2.3 Clinical sensitivity
(proportion of positive tests if the disease is present)
The clinical sensitivity is dependent on variable factors such as age, disease manifestation and family history.
The yield of genetic testing varies considerably, for example, in different geographical regions. On average, the mutation detection rate for the most common ARVC-related genes are: PKP2 11–51%, DSG2 3–20%, DSC2 1–7%, JUP 0.5–16% and DSP 1–7%.5, 12, 13, 17, 18, 19, 20, 21
2.4 Clinical specificity
(proportion of negative tests if the disease is not present)
The clinical sensitivity is dependent on variable factors such as age, disease manifestation and family history.
The penetrance and expression of ARVC is incomplete and variable, respectively, besides being age-dependent, therefore the clinical specificity is <100%.13
2.5 Positive clinical predictive value
(life-time risk to develop the disease if the test is positive)
This is significantly <100% because of incomplete penetrance. The degree of penetrance might depend on the presence or absence of other genetic or exogenous factors, with recent data suggesting a possible role for compound and digenic heterozygosity,13, 18 and steadily increases until advanced age.
2.6 Negative clinical predictive value
(probability not to develop the disease if the test is negative)
Assume an increased risk based on family history for a non-affected person. Allelic and locus heterogeneity may need to be considered.
Index case in that family had been tested:
In the case that a pathogenic mutation has been found in the index patient, the risk of being a carrier for a first-degree relative is 50% (assuming autosomal dominant inheritance). If this mutation is absent in another family member, it is generally assumed that this family member is not at increased risk of developing ARVC. However, as carriership of multiple disease-causing or -modifying mutations in one or more genes has been reported, it cannot be excluded that this mutation-negative family member carries a still unidentified ARVC-related mutation.5, 13, 17, 18
Index case in that family had not been tested:
ARVC is believed to be familial in ∼30–50% of cases.1, 8, 9, 20
Therefore, when an index patient has been clinically diagnosed with ARVC, and no genetic test has been performed, the chance for a first-degree relative to develop ARVC may reach up to 15–25%.
3. CLINICAL UTILITY
3.1 (Differential) diagnostics: The tested person is clinically affected
(To be answered if in 1.9 ‘A' was marked)
3.1.1 Can a diagnosis be made other than through a genetic test?

Comment:
The clinical diagnosis of ARVC is usually made by electrocardiography, signal averaged electrocardiography, echocardiography, holter monitoring and family history/genetic testing with or without magnetic resonance imaging and EMB sampling in accordance with the published revised criteria.1 Because of age-dependent penetrance, a negative clinical test does not exclude the possibility of developing ARVC at a later age. Therefore, repeated cardiological evaluation is recommended.
3.1.2 Describe the burden of alternative diagnostic methods to the patient
Electrocardiography, signal averaged electrocardiography, echocardiography, holter monitoring and magnetic resonance imaging are non-invasive techniques with little risks and inconvenience to the patient when taking the appropriate precautions into consideration. In addition, histological confirmation of ARVC may provide important additional information in patients with suspected ARVC in whom non-invasive evaluation remains inconclusive and in sporadic forms to exclude phenocopies such as sarcoidosis and myocarditis. Evidence of a certain degree of fibro-fatty replacement on EMB is, according to the modified Task Force criteria, considered as a major criterion.1 EMB sampling from predilection areas in patients with ARVC is yet perceived to give an increased risk of perforation, given the reduced thickness of the RV free wall with additional structural alterations due to fibro-fatty replacement.
However, recent data indicate that EMB from these predilection areas in ARVC is not associated with an increased rate of major complications in comparison with other diseases when performed by experienced interventional cardiologists following a precise and dedicated protocol.22
Among the new imaging tools, endocardial voltage mapping has the ability to identify low-amplitude electrical signals, that is, electroanatomic scar areas, which reflect myocardial replaced tissue in the ventricular walls.23 Contrast-enhancement-cardiac magnetic resonance is less sensitive to identify right ventricular scar lesion but is the only tool able to detect early subepicardial LV involvement that can be the first manifestation of the disease in otherwise functionally normal hearts.24
3.1.3 How is the cost effectiveness of alternative diagnostic methods to be judged?
When a pathogenic mutation has been found in the index patient, genetic screening can be offered to first-degree relatives (cascade genetic screening) to determine whether they carry the same mutation and may be at risk of developing the disease in the future.
Regular medical follow-up is advised in this mutation-carrying relative to detect early cardiac disease, and thereby initiate timely treatment. In addition, specific advice to gene carriers regarding physical or occupational activities might be given including reproductive counseling and general lifestyle advices. Pregnancy with volume overload should be carefully assessed individually in affected women.
If relatives do not carry the mutation, they can be reassured and dismissed from regular cardiological follow-up, provided that the severity of the phenotype in any family member does not suggest carriership of multiple mutations. This method has been shown to be cost-effective for HCM,25 but no formal cost-effectiveness studies in ARVC are available yet.
3.1.4 Will disease management be influenced by the result of a genetic test?

3.2 Predictive setting: The tested person is clinically unaffected but carries an increased risk based on family history
(To be answered if in 1.9 ‘B' was marked)
3.2.1 Will the result of a genetic test influence lifestyle and prevention?
When carriership of the familial pathogenic mutation has been confirmed, regular cardiological evaluation to detect signs of ARVC is advised and initiate early medical treatment including estimation of the risk of sudden cardiac death with subsequent indication for ICD implantation. Lifestyle advice and reproductive counseling on the risk of transmission to their offspring can then also be provided. Restriction from competitive sport activity and strenuous physical exercise is at present recommended to prevent disease onset.
If the test result is negative, family members not carrying the mutation can be discharged from regular cardiological follow-up. Nevertheless, because carriership of multiple pathogenic mutations in more than one gene has been reported,5, 13, 17, 18 this mutation-negative family member may carry an yet unidentified ARVC-related mutation.
3.2.2 Which options in view of lifestyle and prevention does a person at-risk have if no genetic test has been done (please describe)?
Recommendations and advice given will be similar to patients with a positive genetic test.
3.3 Genetic risk assessment in family members of a diseased person
(To be answered if in 1.9 ‘C' was marked)
3.3.1 Does the result of a genetic test resolve the genetic situation in that family?
If the phenotype in the affected index patient and family members is in accordance with what is known about a specific pathogenic mutation or gene involved, the genetic situation is resolved.
3.3.2 Can a genetic test in the index patient save genetic or other tests in family members?
Yes, a positive test in the index patient enables targeted genetic testing for the identified mutation in the family. Family members who do not carry the pathogenic mutation can be dismissed from regular cardiological evaluation, with the exception of families in which multiple mutations can be expected, which will enable to cut down on cardiological follow-up costs.
On the other hand, when the test in the index patient is negative, this does not exclude a hereditary nature of the disease, because of which close relatives are still advised to continue regular cardiological follow-up.
3.3.3 Does a positive genetic test result in the index patient enable a predictive test in a family member?
Yes, when a pathogenic mutation is detected, this enables the possibility of a predictive test in family members.
3.4 Prenatal diagnosis
(To be answered if in 1.9 ‘D' was marked)
3.4.1 Does a positive genetic test result in the index patient enable a prenatal diagnosis?
Yes, prenatal diagnostics in ARVC is technically possible when a pathogenic mutation has been identified. However, in late-onset hereditary diseases with options for treatment like ARVC prenatal diagnostic tests are only rarely performed.
4. IF APPLICABLE, FURTHER CONSEQUENCES OF TESTING
Please assume that the result of a genetic test has no immediate medical consequences. Is there any evidence that a genetic test is nevertheless useful for the patient or his/her relatives? (Please describe).
The most striking non-medical consequence of genetic testing is that close relatives of the index patient who do not carry the familial pathogenic mutation can be dismissed from regular cardiological follow-up (provided that the severity of the phenotype in any family member does not suggest carriership of multiple mutations).
Acknowledgments
This work was supported by EuroGentest2 (Unit 2: ‘Genetic testing as part of health care'), a Coordination Action under FP7 (Grant Agreement Number 261469) and the European Society of Human Genetics. CB and GT are supported by CARIPARO Foundation, Padova and the Registry for Cardio-cerebro-vascular Pathology, Veneto Region, Venice, Italy. WPteR was supported by the Interuniversity Cardiology Institute of the Netherlands, (ICIN)—The Netherlands Heart Institute (www.icin.nl) project number 12301 (Stimuleringsfonds).
The authors declare no conflict of interest.
References
- Marcus FI, McKenna WJ, Sherrill D et al: Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force Criteria. Eur Heart J 2010; 31: 806–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiso N, Stephan DA, Nava A et al: Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum Mol Genet 2001; 10: 189–194. [DOI] [PubMed] [Google Scholar]
- Beffagna G, Occhi G, Nava A et al: Regulatory mutations in transforming growth factor-beta3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc Res 2005; 65: 366–373. [DOI] [PubMed] [Google Scholar]
- van der Zwaag PA, Jongbloed JD, van den Berg MP et al: A genetic variants database for arrhythmogenic right ventricular dysplasia/cardiomyopathy. Hum Mutat 2009; 30: 1278–1283. [DOI] [PubMed] [Google Scholar]
- Cox MG, van der Zwaag PA, van der Werf C et al: Arrhythmogenic right ventricular dysplasia/cardiomyopathy: pathogenic desmosome mutations in index-patients predict outcome of family screening: Dutch arrhythmogenic right ventricular dysplasia/cardiomyopathy genotype-phenotype follow-up study. Circulation 2011; 123: 2690–2700. [DOI] [PubMed] [Google Scholar]
- Roberts J, Herkert J, Rutberg J et al: Detection of genomic deletions of PKP2 in arrhythmogenic right ventricular cardiomyopathy. Clin Genet 2012; 83: 452–456. [DOI] [PubMed] [Google Scholar]
- Teekakirikul P, Kelly MA, Rehm HL, Lakdawala NK, Funke BH: Inherited cardiomyopathies: molecular genetics and clinical genetic testing in the postgenomic era. J Mol Diagn 2013; 15: 158–170. [DOI] [PubMed] [Google Scholar]
- Nava A, Bauce B, Basso C et al: Clinical profile and long-term follow-up of 37 families with arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol 2000; 36: 2226–2233. [DOI] [PubMed] [Google Scholar]
- Basso C, Bauce B, Corrado D, Thiene G: Pathophysiology of arrhythmogenic cardiomyopathy. Nat Rev Cardiol 2011; 9: 223–233. [DOI] [PubMed] [Google Scholar]
- Thiene G, Nava A, Corrado D, Rossi L, Pennelli N: Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med 1988; 318: 129–133. [DOI] [PubMed] [Google Scholar]
- Syrris P, Ward D, Asimaki A et al: Clinical expression of plakophilin-2 mutations in familial arrhythmogenic right ventricular cardiomyopathy. Circulation 2006; 113: 356–364. [DOI] [PubMed] [Google Scholar]
- van Tintelen JP, Entius MM, Bhuiyan ZA et al: Plakophilin-2 mutations are the major determinant of familial arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation 2006; 113: 1650–1658. [DOI] [PubMed] [Google Scholar]
- Xu T, Yang Z, Vatta M et al: Compound and digenic heterozygosity contributes to arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol 2010; 55: 587–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Zwaag PA, van Rijsingen IA, Asimaki A et al: Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur J Heart Fail 2012; 14: 1199–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Zwaag PA, Cox MG, van der Werf C et al: Recurrent and founder mutations in the Netherlands: Plakophilin-2 p.Arg79X mutation causing arrhythmogenic right ventricular cardiomyopathy/dysplasia. Neth Heart J 2010; 18: 583–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merner ND, Hodgkinson KA, Haywood AF et al: Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am J Hum Genet 2008; 82: 809–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fressart V, Duthoit G, Donal E et al: Desmosomal gene analysis in arrhythmogenic right ventricular dysplasia/cardiomyopathy: spectrum of mutations and clinical impact in practice. Europace 2010; 12: 861–868. [DOI] [PubMed] [Google Scholar]
- Bauce B, Nava A, Beffagna G et al: Multiple mutations in desmosomal proteins encoding genes in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Heart Rhythm 2010; 7: 22–29. [DOI] [PubMed] [Google Scholar]
- Kapplinger JD, Landstrom AP, Salisbury BA et al: Distinguishing arrhythmogenic right ventricular cardiomyopathy/dysplasia-associated mutations from background genetic noise. J Am Coll Cardiol 2011; 57: 2317–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen-Chowdhry S, Syrris P, Ward D, Asimaki A, Sevdalis E, McKenna WJ: Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into patterns of disease expression. Circulation 2007; 115: 1710–1720. [DOI] [PubMed] [Google Scholar]
- Antoniades L, Tsatsopoulou A, Anastasakis A et al: Arrhythmogenic right ventricular cardiomyopathy caused by deletions in plakophilin-2 and plakoglobin (Naxos disease) in families from Greece and Cyprus: genotype-phenotype relations, diagnostic features and prognosis. Eur Heart J 2006; 27: 2208–2216. [DOI] [PubMed] [Google Scholar]
- Paul M, Stypmann J, Gerss J et al: Safety of endomyocardial biopsy in patients with arrhythmogenic right ventricular cardiomyopathy: a study analyzing 161 diagnostic procedures. JACC Cardiovasc Interv 2011; 4: 1142–1148. [DOI] [PubMed] [Google Scholar]
- Corrado D, Basso C, Leoni L et al: Three-dimensional electroanatomic voltage mapping increases accuracy of diagnosing arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circulation 2005; 111: 3042–3050. [DOI] [PubMed] [Google Scholar]
- Marra MP, Leoni L, Bauce B et al: Imaging study of ventricular scar in arrhythmogenic right ventricular cardiomyopathy: comparison of 3D standard electroanatomical voltage mapping and contrast-enhanced cardiac magnetic resonance. Circ Arrhythm Electrophysiol 2012; 5: 91–100. [DOI] [PubMed] [Google Scholar]
- Wordsworth S, Leal J, Blair E et al: DNA testing for hypertrophic cardiomyopathy: a cost-effectiveness model. Eur Heart J 2010; 31: 926–935. [DOI] [PubMed] [Google Scholar]
- Hodgkinson KA, Connors SP, Merner N et al: The natural history of a genetic subtype of arrhythmogenic right ventricular cardiomyopathy caused by a p.S358L mutation in TMEM43. Clin Genet 2013; 83: 321–331. [DOI] [PubMed] [Google Scholar]