

Précis
Niemann-Pick disease, type C1 (NPC1) is a rare autosomal recessive lysosomal lipidosis resulting in a progressive and fatal neurological deterioration. There is limited evidence suggesting auditory dysfunction is an aspect of the phenotype, but one that is poorly understood and, likely, underreported. Data from 50 patients with NPC1 confirm a prevalent, often high frequency, hearing loss that is progressive in at least some individuals. Retrocochlear involvement is common, and some patients present with a profile of auditory neuropathy spectrum disorder.
Introduction
Niemann-Pick disease, type C1 (NPC1, OMIM #257220) is a rare (1:120,000–150,000), autosomal recessive lysosomal lipidosis with a hallmark neurological deterioration that is fatal in all cases. It is part of a family of metabolic storage disorders with systemic manifestations, and currently there is no definitively effective intervention. Despite major progress over the last century in understanding the metabolic and genetic bases of NPC1, there is much about this disease that remains unknown. Large variability in the age of both onset and diagnosis of the disease, coupled with a broad clinical spectrum, creates significant challenges for researchers investigating the natural history of NPC1. The classical onset of neurological symptoms begins in the late-infantile and juvenile years, with death occurring in the second decade (e.g., Garver et al., 2007). However, age of presentation, and subsequent age of diagnosis, may range from the perinatal period to adulthood (e.g., Vanier and Millat, 2003).
NPC is the result of biallelic mutations in either NPC1 or NPC2 (Carstea et al., 1997; Naureckiene et al., 2000). Over 95% of patients with NPC have mutations in NPC1, although the two complementation groups appear to be biochemically indistinguishable (e.g., Ikonen and Hölttä-Vuori, 2004). The ensuing metabolic defect is an over-accumulation of exogenous and unesterified cholesterol within cells and tissues throughout the body (Liscum and Faust, 1987; Liscum et al.,1989), including brain areas with high concentrations of lipids, such as myelin and neural plasma membranes (Vincent, Bu, and Erickson, 2003). Early symptoms of NPC often involve the hepatic system; however, diagnosis is usually delayed until the onset of neurological manifestations, which may include, among other complications, cerebellar ataxia, dysarthria, dysphagia, dystonia/hypotonia, cataplexy, and seizures. In some cases, neurological involvement may progress to states of psychosis and dementia.
There is a limited literature suggesting the auditory system is affected during the course of this disease (e.g., Pikus, 1989), although the full natural history of the auditory phenotype is unknown. Indeed, auditory manifestations have likely been underreported given the difficulty in obtaining behavioral audiological evaluations in a predominantly pediatric, neurologically compromised population and the inability of many affected individuals to self-report hearing difficulties. Select case studies (Aisen et al., 1985; Palmeri et al., 2005) and reports that mention hearing in NPC (Pikus, 1989; Fink et al. 1989; Higgins et al., 1992; Garver et al., 2007) include sporadic descriptions of high-frequency hearing loss, acoustic reflex abnormalities, and auditory brainstem response (ABR) disturbance, which together suggest possible widespread auditory dysfunction. There remains a need to examine comprehensively the auditory phenotype in NPC1, characterizing both how the dysfunction manifests and if, like the global neurological phenotype, it is progressive.
Materials and Methods
Participants were those patients admitted to a natural history protocol investigating NPC1 at the National Institutes of Health Clinical Center between August 2006 and December 2010. All patients with an established diagnosis of NPC1 were considered for study; however, patients who were non-ambulant with vegetative disturbances were not enrolled. This protocol was approved by the Institutional Review Board of the National Institute of Child Health and Human Development. Written informed consent and, when possible, assents were obtained and documented in the medical record.
Fifty patients (25 males, 25 females) with NPC1 were enrolled in this study between 8/14/2006 and 12/27/2010. Biallelic NPC1 mutations were confirmed via molecular testing in all but two participants at the time of study. Although the molecular status of the remaining two participants cannot yet be confirmed, neither showed clinical evidence of significant respiratory involvement, which is more often associated with NPC2. Five sibling pairs, including one set of monozygotic twins, are included in the cohort. The mean age at enrollment was 9.3 years (SD =6.5 years), ranging from 4 months to 21.8 years. Over half (58%) of the cohort was less than 10 years of age, and the majority (88%) fell within a pediatric range of less than 18 years. The average age of disease onset (age at first symptom) was 3.3 years.
Age- and ability-appropriate behavioral audiological data, as well as physiological and electrophysiological auditory data were collected in sound suites meeting American National Standards Institute criteria (ANSI, 2010). For patients unable to cooperate for behavioral and/or physiological/electrophysiological analyses while awake, sedated testing took place in either an operating room or a procedure room where ambient noise levels were kept to a minimum.
Behavioral hearing tests were conducted using a Grason-Stadler (Edin Prairie, MN, USA) diagnostic audiometer (GSI-61). Whenever possible, ear-specific data were obtained for air conduction (250–8000 Hz) and bone conduction (250–4000 Hz) stimuli. Middle ear function was measured using an acoustic immittance unit (GSI Tymp Star, GSI-33) and included 226 Hz tympanometry, measurement of ipsilateral and contralateral acoustic stapedial reflex thresholds (500, 1000, and 2000 Hz) and acoustic reflex decay (500, 1000 Hz). Distortion product otoacoustic emissions (DPOAEs) were evaluated using an Otodynamics (Hatfield, Hertfordshire, UK) Echoport Otoacoustic Emission System supported by ILO V6 Clinical OAE software. DPOAEs were evaluated in one-quarter octave decrements from 7996-842 Hz, using a frequency (f) ratio of f2/f1 = 1.2, with levels (L1 and L2) set at 65 dB SPL and 55 dB SPL, respectively. Emissions were considered present if they achieved a 6 dB signal-to-noise ratio.
Neurotologic auditory brainstem response (ABR) data were collected using Grason-Stadler Audera software. Disposable surface electrodes were placed in an Fz to earlobe configuration, with a grounding electrode placed at Fpz. Broadband click stimuli were presented via insert earphones (Ear Tone ER-3A) at a high level (85 or 95 dB nHL) using single polarity rarefaction and condensation clicks delivered at a rate of 8.3 per second. Test paradigms including a fast click rate of 63.3 per second (85/95 dB nHL, rarefaction) and a lower intensity stimulus (55 or 65 dB nHL, 8.3/second, rarefaction) were administered to confirm a neural origin of the response (Hall, 1992; Hood, 1998). A minimum of 1000 stimulus presentations was averaged for each ABR test run, and every condition was replicated to determine repeatability of the waveform.
Statistical Analyses
Data were maintained and analyzed using Microsoft Excel, the Statistical Package for Social Science Software (SPSS, v15), and the Proc MIXED SAS 9.1 software packages. Data were evaluated as a single-group design for analysis on an individual level and across the group of NPC patients. For those individuals with longitudinal data, their baseline audiological exam served as the reference to which all subsequent evaluations were compared.
To identify potential independent variables that may affect hearing, a mixed model linear regression analysis was used to evaluate effects of gender, use of the experimental therapeutic drug miglustat, and age. Three variables related to age were examined: age at disease onset (defined as age at first symptom), age at baseline testing, and time from disease onset to test date. Because all three age variables are related, they were not included in the model together at any one time, but rather evaluated independently and with other non-age-related variables. The dependent variables used to quantify hearing were pure-tone averages: .5/1/2 kHz, .5/1/2/4 kHz, .25/.5 kHz, and 4/8 kHz. For this analysis, left and right ears were included together. For all statistical analyses α = .05.
Disease severity scores were assigned using a Likert-like scale developed by Yanjanin et al. (2009) for use with the NPC1 population and compared to the 4 kHz and 8 kHz air conduction pure-tone thresholds. Notably, this scale includes pure-tone sensorineural hearing loss and neurotologic ABR outcomes as part of its nine major and eight minor clinical domains, respectively. As such, rigorous statistical analysis correlating the two outcomes is not appropriate. Total possible severity scores range from 0 to 61, where a higher score indicates a more severe clinical impairment.
Longitudinal changes in hearing were evaluated for individual patients, and cross-sectional data were used to evaluate hearing and age-related change. Interpretation of ABR absolute and interpeak latencies was based on normative data published by Schwartz et al. (1989) for those greater than 3 years of age and Issa and Ross (1995) for those less than or equal to 3 years. Descriptive statistical group data, including categorical interpretation (normal versus abnormal) for absolute and interpeak latencies are reported.
All data collected were prospectively obtained with the exception of one patient who had longitudinal pure-tone and ABR data available from participation in an earlier protocol at the NIH. These data were also included in the longitudinal data analyses.
Results
Thirty-one (62%) patients were able to participate to some degree for behavioral testing, which resulted in data that varied from a single pure-tone threshold to a complete audiological evaluation. Alternatively, 19 patients, ranging in age from six months to 21.5 years, were unable to provide any behavioral hearing data, which is a reflection of both young age and advanced disease status.
Baseline audiometry findings
Pure-tone averages for air- and bone-conduction at baseline for 31 patients ranging in age from three to 21 years (x̄ =10.9, SD = 5.6) are presented in Table 1. Because this is predominantly a pediatric population, 15 dB HL was used as the normative cut-off for hearing loss (Clark, 1981). With these criteria, over half (53% of ears, Figure 1; 64% of patients) of the sample for whom thresholds could be reliably established exhibited hearing loss at 8000 Hz. Moreover, of the 31 patients with behavioral data, 23 (74%) presented with clinically significant hearing loss involving the frequencies most important to speech understanding (500–4000 Hz). The hearing loss was bilateral in 17 of these patients, although the five patients with unilateral hearing loss did not include any asymmetries in hearing larger than 15 dB (x̄ = 8 dB, SD = 4 dB), and one patient had behavioral data available from only one ear. Fifteen patients (48%) were newly identified with hearing loss at their baseline evaluation from behavioral data. Another 10 unable to participate in behavioral evaluations showed evidence of hearing loss via sedated auditory evoked potential assessments of hearing (data not shown).
Table 1.
Pure-tone averages for air- and bone-conduction at baseline from 31 patients with NPC1
| .5/1/2 AC (n=52) | .5/1/2 BC (n=31) | .5/1/2/4 AC (n=52) | .5/1/2/4 BC (n=30) | .25/.5 AC (n=40) | .25/.5 BC (n=28) | 4/8 AC (n=49) | |
|---|---|---|---|---|---|---|---|
| Mean | 13.1 | 10 | 14.9 | 12.2 | 14.1 | 9.5 | 20.7 |
| SD | 6.6 | 6.5 | 8.1 | 8.1 | 5.1 | 5.2 | 15.8 |
| Range | 1.7 – 28.3 | −1.7 – 26.7 | 2.5 – 37.5 | 0 – 35 | 5 – 22.5 | 0 – 20 | 0 – 60 |
Note. Data are presented in dB HL, based on ears (n). AC, air-conduction; BC, bone-conduction.
Figure 1.

Percent of ears with hearing loss (>15 dB HL) by frequency at baseline based on data from 31 patients.
A mixed model linear regression analysis failed to reveal any relationships between hearing and the independent variables age (baseline, disease onset, time from disease onset), gender, or use of miglustat, an off-label drug therapy taken by many patients with NPC1 (data not shown). Cross-sectional data showing the distribution of hearing (.5/1/2/4 and 4/8 kHz pure-tone averages) by age at baseline are shown in Figure 2.
Figure 2.
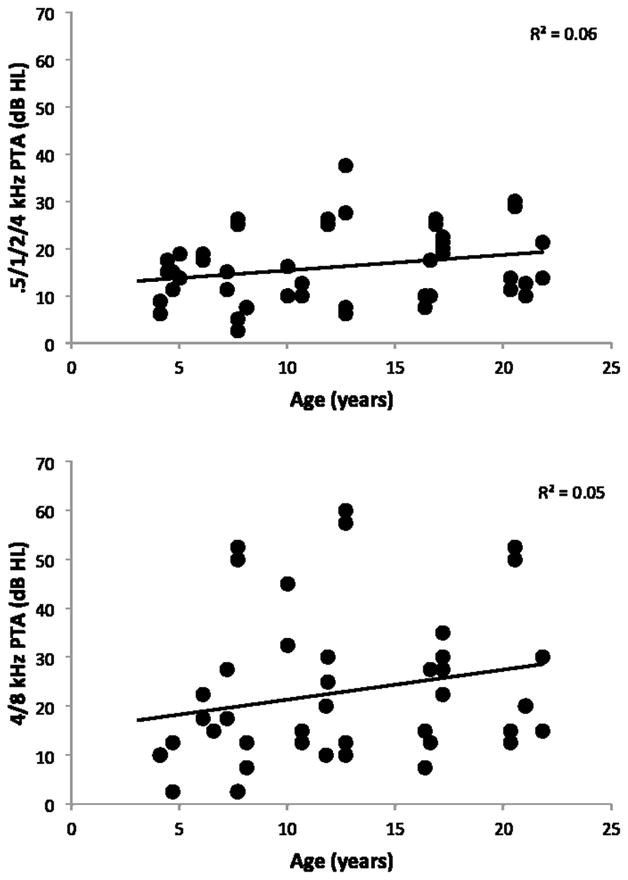
Air-conduction hearing thresholds (ears) at baseline for both a four-frequency (top panel) and high-frequency (bottom panel) pure-tone average (PTA) by age at the time of test. Trend line plotted.
Immittance findings
Immittance data were collected on 49 patients. Table 2 shows the percentage of normal and abnormal findings for tympanometry and the acoustic reflex. Overall, middle ear status fell within normal limits for tympanometry (Margolis and Heller, 1987). The most common abnormality observed involved tympanometric peak pressure, which included both positive and negative pressure findings.
Table 2.
Immittance findings for 49 patients with NPC1
| Tympanometry Findings (n=98 ears)
| ||||
|---|---|---|---|---|
| WNL | Abnormal | CNT | ||
|
|
||||
| High | Low | |||
| Vec (cm3) | 79% (77) | 17% (17)* | 4% (4) | |
| Admittance (mmhos) | 80% (78) | 10% (10) | 10% (10) | |
| Negative | Positive | |||
| TPP (daPa) | 50% (49) | 18% (18) | 25% (24) | 7% (7)* |
| Acoustic Reflex Findings (n=22 patients)
| |||
|---|---|---|---|
| WNL | Abnormal | CNT | |
| Acoustic Reflex Thresholds | 9% (2) | 91% (20)** | |
| Acoustic Reflex Adaptation | 27% (6) | 14% (3) | 59% (13) |
Note. WNL, within normal limits; CNT, could not test; Vec, equivalent ear canal volume; TPP, tympanometric peak pressure.
Includes four ears with intact, patent pressure equalization tubes
60% (12/20) cannot be explained by middle ear status or peripheral hearing thresholds.
Acoustic reflex thresholds were abnormal (elevated or absent, Gelfand et al., 1990) in 91% (20/22) of patients in whom these data could be collected. Because of the binaural relationship in contralateral acoustic reflex measurement, and because not every patient was able to cooperate for threshold assessment at every frequency, acoustic reflex abnormalities were analyzed by patient (versus ear) and an abnormality at any test frequency was sufficient for inclusion in this category. Sixty percent (12/20) of these cases could not be explained by either middle ear status or peripheral hearing thresholds. Although acoustic reflex adaptation could only be evaluated in a small number of individuals (n=9), adaptation was abnormal (positive) in three (33%) of these cases.
ABR findings
Baseline neurotologic ABRs were collected in 49 patients (97 ears). Table 3 shows the number of ears with normal and abnormal findings for both absolute and interpeak latencies. Results reflect latencies from the averaged (condensation and rarefaction) responses to ipsilateral stimulation of each ear. The most common ABR abnormalities observed were poor waveform morphology, characterized by absent waves I and III, and prolongation of the interpeak interval between waves I–III and I–V. Thirty-seven of 49 patients (76%) had an ABR abnormality in at least one ear. Of these, nine had normal hearing across test frequencies, 10 had peripheral hearing loss, but ABR abnormalities were disproportionate to the degree of loss (e.g., mild high frequency hearing loss, grossly abnormal waveform morphology), three patients had hearing loss that could account for the ABR abnormalities, and 15 patients had no behavioral peripheral hearing data with which to compare their ABR abnormalities.
Table 3.
Absolute and interpeak latency data for ABRs collected on 49 patients (97 ears) with NPC1
| Absolute Latencies | Interpeak Latencies | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| I | III | V | I–III | III–V | I–V | |
|
|
||||||
| Normal latencies | 60% (58) | 49% (48) | 72% (70) | 70% (37) | 100% (72) | 72% (42) |
| Prolonged Latencies | 0% (0) | 26% (25) | 23% (22) | 30% (16) | 0% (0) | 28% (16) |
| Absent Waves | 40% (39) | 25% (24) | 5% (5) | * | * | * |
An additional salient finding on ABRs collected in this cohort were large and prolonged cochlear microphonics (CM), characterized by sinusoidal activity occurring in the first few milliseconds (e.g., 1–5 ms) of the recording that reversed polarity with changes in stimulus polarity. Electrical artifact was ruled out via a control run during which the same stimuli were delivered and the tubing of the insert earphone was clamped. Of 79 ears in which a CM was visible during the ABR recording, 35 (44%) were prolonged well beyond 1 ms and many of these responses were noted have a large amplitude as well. These 35 cases occurred in 19 individuals (16 bilateral, 3 unilateral).
Site of lesion
When each patient’s collective findings are considered, an individual profile of a cochlear, retrocochlear, or combined site of lesion is available for 41 patients. The remaining 9 patients lacked sufficient data to determine a site of lesion. Results supporting a principally cochlear site of lesion were observed in only 5% (2/41) of patients. Retrocochlear dysfunction, with or without cochlear involvement, was the most common finding identified in 22% (9/41) and 44% (18/41) of patients, respectively. Twelve (29%) patients had audiologic findings consistent with a normal auditory system, based on data that were obtained. Seven of these patients were aged five years or under and, although the combination of limited behavioral and objective assessments predicted normal auditory function, comprehensive examinations were not completed (e.g., acoustic reflex measures) and, consequently, subtle or subclinical auditory dysfunction could not be ruled out completely.
Disease severity and hearing
Figure 3 plots hearing sensitivity at 4k and 8k Hz by total disease severity score for 25 and 22 patients, respectively, for whom these data were available. The threshold of the poorer-hearing ear is plotted, although the effect sizes between these data and mean left/right thresholds are comparable (data not shown). There is a trend for elevated thresholds with higher overall disease severity scores.
Figure 3.

Air-conduction thresholds of the poorer-hearing ear plotted against total disease severity score (Yanjanin et al., 2009) at 4 kHz (top panel) and 8 kHz (bottom panel) for 25 and 22 patients, respectively, for whom these data were available.
Longitudinal data
Longitudinal hearing data were available for 30 patients; 17 had longitudinal pure-tone data and 26 had longitudinal ABR data. The majority of patients in both subsets were followed for two or more years. Young age and progression of disease influenced testing that could be completed on follow-up. Rigorous statistical analysis of the longitudinal data is not possible given the variability in duration of follow-up and type of data collected across individuals
Longitudinal pure-tone findings
Longitudinal pure-tone data for the .5/1/2/4 and 4/8 kHz pure-tone averages, as well as for 8 kHz are plotted in Figure 4. Ears that underwent a clinically significant change in hearing attributable to a concurrent change in middle ear status are not plotted (3 ears). Patients followed longitudinally ranged in age from four to 21 years at the time of their final follow-up audiogram. Minimal significant change in averaged mid-frequency (.5/1/2/4 kHz) hearing is noted (Figure 4, top panel); of 27 ears with data, only three ears from two individuals had a clinically significant (>10 dB) but small (<13 dB) decline in hearing. However, the high-frequency average identifies eight ears (32%, 8/25) from five people that had a clinically significant decline in hearing sensitivity from baseline (Figure 4, middle panel). The largest change in hearing for the high-frequency average (32.5 dB and 27.5 dB for the right and left ears, respectively) occurred in the individual followed for the longest period of time (135 months); she was 21 years at the time of her final audiogram. When only the 8k Hz threshold is considered, seven ears from five people (27%, 7/26) showed clinically significant decline in hearing (Figure 4, bottom panel), with an average change of 31 dB.
Figure 4.

Individual change in air-conduction thresholds from baseline to the final audiologic assessment, as a function of duration of follow-up (months) for a four-frequency (top panel) and high-frequency pure-tone average (PTA) (middle panel), and 8 kHz (bottom panel). Dashed line at 0 dB indicates no change in threshold; positive values depict a decline in threshold and negative values depict an improvement in threshold. Solid lines demarcate the range of test/retest variability standardly associated with pure-tone threshold assessment. Grayed symbols indicate overlapping data points.
Longitudinal ABR findings
Twenty-six patients provided longitudinal ABR data, with an average duration of follow-up of 18 months. Clinically significant change (> 1 ms, e.g., Hood, 1998, Ornitz and Walter, 1975) from baseline was not observed in either ear of any individual for absolute (I, III, V) or interpeak (I-III, III-V, I-V) latency (data not shown). There was no observable trend in the data, in either direction, to suggest an overall pattern of change in the cohort. However, there appeared to be more stability in the response for the absolute latency of waves I and III, and a less stable response (both positive and negative change) for the absolute latency of wave V. A similar pattern was not observed for interpeak latencies.
Ten individuals underwent categorical change in their ABR interpretation, either from a present to absent waveform, or a normal to abnormal latency. The most common finding was a loss (present to absent) of either wave I or wave III, which occurred in 5 and 3 people, respectively, and never in the same individual.
Summary and Discussion
The data presented here represent the largest cohort of patients with NPC1 in whom auditory function has been examined comprehensively. Results of the baseline auditory assessment support an auditory phenotype in humans with NPC1. Data from patients able to participate in behavioral testing reveal a common high frequency peripheral hearing loss (Figure 1, Table 1). Insert earphones were used as the standard for data collection whenever possible, which mitigates the risk that findings represent an artifact from standing waves or collapsing external auditory canals (Gelfand, 2009). These findings corroborate previous reports from a small cohort of patients with NPC in whom high frequency hearing loss was reported (Fink et al., 1989; Pikus, 1991). Additionally, 74% of patients in this cohort in whom behavioral thresholds could be established exhibited hearing loss in the frequencies most critical to speech understanding (.5, 1, 2, 4 kHz). Twenty-five patients were newly identified with hearing loss, which supports the hypothesis that hearing loss is an underestimated component of the NPC1 phenotype. Indeed, early-onset hearing loss in this population may be missed easily, as common screening procedures for hearing in medical offices and academic settings typically do not test above 4000 Hz. Behavioral hearing evaluations with this difficult-to-test population should emphasize collection of high frequency information, when possible, with the knowledge that a complete behavioral audiogram may not be feasible. Furthermore, use of objective measures of hearing sensitivity should be incorporated in the evaluation of these patients and consideration given to the use of sedation, if necessary, to complete data collection. Patients with NPC1 with peripheral hearing loss can benefit from re/habilitation and, therefore, evaluation of auditory function should be a priority.
The tremendous heterogeneity and relatively small size of the sample made identifying contributions of specific independent variables to observed hearing loss challenging. Only eight patients (15%) were able to participate in every component of the test battery. This small number precluded a meaningful multivariate regression analysis. Our cohort contained five biological sibling pairs, for whom data are not independent; however, three of these five pairs did not have behavioral data, which limits the potential underlying effect of this observation. Although gender, age, and use of miglustat were not identified as contributing to the variability in hearing sensitivity among patients with NPC1 in the current study, we acknowledge the small size of our sample as a statistical limitation in this analysis. Furthermore, evaluating age and hearing alone may not be the best way that characterize disease progression given the clinical variability of the disease. There is evidence for an association between hearing and disease severity (Figure 3). The severity scale applied is the most comprehensive rating system developed for use with the NPC1 population, although we acknowledge some intrinsic association between hearing sensitivity and the derived severity score. We chose to plot 4k and 8k Hz data to mitigate this bias as the scale predominantly uses a three frequency pure-tone average to classify sensorineural hearing impairment, and in order to best capture the dominant effect of disease on high frequency hearing. For patients able to participate in behavioral pure-tone audiometry, there is a trend for hearing sensitivity to decline as disease severity increases; however, it is reasonable to anticipate that patients with higher severity scores may be less able to cooperate in behavioral measures of hearing. Twelve of the 19 patients with no behavioral data were very young children (mean age 2.3 years). The remaining seven individuals (mean age 9.3 years) had disease severity scores higher than any patient with behavioral data plotted in figure 3 (mean score 32.6). Those patients who were most affected in the cohort had no behavioral hearing data because of their advanced disease status, which renders a more definitive picture of the relationship between disease severity and hearing incomplete.
Auditory dysfunction associated with NPC1 can occur within the cochlea and neural auditory pathway, which supports the pathogenesis of NPC1 having a widespread and complex role within the auditory system for some affected individuals. While ear-specific bone conduction thresholds were difficult to obtain, middle ear disease was not a prevalent finding (Table 2). Retrocochlear dysfunction with or without evidence for cochlear involvement was observed in 66% of patients with sufficient data to predict site of lesion. Sixty percent of patients tested had abnormal acoustic reflex patterns that could not be explained by the status of the peripheral auditory system, and peripheral hearing thresholds could not account for almost half of the ABR abnormalities observed. Previous investigators have reported a disturbance in the acoustic reflex arc (Fink et al. 1989, Pikus, 1991) and, although descriptions of specific ABR abnormalities in prior reports are limited, our current data are consistent with findings that implicate disruption in early components of the ABR waveform (Aisen et al., 1985; Fink et al., 1989; Palmeri et al., 2005; Pikus, 1991). In addition to absent waves I and III, we observed prolongation of interpeak latencies I–III and I–V. While absence of early waves may be attributed to high frequency peripheral hearing loss, prolongation in interpeak latencies indicates interference in neural transmission. The totality of these findings implicates potential disease-related involvement in cranial nerves VII and VIII, as well as the lower auditory brainstem.
A pattern of disproportionate neural findings that cannot be explained by degree of peripheral hearing loss suggests that some patients with NPC1 fit the diagnostic criteria for auditory neuropathy spectrum disorder (ANSD). Four patients in our cohort clearly meet the criteria for ANSD, which is now widely accepted to encompasses a continuum of clinical findings and a varying range of etiologies (e.g., syndromic and nonsyndromic, neonatal hyperbilirubinemia, idiopathic) that result in a depletion and/or dyssynchrony of afferent auditory nerve fibers. It is important, however, to consider the limitations associated with assigning site of lesion in this population. Because of young age and disease status, patients are only loosely placed in these categories based on interpretation of the testing they were capable of completing. Therefore, the estimation of cochlear and retrocochlear dysfunction, and a concurrent diagnosis of ANSD, may be less than the true prevalence associated with this disease. The clinical implication of this supports conservative management for possible central auditory processing difficulties in these patients, many of whom are unable to self-report difficulty hearing, even if they pass basic screenings for peripheral hearing loss.
Middle ear measures and OAEs can serve as especially useful tools to either screen for peripheral hearing loss or help confirm behavioral test results in a young, neurologically compromised population. Moreover, the ABR is a useful method for monitoring auditory neural status and should be considered for patients with NPC, especially those unable to participate fully in behavioral evaluations. It is important to consider, however, given the evidence that some patients can present with an ANSD-like profile, that electrophysiological estimates of peripheral hearing sensitivity may be inaccurate in some cases (Attias et al., 2006).
The auditory phenotype associated with NPC1 can be progressive, which is supported here by a subset of longitudinal data identifying clinically significant (>10 dB) change in high frequency hearing in some individuals. Of seven patients followed for 23 months or more, six experienced a significant decline in hearing, and the person experiencing the largest deterioration (>27 dB) was followed for the longest period of time (11 years). These data reveal that, like the global neurological phenotype, hearing loss in at least some patients with NPC1 is progressive. This finding supports the importance of regular audiologic monitoring in all patients with NPC1 regardless of a history of normal hearing.
ABR data failed to reveal clinically significant change in wave latency (> 1 ms) for patients followed longitudinally. However, the ABRs of several patients underwent decline in waveform morphology and evidenced categorical change from normal to abnormal. The most common longitudinal change observed across the average duration of follow-up (18 months) was a loss of wave I or III. This suggests that detrimental longitudinal change in ABR morphology may occur in patients with NPC1, although it remains to be seen whether ABR latencies ultimately will show progressive deterioration and, if so, what the time course of the change may be.
The data from this comprehensive study provide a baseline for the natural history of hearing in NPC1, which should be valuable when evaluating the efficacy and toxicity of future therapeutic interventions. 2-Hydroxypropyl-β-Cyclodextrin (HP-β-CD) is being considered for the treatment of NPC1, and has shown promising results in animal models. Weekly administration of HP-β-CD significantly increased the lifespan and ameliorated the cerebellar neurodegeneration in Npc1 mutant mice (Ramirez et al. 2010). However, work with the feline model for NPC1 revealed a dose-dependent effect of HP-β-CD that decline in hearing thresholds in affected cats and normal controls (Ward et al. 2010); those animals treated intrathecally with HP-β-CD (4000 mg/kg brain weight every two weeks) experienced a profound and irreversible elevation in ABR thresholds. Experimental trials with HP-β-CD are being extended to humans with NPC1, and toxicity at this point is unknown. Without natural history data of the auditory phenotype it is impossible to discriminate an expected change in hearing due to disease progression versus a direct adverse event from a drug. The results presented here contribute to addressing this important issue.
In summary, these data confirm that auditory dysfunction is a prevalent finding in NPC1 and that patients with this disease are at risk for developing functionally significant and progressive hearing loss. With over 240 disease-causing mutations associated with NPC1 (Runz, 2009), specific phenotype-genotype correlations in a human population are not possible. It is probable that this molecular heterogeneity contributed to the variability in auditory function we observed. Clinicians and researchers should be aware of the involvement of the auditory system, which historically has been an overlooked component of the disorder. Hearing loss in some patients will affect daily communication, and some individuals with NPC1 in our cohort have successfully benefited from the use of hearing aids, per parent and patient report. An awareness and appreciation of the potential for retrocochlear involvement and possible ANSD should also be considered, as some forms of ANSD do not respond well to conventional amplification.
To elucidate further the pathogenesis of the hearing loss in NPC1 and perhaps understand better the effect of potential treatments, evaluation of the auditory system in animal models of NPC1, including histology of the cochlea and auditory brainstem pathways, is necessary.
Acknowledgments
This work was supported, in part, by a National Institutes of Health (NIH) training grant (T32DC000046) to KAK, and by the intramural divisions of the National Institute on Deafness and Other Communication Disorders and the Eunice Kennedy Shriver National Institute of Child Health and Human Development. Further support came from a Bench-to-Bedside award from the NIH Office of Rare diseases and the NIH Clinical Research Center, and from both the Ara Parseghian Medical Research Foundation and Dana’s Angels Research Trust. The authors are grateful to Lisa Cunningham, Robert Dooling, Tracy Fitzgerald, Andrew Griffith, and Arthur Popper for their insightful feedback on methodology and careful review of the manuscript. We extend our sincere thanks to our participants and their families for their time and energy.
References
- Aisen M, Rapoport S, Solomon G. Brain stem auditory evoked potentials in two siblings with Niemann-Pick disease. Brain Dev. 1985;7(4):431–433. doi: 10.1016/s0387-7604(85)80142-x. [DOI] [PubMed] [Google Scholar]
- ANSI. S3.1-1999 American National Standard Maximum Permissible Ambient Noise Levels for Audiometric Test Rooms. New York: American National Standards Institute; 2010. (Standard S3.1) [Google Scholar]
- Attias J, Buller N, Rubel Y, et al. Multiple auditory steady-state responses in children and adults with normal hearing, sensorineural hearing loss, or auditory neuropathy. Annals of Otology, Rhinology, & Laryngology. 2006;115(4):268–276. doi: 10.1177/000348940611500404. [DOI] [PubMed] [Google Scholar]
- Carstea ED, Morris JA, Coleman KG, et al. Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science. 1997;277(5323):228–231. doi: 10.1126/science.277.5323.228. [DOI] [PubMed] [Google Scholar]
- Clark JG. Uses and abuses of hearing loss classification. American Speech Language Hearing Association. 1981;23:493–500. [PubMed] [Google Scholar]
- Fink JK, Filling-Katz MR, Sokol J, et al. Clinical spectrum of Niemann-Pick disease type C. Neurology. 1989;39(8):1040–1049. doi: 10.1212/wnl.39.8.1040. [DOI] [PubMed] [Google Scholar]
- Garver WS, Francis GA, Jelinek D, et al. The National Niemann-Pick C1 disease database: report of clinical features and health problems. Am J Med Genet A. 2007;143(11):1204–1211. doi: 10.1002/ajmg.a.31735. [DOI] [PubMed] [Google Scholar]
- Gelfand SA. Essentials of Audiology. 3. New York, NY: Thieme Medical Publishers, Inc; 2009. [Google Scholar]
- Gelfand SA, Schwander T, Silman S. Acoustic reflex thresholds in normal and cochlear-impaired ears: Effects of no-response rates on 90th percentiles in a large sample. Journal of Speech and Hearing Disorders. 1990;55:198–205. doi: 10.1044/jshd.5502.198. [DOI] [PubMed] [Google Scholar]
- Hall JW. Test protocols and procedures. In: Hall JW, editor. Handbook of auditory evoked responses. 1. Needham Heights, MA: Allyn and Bacon; 1992. pp. 277–304. [Google Scholar]
- Higgins JJ, Patterson MC, Dambrosia JM, et al. A clinical staging classification for type C Niemann-Pick disease. Neurology. 1992;42(12):2286–2290. doi: 10.1212/wnl.42.12.2286. [DOI] [PubMed] [Google Scholar]
- Hood LJ. Clinical applications of the ABR in neurological testing. In: Hood LJ, editor. Clinical applications of the auditory brainstem response. Clifton Park, NY: Singular Publishing Group; 1998. pp. 67–91. [Google Scholar]
- Ikonen E, Hölttä-Vuori M. Cellular pathology of Niemann-Pick type C disease. Semin Cell Dev Biol. 2004;15(4):445–454. doi: 10.1016/j.semcdb.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Issa A, Ross HF. An improved procedure for assessing ABR latency in young subjects based on a new normative data set. International Journal of Pediatric Otorhinolaryngology. 1995;32:35–47. doi: 10.1016/0165-5876(94)01110-j. [DOI] [PubMed] [Google Scholar]
- Liscum L, Faust JR. Low density lipoprotein (LDL)-mediated suppression of cholesterol synthesis and LDL uptake is defective in Niemann-Pick type C fibroblasts. J Biol Chem. 1987;262(35):17002–17008. [PubMed] [Google Scholar]
- Liscum L, Ruggiero RM, Faust JR. The intracellular transport of low density lipoprotein-derived cholesterol is defective in Niemann-Pick type C fibroblasts. J Cell Biol. 1989;108(5):1625–1636. doi: 10.1083/jcb.108.5.1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis RH, Heller JW. Screening tympanometry: criteria for medical referral. Audiology. 1987;26:197–208. doi: 10.3109/00206098709081549. [DOI] [PubMed] [Google Scholar]
- Naureckiene S, Sleat DE, Lackland H, et al. Identification of HE1 as the second gene of Niemann-Pick C disease. Science. 2000;290(5500):2298–2301. doi: 10.1126/science.290.5500.2298. [DOI] [PubMed] [Google Scholar]
- Ornitz EM, Walter DO. The effect of sound pressure waveform on human brain stem evoked responses. Brain Research. 1975;92:490–498. doi: 10.1016/0006-8993(75)90336-4. [DOI] [PubMed] [Google Scholar]
- Palmeri S, Tarugi P, Sicurelli F, et al. Lung involvement in Niemann-Pick disease type C1: improvement with bronchoalveolar lavage. Neurol Sci. 2005;26(3):171–173. doi: 10.1007/s10072-005-0456-z. [DOI] [PubMed] [Google Scholar]
- Pikus A. Audiologic profile in Niemann-Pick C. Ann N Y Acad Sci. 1991;630:313–314. doi: 10.1111/j.1749-6632.1991.tb19618.x. [DOI] [PubMed] [Google Scholar]
- Ramirez CM, Liu B, Taylor AM, et al. Weekly cyclodextrin administration normalizes cholesterol metabolism in nearly every organ of the Niemann-Pick type C1 mouse and markedly prolongs life. Pediatric Research. 2010;68(4):309–315. doi: 10.1203/PDR.0b013e3181ee4dd2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runz H. Niemann-Pick Type C Disease Gene Variation Database. 2009. [DOI] [PubMed] [Google Scholar]
- Schwartz DM, Pratt RE, Schwartz JA. Auditory brain stem responses in preterm infants: Evidence of peripheral maturity. Ear and Hearing. 1989;10:14–22. doi: 10.1097/00003446-198902000-00003. [DOI] [PubMed] [Google Scholar]
- Vanier MT, Millat G. Niemann-Pick disease type C. Clin Genet. 2003;64(4):269–281. doi: 10.1034/j.1399-0004.2003.00147.x. [DOI] [PubMed] [Google Scholar]
- Vincent I, Bu B, Erickson RP. Understanding Niemann-Pick type C disease: a fat problem. Curr Opin Neurol. 2003;16(2):155–161. doi: 10.1097/01.wco.0000063764.15877.1c. [DOI] [PubMed] [Google Scholar]
- Ward S, O’Donnell P, Fernandez S, et al. 2-Hydroxypropyl-β-Cyclodextrin raises hearing threshold in normal cats and in cats with Niemann-Pick type C disease. Pediatric Research. 2010;68(1):52–56. doi: 10.1203/PDR.0b013e3181df4623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanjanin NM, Vélez JI, Gropman A, et al. Linear clinical progression, independent of age of onset, in Niemann-Pick Disease, Type C. American Journal of Medical Genetics. 2009;5(153B):132–140. doi: 10.1002/ajmg.b.30969. [DOI] [PMC free article] [PubMed] [Google Scholar]