

Abstract
Introduction
O-(2-[18F]fluoroethyl)-L-tyrosine (FET, [18F]1) is a useful amino-acid-based imaging agent for brain tumors. This paper reports the synthesis and evaluation of three FET prodrugs, O-(2-[18F]fluoroethyl)-L-tyrosyl-L-glycine (FET-Gly, [18F]2), O-(2-[18F]fluoroethyl)-L-tyrosyl-L-alanine (FET-Ala, [18F]3) and N-acetyl O-(2-[18F]fluoroethyl)-L-tyrosine (AcFET, [18F]4), which could be readily hydrolyzed to FET in vivo for tumor imaging. We investigated their metabolism in the blood and imaging properties in comparison to FET ([18F]1).
Methods
Three new [18F]FET derivatives, 2 – 4, were prepared from their corresponding tosylate-precursors through nucleophilic fluorination and subsequent deprotection reactions. In vitro uptake studies were carried out in 9L glioma cancer cell lines. In vitro and in vivo hydrolysis studies were conducted to evaluate the hydrolysis of FET prodrugs in blood and in Fisher 344 rats. Biodistribution and PET imaging studies were then performed in rats bearing 9L tumors.
Results
New FET prodrugs were prepared with 3 – 28 % decay corrected radiochemical yields, good enantiomeric purity (> 95 %) and high radiochemical purity (> 95 %). FET-Gly ([18F]2), FET-Ala ([18F]3), and AcFET ([18F]4) exhibited negligible uptake in comparison to the high uptake of FET ([18F]1) in 9L cells. Metabolism studies of FET-Gly ([18F]2), FET-Ala ([18F]3), and AcFET ([18F]4) in rat and human blood showed that FET-Ala ([18F]3) was hydrolyzed to FET ([18F]1) faster than FET-Gly ([18F]2) or AcFET ([18F]4). Most of the FET-Ala (79 %) was converted to FET ([18F]1) within 5 min in blood in vivo. Biodistribution studies demonstrated that FET-Ala ([18F]3) displayed the highest tumor uptake. The tumor-to-background ratios of FET-Ala ([18F]3) and FET ([18F]1) were comparable and appeared to be better than those of FET-Gly ([18F]2) and AcFET ([18F]4). PET imaging studies showed that both FET ([18F]1) and FET-Ala ([18F]3) could visualize tumors effectively, and that they share similar imaging characteristics.
Conclusions
FET-Ala ([18F]3) demonstrated promising properties as a prodrug of FET ([18F]1), which could be used in PET imaging of tumor amino acid metabolism.
Keywords: 18F, FET, PET, Peptide, Prodrug, Imaging agent
1. Introduction
Positron emission tomography (PET) imaging is an important noninvasive technique that facilitates cancer diagnosis and treatment planning. FDG is currently the most available PET imaging agent targeting upregulated glycolysis in tumor cells [1]. There is a growing infrastructure of local radiopharmacies supplying FDG, thus circumventing the need for an on-site cyclotron in nuclear medicine clinics. However, due to a relatively high uptake of FDG by normal brain and inflammatory tissues, FDG/PET is not an ideal tracer for detecting glioblastomas. These require imaging agents that target a different aspect of tumor metabolism.
The continuous growth and proliferation of cancer cells require a high rate of protein synthesis and the increased uptake of amino acids to be used as an energy source, as well as a source of carbon and nitrogen for the synthesis of macromolecules [2]. In mammalian cells, amino acids mainly enter using the amino acid transport systems A (alanine preferring), ASC (alanine-serine-cysteine preferring), and L (leucine preferring) [3]. In particular, the system L subtype LAT1 and system ASC subtype ASCT2, are found to correlate with the progression and prognosis of various cancers, including glioma, breast carcinoma, and hepatoma [4]. This provides a strong basis for developing labeled amino acids as imaging agents.
A number of 11C and 18F labeled amino acids (Fig. 1) have been used as PET agents for imaging tumors and have demonstrated improved imaging results in comparison to FDG, especially for glioblastoma [5, 6]. They include 11C labeled L-[11C]-methionine (MET) [7], which has been widely used, and the 18F labeled non-natural amino acids: L-6-[18F]fluorodopa (FDOPA) [8], L-3-[18F]-α-methyl-L-tyrosine (FMT) [9], O-(2-[18F]fluoroethyl)-L-tyrosine (FET, [18F]1) [10], and Anti-1-amino-3-[18F]fluorocyclobutyl carboxylic acid (FACBC) [11]. FET is one of the most promising and widely evaluated agents for imaging brain tumors. It could play a significant role in diagnosing intracranial tumors, identifying the location and extent of the tumors, guiding biopsies, detecting tumor recurrences, and differentiating recurrences from radionecrosis. [10, 12–15]. It is a promising tracer for routine clinical practice due to several advantages: A) It is an 18F labeled tracer which can be prepared by an efficient radiosynthesis using a nucleophilic reaction with high radiochemical yield. This allows for distribution by radiopharmacies, similar to FDG [16]; B) In contrast to FDG, it is not taken up by inflammatory tissues [17]; C) FET is also metabolically stable in vivo and exhibits excellent uptake kinetics for clinical imaging. It is not incorporated into proteins, and catecholamine metabolism and melanin synthesis do not interfere with FET uptake in tumors. The high uptake of FET ([18F]1) in tumors is closely related to the upregulated activity of sodium-independent amino acid transport system L (LAT) [12, 18–20].
Fig. 1.
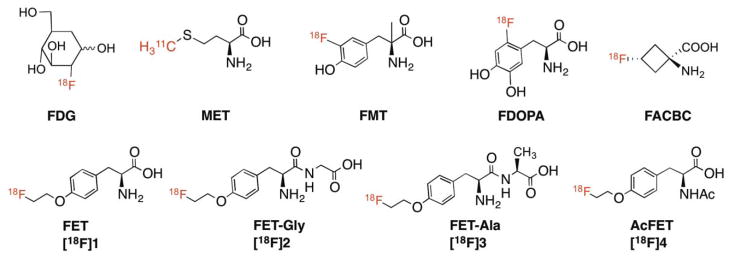
Chemical structures of FDG and amino acid tracers, FET, [18F]1 and its derivatives, FETGly ([18F]2), FET-Ala ([18F]3) and AcFET ([18F]4).
However, the major drawback of FET ([18F]1) is that it is not an FDA approved imaging agent and thus it is not available for routine clinical practice in the United States. Here we propose to prepare and test three FET prodrugs, and when approved by the FDA, they may have a better chance of being developed commercially, thereby benefiting a large number of cancer patients. Using “prodrugs” as imaging agents is not entirely new in the field of radiopharmaceuticals. An early example is the prodrug of FDOPA, 6-[18F]fluoro-O-pivaloyl-L-dopa, which is used for imaging Parkinson’s disease as well as neuroendocrine tumors [21]. The prodrugs were able to overcome the susceptibility of FDOPA to peripheral metabolism, thus improving the accuracy of quantitative studies of presynaptic dopamine metabolism by FDOPA/PET [21, 22]. The esters of [18F]fluoroactetate, which have been prepared and tested in rats, have increased lipophilicity and therefore facilitate penetration through the blood-brain barrier for measuring glia metabolism [23–25]. Reported herein is the synthesis and characterization of three FET dipeptides O-(2-[18F]fluoroethyl)-L-tyrosyl-L-glycine (FET-Gly, [18F]2), O-(2-[18F]fluoroethyl)-L-tyrosyl-Lalanine (FET-Ala, [18F]3), and N-acetyl O-(2-[18F]fluoroethyl)-L-tyrosine (AcFET, [18F]4) as FET prodrugs for targeting brain tumors as shown in Fig. 1. The proposed FET prodrugs could be readily hydrolyzed in vivo by the dipeptidases and acylases in the blood resulting in “free” FET for tumor imaging.
2. Materials and methods
2.1. General
All chemicals were purchased from Aldrich Chemical (St. Louis, MO) or TCI America (Portland, OR). The commercially available materials were used without further purification, unless otherwise indicated. Solvents were dried through a molecular sieve system (Pure Solve Solvent Purification System; Innovative Technology, Inc.). 1H spectra and 13C NMR were recorded by a Bruker DPX spectrometer at 200 MHz and 50 MHz, respectively, and referenced to NMR solvents as indicated. Chemical shifts are reported in ppm (δ), coupling constant J in Hz. Multiplicity is defined by s (singlet), d (doublet), t (triplet), br (broad), or m (multiplet). High-resolution mass spectrometry (HRMS) data were obtained with an Agilent (Santa Clara, CA) G3250AA LC/MSD TOF system. Thin-layer chromatography (TLC) analyses were performed using Merck (Darmstadt, Germany) silica gel 60 F254 plates. Crude compounds generally were purified by the CombiFlash® Rf system (Teledyne Isco Inc, Lincoln, NE) using RediSep Rf silca columns. [18F]Fluoride was purchased from IBA Molecular (Somerset, NJ) as an [18O]enriched aqueous solution of [18F]fluoride. Solid-phase extraction (SPE) cartridges such as Sep-Pak QMA Light and Oasis HLB cartridges were purchased from Waters (Milford, MA). High performance liquid chromatography (HPLC) was performed on an Agilent 1100 series system. [18F]radioactivity was measured by a gamma counter (Cobra II auto-gamma counter D5003 spectrometer, Canberra-Packard) in the 400 – 1600 keV energy range. The animal experiments were carried out in compliance with ethics and animal welfare according to regulation requirements.
2.2. Chemistry
2.2.1. (S)-tert-butyl 2-(2-((tert-butoxycarbonyl)amino)-3-(4-hydroxyphenyl)propanamido)acetate (6a)
To a solution of N-(tert-butoxycarbonyl)-L-tyrosine 5 (844 mg, 3.0 mmol) in THF (30 mL) at 0 °C, glycine t-butyl ester hydrochloride (553 mg, 3.3 mmol), 1-ethyl-3-(3′-dimethylaminopropyl) carbodiimide (EDCI, 422 mg, 2.2 mmol), N-Hydroxybenzotriazole (HOBt, 338, 2 mmol), and N-methylmorpholine (405 mg, 4.0 mmol) were added sequentially. The reaction mixture was kept at 0 °C for 30 min and then stirred overnight at room temperature. Upon completion, the solvent was removed under vacuo. The residue was diluted with ether (50 mL) and washed with 1N HCl (5 mL), 1 N NaHCO3 (5 mL) and then brine (5 mL). The organic layer was dried over Na2SO4. After removal of the solvent, the residue was purified by the CombiFlash®Rf system eluting with Hexane/EtOAc (gradient) to afford the titled compound as a white foam in 78 % yield. [α]24D = +30.3 (c 1.0, CHCl3). 1H NMR (200 MHz, CDCl3) δ = 7.04 (d, 2H, J = 8.4 Hz), 6.73 (d, 2H, J = 8.6 Hz), 6.42 - 6.37 (m, 1H), 5.70 (s, br, 1H), 5.06 (s, br, 1H), 4.37 - 4.34 (m, 1H), 3.98 (dd, 1H, J1 = 18.2 Hz, J2 = 5.2 Hz), 3.81 (dd, 1H, J1 = 18.2 Hz, J2 =4.8 Hz), 3.01 (d, 2H, J = 6.6 Hz), 1.47 (s, 9H), 1.42 (s, 9H). HRMS calcd for C20H30N2O6 ([M+H]+) 395.2182, found 395.2184.
2.2.2. (S)-tert-butyl 2-((S)-2-((tert-butoxycarbonyl)amino)-3-(4-hydroxyphenyl)propanamido) propanoate (6b)
Followed the same procedure as 6a. Yield 72 %. [α]24D = +37.8 (c 1.0, CHCl3). 1H NMR (200 MHz, CDCl3) δ = 7.02 (d, 2H, J = 8.4 Hz), 6.73 (d, 2H, J = 8.4 Hz), 6.60 - 6.50 (m, 1H), 5.07 (s, br, 1H), 4.42 - 4.30 (m, 2H), 2.98 (d, 2H, J = 6.4 Hz), 1.45 (s, 9H), 1.42 (s, 9H), 1.32 (d, 3H, J = 6.8 Hz). HRMS calcd for C21H33N2O6 ([M+H]+) 409.2339, found 409.2330.
2.2.3. (S)-tert-butyl 2-(2-((tert-butoxycarbonyl)amino)-3-(4-(2-(tosyloxy)ethoxy)phenyl) propanamido) acetate (7a)
To the solution of 6a (79 mg, 0.2 mmol) in 2 mL of dry DMF, K2CO3 (83 mg, 0.6 mmol) and 1,2-bis(tosyloxy)ethane (148 mg, 0.4 mmol) were added sequentially. The mixture was stirred at 70 °C for 6 h. After completion of the reaction, the solution was diluted with 40 mL ether, and then washed with 5 mL water and 5 mL brine. The organic layer dried over Na2SO4 and evaporated. The residue was purified via CombiFlash®Rf system eluting with Hexane/EtOAc (gradient) to afford the titled compound 7a as a white solid in 64 % yield. [α]24D = +22.5 (c 1.0, CHCl3). 1H NMR (200 MHz, CDCl3) δ = 7.81 (d, 2H, J = 8.2 Hz), 7.34 (d, 2H, J = 8.0 Hz), 7.08 (d, 2H, J = 8.6 Hz), 6.71 (d, 2H, J = 8.4 Hz), 6.34 - 6.31 (m, 1H), 4.95 (s, br, 1H), 4.37 - 4.32 (m, 2H), 4.13 - 4.08 (m, 2H), 3.96 - 3.76 (m, 2H), 3.10 (d, J = 6.4 Hz), 2.45 (s, 3H), 1.45 (s, 9H), 1.40 (s, 9H). HRMS calcd for C29H40N2O9S ([M+H]+) 593.2533, found 593.2553.
2.2.4. (S)-tert-butyl 2-((S)-2-((tert-butoxycarbonyl)amino)-3-(4-(2-(tosyloxy)ethoxy)phenyl) propanamido) propanoate (7b)
Followed the same procedure as 7a. Yield: 67 %. [α]24D = +28.4 (c 1.0, CHCl3). 1H NMR (200 MHz, CDCl3) δ = 7.82 (d, 2H, J = 8.4 Hz), 7.35 (d, 2H, J = 8.2 Hz), 7.08 (d, 2H, J = 8.6 Hz), 6.71 (d, 2H, J = 8.6 Hz), 6.48 - 6.43 (m, 1H), 4.97 (s, br, 1H), 4.38 - 4.32 (m, 4H), 4.13 - 4.08 (m, 2H), 2.99 (d, J = 6.6 Hz), 2.46 (s, 3H), 1.44 (s, 9H), 1.41 (s, 9H), 1.32 (d, 2H, J = 7.0 Hz). HRMS calcd for C30H43N2O9S ([M+H]+) 607.2689, found 607.2696.
2.2.5. (S)-tert-butyl 2-(2-((tert-butoxycarbonyl)amino)-3-(4-(2-fluoroethoxy)phenyl) propanamido) acetate (8a)
To the solution of 6a (100 mg, 0.256 mmol) dissolved in 2.5 mL DMF, K2CO3 (106 mg, 0.77 mmol) and 1-bromo-2-fluoroethane (65 mg, 0.512 mmol) were added sequentially. The mixture was stirred at 70 °C overnight. After the completion of the reaction, the mixture was diluted with 40 mL of ether and washed with 5 mL water and 5 mL brine. The organic layer was dried over Na2SO4 and the solvent was removed under vacuo. The crude product was purified via CombiFlash®Rf system eluting with Hexane/EtOAc (gradient) to afford the titled compound 8a as white foam in 80 % yield. [α]24D = +25.2 (c 1.0, CHCl3). 1H-NMR (200 MHz, CDCl3): δ = 7.12 (d, 2H, J = 8.6 Hz), 6.85 (d, 2H, J = 8.6 Hz), 6.36 - 6.31 (m, 1H), 4.96 (s, br, 1H), 4.73 (dt, 2H, J1 = 47.4 Hz, J2 = 4.2 Hz), 4.36 - 4.27 (m, 1H), 4.18 (dt, 2H, J1 = 27.8 Hz, J2 =4.2 Hz), 3.99 - 3.74 (m, 2H), 3.02 (d, 2H, J = 6.6 Hz), 1.45 (s, 9H), 1.40 (s, 9H). HRMS calcd for C22H33FN2O6 ([M+H]+) 441.2401, found 441.2387.
2.2.6. (S)-tert-butyl 2-((S)-2-((tert-butoxycarbonyl)amino)-3-(4-(2-fluoroethoxy)phenyl) propanamido propanoate (8b)
Followed the same procedure as 8a. Yield: 72 %. [α]24D = +28.7 (c 1.0, CHCl3). 1H-NMR (200 MHz, CDCl3): δ = 7.13 (d, 2H, J = 8.6 Hz), 6.85 (d, 2H, J = 8.6 Hz), 6.47 - 6.43 (m, 1H), 4.98 (s, br, 1H), 4.75 (dt, 2H, J1 = 47.4 Hz, J2 = 4.0 Hz), 4.41 - 4.33 (m, 2H), 4.18 (dt, 2H, J1 = 27.8 Hz, J2 =4.0 Hz), 3.10 - 2.91 (m, 2H), 1.45 (s, 9H), 1.40 (s, 9H), 1.32 (d, 3H, J = 7.0 Hz). HRMS calcd for C23H36FN2O6 ([M+H]+) 455.2557, found 455.2580.
2.2.7. (S)-2-(2-amino-3-(4-(2-fluoroethoxy)phenyl)propanamido)acetic acid (2, FET-Gly)
Compound 8a (35 mg, 0.08 mmol) was dissolved in 90 % trifluoroacetic acid (TFA) solution (2 mL) containing anisole (0.2 mL). The solution was stirred at room temperature for 4 h. After the reaction was complete, the solution was concentrated under vacuo. The residue was washed with diethyl ether three times to get the final product in 60 % yield. [α]24D = +12.3 (c 0.1, MeOH). 1H NMR (200 MHz, MeOD): δ = 7.23 (d, 2H, J = 8.6 Hz), 6.95 (d, 2H, J = 8.6 Hz), 4.71 (dt, 2H, J1 = 47.6 Hz, J2 = 4.0 Hz), 4.28 (t, 1H, J = 4.0 Hz), 4.16 - 4.04 (m, 2H), 4.00 - 3.95 (m, 2H), 3.21 (dd, 1H, J1 = 14.4 Hz, J2 = 6.2 Hz), 3.00 (dd, 1H, J1 = 14.2 Hz, J2 = 8.0 Hz). 13C NMR (50 MHz, MeOD): δ = 173.7, 166.2, 156.6, 130.5, 129.1, 115.1, 82.8 (d, J = 168.0 Hz), 67.8 (d, J = 19.0 Hz), 53.7, 42.1, 37.8. HRMS calcd for C13H17FN2O4 ([M+H]+) 285.1251, found 285.1268.
2.2.8. (S)-2-((S)-2-amino-3-(4-(2-fluoroethoxy)phenyl)propanamido)propanoic acid (3, FET-Ala)
Followed the same procedure as 2. Yield 36 %. [α]24D = +22.8 (c 0.2, MeOH). 1H NMR (200 MHz, MeOD): δ = 7.26 (d, 2H, J = 8.6 Hz), 6.95 (d, 2H, J = 8.6 Hz), 4.71 (dt, 2H, J1 = 47.8 Hz, J2 = 4.0 Hz), 4.43 (q, 1H, J = 7.4 Hz), 4.21 (dt, 2H, J1 = 28.6 Hz, J2 = 4.0 Hz), 4.06 (dd, 1H, J1 = 8.4 Hz, J2 = 5.4 Hz), 3.24 (dd, 1H, J1 = 14.2 Hz, J2 = 5.6 Hz), 2.96 (dd, 1H, J1 = 14.2 Hz, J2 = 8.4 Hz), 1.44 (d, 3H, J = 7.4 Hz). 13C NMR (50 MHz, MeOD): δ = 171.8, 170.9, 155.7, 130.6, 127.5, 115.8, 82.0 (d, J = 169.5 Hz), 67.3 (d, J = 20 Hz), 55.6, 49.0, 37.8, 18.6. HRMS calcd for C14H20FN2O4 ([M+H]+) 299.1407, found 299.1403.
2.2.9. (S)-tert-butyl 2-acetamido-3-(4-hydroxyphenyl)propanoate (10) [26]
Dropwise was added to a solution of L-tyrosine t-butyl ester 1 (712 mg, 3 mmol) and triethylamine (0.63 mL, 4.5 mmol) in dichloromethane (15 mL) at 0 °C, acetyl chloride (0.23 mL, 3.3 mmol). The reaction mixture was stirred overnight at room temperature. Upon completion, the solvent was removed under vacuo. The residue was diluted with H2O and extracted three times with EtOAc. The organic layer was combined, dried over Na2SO4, and evaporated. The residue was purified using the CombiFlash®Rf system (Hexane/EtOAc, gradient) to afford pure titled compound as clear liquid in 49 % yield. [α]23D = −0.5 (c 1.0, CHCl3). 1H NMR (200 MHz, CDCl3) δ = 7.01 (d, 2H, J = 8.6 Hz), 6.74 (d, 2H, J = 8.6 Hz), 6.10 (d, 1H, J = 8.4 Hz), 4.77 - 4.29 (m, 1H), 3.12 - 2.91 (m, 2H), 1.99 (s, 3H), 1.44 (s, 9H). 13C NMR (50 MHz, CDCl3): 171.5, 170.3, 155.0, 130.2, 126.9, 115.0, 82.2, 53.1, 37.0, 27.3, 22.9. HRMS calcd for C15H22NO4 ([M+H]+) 280.1549, found 280.1497
2.2.10. (S)-tert-butyl 2-acetamido-3-(4-(2-(tosyloxy)ethoxy)phenyl)propanoate (11)
The procedure described above for 7a was employed to convert 10 into tosylate precursor 11. [α]23D = +10.5 (c 1.0, CHCl3). Yield: 62 %. 1H NMR (200 MHz, CDCl3) δ = 7.82 (d, 2H, J = 8.4 Hz), 7.35 (d, 2H, J = 8.2 Hz), 7.03 (d, 2H, J = 8.6 Hz), 6.71 (d, 2H, J = 8.6 Hz), 5.94 (d, 1H, J = 7.6 Hz), 4.76 - 4.65 (m, 1H), 4.38 - 4.32 (m, 2H), 4.15 - 4.09 (m, 2H), 3.02 (d, J = 5.6 Hz), 2.46 (s, 3H), 1.98 (s, 3H), 1.42 (s, 9H). HRMS calcd for C24H32NO7S ([M+H]+) 478.1899, found 478.1917.
2.2.11. (S)-2-acetamido-3-(4-(2-fluoroethoxy)phenyl)propanoic acid (12)
The procedure described above for 8a was employed to convert 10 into tosylate precursor 12. Yield: 57 %. [α]23D = +15.4 (c 1.0, CHCl3). 1H-NMR (200 MHz, CDCl3): δ = 7.07 (d, 2H, J = 8.6 Hz), 6.85 (d, 2H, J = 8.6 Hz), 6.02 - 5.92 (m, 1H), 4.75 (dt, 2H, J1 = 47.8 Hz, J2 = 4.2 Hz), 4.77 - 4.65 (m, 1H), 4.19 (dt, 2H, J1 = 27.8 Hz, J2 =4.2 Hz), 3.03 (d, 2H, J = 5.8 Hz), 1.99 (s, 3H), 1.43 (s, 9H). HRMS calcd for C17H25FNO4 ([M+H]+) 326.1768, found 326.1764.
2.2.12. (S)-2-((S)-2-amino-3-(4-(2-fluoroethoxy)phenyl)propanamido)propanoic acid (4, AcFET)
Followed a similar procedure as used for 2, except for purification of the compound, the CombiFlash®Rf system (MeOH/CH2Cl2, gradient) was used. Yield: 77 %. [α]23D = − 6.2 (c 1.0, MeOH). 1H NMR (200 MHz, MeOD): δ = 7.15 (d, 2H, J = 8.6 Hz), 6.87 (d, 2H, J = 8.6 Hz), 4.81 (t, 1H, J = 4.0 Hz), 4.54 - 4.64 (m, 2H), 4.17 (dt, 2H, J1 = 47.6 Hz, J2 = 4.0 Hz), 3.18 - 2.81 (m, 2H), 1.91 (s, 3H). 13C NMR (50 MHz, MeOD): δ = 174.9, 173.3, 159.1, 131.5, 131.1, 115.7, 83.3 (d, J = 168 Hz), 68.7 (d, J = 19.5 Hz), 55.4, 37.8, 22.5. HRMS calcd for C13H17FNO4 ([M+H]+) 270.1142, found 270.1118.
2.3. Radiosynthesis of FET derivatives
A similar process was applied for the radiolabeling of FET-Gly ([18F]2), FET-Ala ([18F]3), and AcFET ([18F]4). The 18F activity trapped on a Sep-Pak light QMA cartridge (Waters, preconditioned with 10 mL 1 N NaHCO3, 10 mL water and dried with argon) was eluted with 1 mL TBAHCO3 solution (21.5 mg TBAHCO3 in 0.5 mL CH3CN and 0.5 mL H2O) and azeotropically dried twice under argon at 110 °C. Then, 5 mg of precursor was dissolved in 1 mL anhydrous CH3CN and added to the 18F. The fluorination reaction continued for 5 min at 80 °C. Upon completion, the reaction mixture was diluted with 10 mL water, loaded on an HLB oasis cartridge (preconditioned with 10 mL ethanol and 10 mL water) and eluted with 1mL ethanol. The crude intermediates were further purified using a semi-preparative HPLC system on the Phenomenex Gemini C-18 column (10 × 250 mm, 5 μm) with a mobile phase containing CH3CN and 0.1 % formic acid buffer at flow rate of 3mL/min. The desired fraction was concentrated via HLB cartridges and eluted with ethanol. After removal of ethanol, 0.3 mL TFA containing 5 % anisole was added to the purified intermediates. The reaction mixture was kept at room temperature for 10 min. Afterwards, TFA was removed under argon flow. The residue was dissolved in phosphate buffer saline (PBS) and passed through a 0.45 μ filter to give the final labeled product. Chemical and radiochemical purity (RCP) were determined by analytical HPLC performed on a Luna C-18 column (4.6 × 250 mm, 5 μm) with the mobile phase consisting of CH3CN and 0.1 % formic acid at a flow rate of 1 mL/min and TLC (NH4OH/MeOH/CH2Cl2, 1/6/14). Enantiomeric purity was determined via analytical HPLC using Daicel OD-H column (5μ, 250 × 4.6 mm) eluting with hexane and isopropanol at a flow rate of 1 mL/min.
2.4. In vitro uptake studies in 9L cells
Cells from the brain gliosarcoma cell line 9L were purchased from ATCC. The 9L cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10 % fetal bovine serum. Cells were maintained in T-75 culture flasks under humidified incubator conditions (37 °C, 5 % CO2) and were routinely passaged at confluence.
Tumor cells were plated in culturing media at a concentration of 2.0 × 105 cells/well, 24 hours prior to cell uptake studies. On the day of the experiment, the media was aspirated and the cells were washed 3 times with 1 mL of warm PBS (containing 0.90 mM of Ca2+ and 1.05 mM of Mg2+). Each assay condition was performed in triplicate. FET derivatives were mixed within PBS solution and then added to each well (~500,000 cpm/mL/well). The cells were incubated at 37 °C for 5, 30, 60, and 120 minutes. At the end of the incubation period, the wells were aspirated free of ligands and then the residual cells were washed 3 times with 1 mL ice-cold PBS without Ca2+ and Mg2+. After washing, 350 μL 0.1N NaOH was used to lyse the cells. The lysed cells were collected onto filter paper and counted using a gamma counter. 100 μL of the cell lysate was used for the determination of protein concentration by a modified Lowry method. The data was normalized as percentage uptake relative to 100 μg protein content (%uptake/100 μg protein).
2.5. In vitro metabolism studies of FET prodrugs
Rat blood was freshly drawn from anesthetized Fisher 344 rats into heparinized tubes (100 unit/mL). Heparinized human blood samples were provided by Dr. Dan Pryma from the Hospital of the University of Pennsylvania and kept at −70 °C. Incubations were carried out in a 37 °C water bath. The hydrolysis reactions were initiated by adding FET derivatives (~3 MBq) in 50 μL PBS to preheated blood samples (1.0 mL) or PBS (as a control). Aliquots (50 μL) were taken from blood at 5, 10, 15, 30, 60, and 120 min and were deproteinized by mixing with 1 % picric acid (250 μL). After centrifuging for 5 min at 12,000g, the supernatant layer and pellet were counted with a gamma counter. 10 μL of the supernatant layer was then analyzed by analytical HPLC using the Luna C-18 column (250 × 4.6 mm, 5 μ) with CH3CN/0.1 % formic acid (10/90 for FET-Gly and FET-Ala, 25/75 for AcFET) at a flow rate of 1 mL/min. Fractions of radiotracers were collected and counted with a gamma counter for analysis of radio-metabolites. FET derivatives and their radio-metabolites were identified on the basis of retention time. The amount of remaining intact ligands (% residual) was plotted as a function of incubation time for kinetic analysis [22].
2.6. In vivo metabolism studies of FET prodrugs
The FET derivatives (~ 37 MBq) were injected into Fisher 344 rats intravenously. The blood samples were drawn from the heart 5 min after injection of activity. Aliquots (50 μL) of the blood samples were processed and analyzed as described above.
2.7. In vivo biodistribution in Fisher rats with 9L glioma tumors
Fischer 344 rats were purchased from Charles River Laboratories (Malvern, PA). 9L tumor cells (~107 per allograft) in PBS (0.2 mL) were injected subcutaneously into the right thigh of Fischer 344 rats. Rats were monitored on a daily basis. The tumors took 12 – 15 days to reach appropriate size (1 cm diameter). For the biodistribution study, 3 – 6 rats per group were used. All animals were fasted for 12 – 18 hours prior to the study. The rats were then anesthetized (2 – 3 % isoflurane in oxygen) and a 0.25 mL saline solution, containing 925 kBq activity, was injected intravenously. The rats were sacrificed at selected time points post-injection by cardiac excision while under isoflurane anesthesia. The organs of interest were removed, weighed and the radioactivity was counted with a gamma counter. The percent dose per gram was calculated by a comparison of the counts of tissue activity to the counts of 1.0 % of the initial dose. Results were expressed as the percentage of the injected dose per gram (% dose/gram) of tissue. Each value represents the mean SD of six rats unless otherwise noted.
2.8. Small animal imaging with a microPET
Dynamic small animal PET imaging studies were conducted on Fisher rats bearing 9L tumor models. PET imaging studies were performed on a Phillips Mosaic small animal PET scanner, which has an imaging field of view of 11.5 cm. Under isoflurane anesthesia (1 – 2 %, 1 L/min oxygen), the tumor-bearing F344 rats were injected with 19 – 37 MBq activity by an intravenous injection into the lateral tail vein. Data acquisition began immediately following the injection. Dynamic scans were conducted over a period of 2 h (5 min/frame; image voxel size 0.5 mm3). Rats were visually monitored for breathing, and a heating pad was used to maintain body temperature throughout the entire procedure. Images were reconstructed and a region of interest (ROI) analysis was performed using AMIDE software (http://amide.sourceforge.net/).
3. Results
3.1. Chemistry
An efficient synthesis of FET dipeptides 2 and 3 was carried out as shown in scheme 1. Boc-L-tyrosine 5 was conjugated with either L-glycine or alanine to give dipeptide intermediates [27]. The precursors could be obtained from 6a (or 6b) via alkylation with ethylene glycol ditosylate. The final products were synthesized through alkylation and deprotection in TFA. Synthesis of N-Acetyl derivative 4 (as shown in scheme 2) is similar to FET dipeptides, except for the first step—acylation of L-tyrosine t-butyl ester.
Scheme 1.
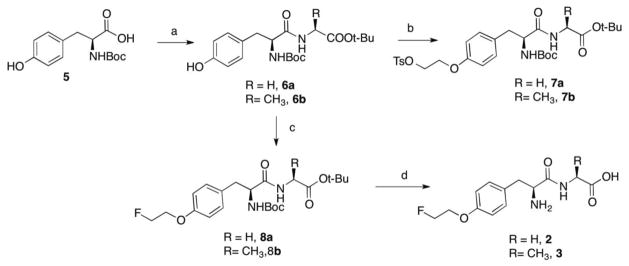
Reagents and conditions: a) L-glycine (or L-Alanine), N-Hydroxybenzotriazole, N-Methyl morpholine, EDCI, THF, rt, overnight, 72 – 78 %; b) TsO(CH2)2OTs, K2CO3, DMF, 70 °C, 6 h, 64 – 67 %; c) K2CO3, DMF, FCH2CH2Br, 70 °C, overnight, 71 – 80 %; d) TFA/anisole, rt, 4 h, 36 – 60 %.
Scheme 2.
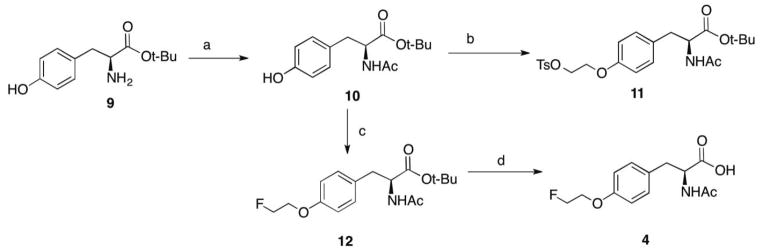
Reagents and conditions: a) CH3COCl, Et3N, CH2Cl2, 0 °C to rt, overnight, 49 %; b) TsO(CH2)2OTs, K2CO3, DMF, 70 °C, 6 h, 62 %; c) K2CO3, DMF, FCH2CH2Br, 70 °C, 5h, 57 %; d) TFA/anisole, rt, 4 h, 77 %.
3.2. Radiosynthesis of FET prodrugs
Radiolabeling of the three FET prodrugs was carried out by a two-step reaction as shown in scheme 3. The first step was a no-carrier-added (NCA) nucleophilic fluorination with dried TBA18F and TBAHCO3 in acetonitrile. The 18F labeled intermediate was then subjected to semipreparative HPLC purification. The second step was the removal of protecting groups in TFA. Enantiomerically pure (>95 %) ligands could be prepared in 70 min with good radiochemical purity (>95 %) as measured by analytical HPLC and TLC. Decay corrected radiochemical yields of FET-Gly ([18F]2), FET-Ala ([18F]3), and AcFET ([18F]4) were 3.3 ± 0.4 (n = 6), 7.7 ± 2.0 (n = 6), 28.0 ± 5.3 (n = 4).
Scheme 3.
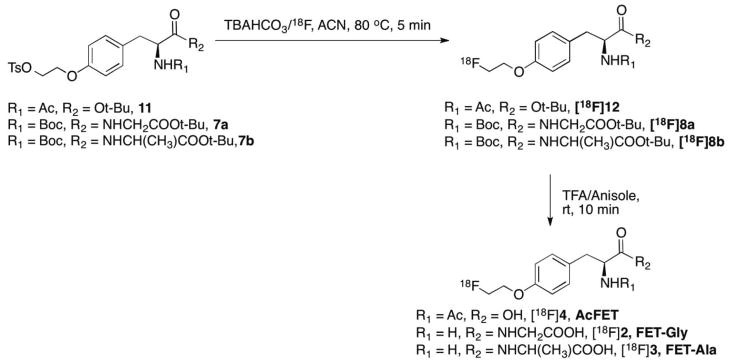
Radiosynthesis of FET-Gly ([18F]2), FET-Ala ([18F]3), and AcFET ([18F]4).
3.3. Cell uptake studies
Time dependent uptake of FET-Gly ([18F]2), FET-Ala ([18F]3), and AcFET ([18F]4) in comparison to FET ([18F]1) in 9L glioma cells is shown in Fig. 2. Uptake of FET-Gly, FET-Ala, and AcFET is less than 1 % than that of FET. This indicates these FET prodrugs could not enter 9L tumor cells in their native forms. Cells can take up these prodrugs only after being hydrolyzed to FET.
Fig. 2.
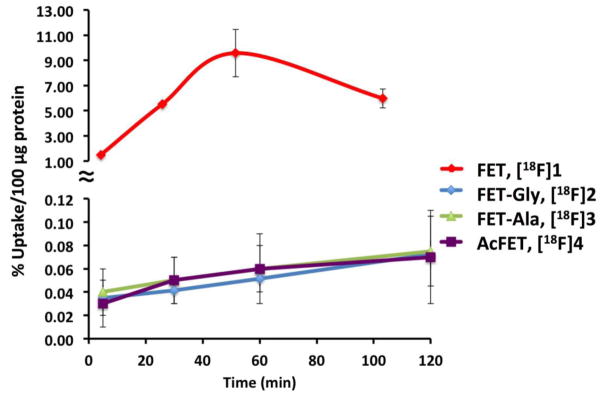
Time-dependent uptake of FET, [18F]1 and its derivatives, FET-Gly ([18F]2), FET-Ala ([18F]3) and AcFET ([18F]4), in 9L glioma cells were measured. They are expressed as percentage of uptake in 100 μg protein (mean ± SD, n = 3). The cell uptake for FET, [18F]1, was much higher than its derivatives. It is likely that the amino acid transporter on the tumor cell surface would only recognize the FET, [18F]1; while all of the derivatives displayed low cell uptake.
3.4. In vitro and in vivo metabolism studies of FET derivatives
The in vitro metabolism studies were carried out at 37 °C in rat and human blood. The metabolites at selected time points were characterized by using HPLC. There were no radio-metabolites observed other than FET and the native ligands. The kinetics of hydrolysis were determined and the first-order kinetic plots for hydrolysis of the FET prodrugs are shown in Fig. 3. The half-lives of these ligands are shown in table 1. The results indicate that AcFET ([18F]4) is the most stable ligand. It underwent hydrolysis in rat blood, but remained completely stable in human blood. This suggests the lack of the corresponding amino acid deacylase in human blood. FET-Ala ([18F]3) had the fastest hydrolysis rate with half-lives of 5.8 min and 0.8 min in rat and human blood, respectively. Although FET-Gly is not as fast as FET-Ala, it could still be readily metabolized to FET with half-lives of 17.3 and 22.7 min, respectively.
Fig. 3.
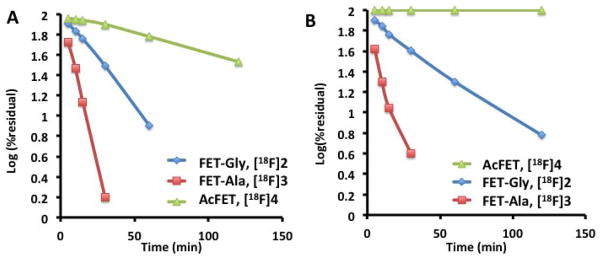
First-order kinetic plots were obtained in vitro for hydrolysis of FET prodrugs in rat (A) and human (B) blood at 37 °C. It was clearly demonstrated that FET-Gly ([18F]2) and FET-Ala ([18F]3) showed excellent in vitro hydrolysis – releasing FET ([18F]1) rapidly. However, the Nacetyl derivative of FET, AcFET ([18F]4), exhibited a very slow rate.
Table 1.
*Rate constant for the hydrolysis of FET prodrugs in rat and human blood at 37 °C
| Compound | t1/2 (min) | |
|---|---|---|
| Rat blood | Human blood | |
| FET-Gly ([18F]2) | 17.3 | 22.7 |
| FET-Ala ([18F]3) | 5.8 | 0.8 |
| AcFET ([18F]4) | 77.5 | n/a |
Data obtained from average of two experiments
In vivo hydrolysis studies in Fisher rats demonstrated that 79 % of FET-Ala ([18F]3), 72 % of FET-Gly ([18F]2), and 66 % of AcFET ([18F]4) was metabolized to FET at 5 min. This indicated that the hydrolysis rate of FET-Ala ([18F]3) was faster than that of FET-Gly ([18F]2) and AcFET ([18F]4), which is consistent with the in vitro results.
3.5. Biodistribution studies
The in vivo biodistribution data of FET-Gly ([18F]2), FET-Ala ([18F]3), and AcFET ([18F]4) at 30 and 60 min after intravenous injection in Fisher rats bearing 9L tumors is presented in Table 2. No considerable changes were observed from 30 min to 60 min in the blood, heart, lung, kidney, spleen, liver, and bone. Relatively low bone uptake indicates that there was no significant defluorination. The highest uptake of all three ligands was in the pancreas, which is consistent with the action of FET and other reported amino acid tracers [28, 29]. The uptake of the FET prodrugs in muscle and brain increased from 30 to 60 min.
Table 2.
Tissue distribution (% ID/g) of radioactivity in Fisher 344 rats bearing 9L tumors models after intravenous injection of FET prodrugs (three to six rats per time point).
| Organ | FET-Gly ([18F]2) | FET-Ala([18F]3) | AcFET([18F]4) | |||
|---|---|---|---|---|---|---|
|
| ||||||
| 30 min | 60 min | 30 min | 60 min | 30 min | 60 min | |
| Blood | 0.72 ± 0.02 | 0.74 ± 0.06 | 0.72 ± 0.03 | 0.72 ± 0.03 | 0.69 ± 0.03 | 0.62 ± 0.01 |
| Heart | 0.67 ± 0.03 | 0.68 ± 0.05 | 0.67 ± 0.03 | 0.66 ± 0.03 | 0.60 ± 0.03 | 0.56 ± 0.02 |
| Muscle | 0.52 ± 0.04 | 0.67 ± 0.06 | 0.54 ± 0.01 | 0.60 ± 0.03 | 0.45 ± 0.02 | 0.49 ± 0.01 |
| Lung | 0.59 ± 0.01 | 0.62 ± 0.06 | 0.62 ± 0.02 | 0.61 ± 0.03 | 0.55 ± 0.04 | 0.51 ± 0.01 |
| Kidney | 0.57 ± 0.01 | 0.57 ± 0.05 | 0.58 ± 0.02 | 0.57 ± 0.02 | 0.70 ± 0.07 | 0.57 ± 0.05 |
| Pancreas | 2.74 ± 0.39 | 2.92 ± 0.44 | 3.06 ± 0.38 | 3.52 ± 0.26 | 2.54 ± 0.11 | 2.23 ± 0.51 |
| Spleen | 0.76 ± 0.05 | 0.80 ± 0.12 | 0.74 ± 0.06 | 0.71 ± 0.03 | 0.66 ± 0.02 | 0.65 ± 0.06 |
| Liver | 0.67 ± 0.02 | 0.66 ± 0.05 | 0.66 ± 0.02 | 0.65 ± 0.02 | 0.64 ± 0.01 | 0.56 ± 0.01 |
| Skin | 0.44 ± 0.03 | 0.46 ± 0.01 | 0.69 ± 0.20 | 0.73 ± 0.16 | 0.48 ± 0.01 | 0.44 ± 0.01 |
| Brain | 0.33 ± 0.02 | 0.47 ± 0.06 | 0.31 ± 0.01 | 0.38 ± 0.02 | 0.26 ± 0.01 | 0.36 ± 0.03 |
| Bone | 0.41 ± 0.01 | 0.45 ± 0.04 | 0.41 ± 0.03 | 0.46 ± 0.03 | 0.32 ± 0.01 | 0.33 ± 0.01 |
| Tumor | 0.81 ± 0.30 | 0.56 ± 0.33 | 1.18 ± 0.10 | 1.10 ± 0.29 | 0.61 ± 0.13 | 0.85 ± 0.09 |
|
| ||||||
| Tumor/Muscle | 1.56 ± 0.59 | 0.84 ± 0.50 | 2.18 ± 0.19 | 1.83 ± 0.49 | 1.36 ± 0.30 | 1.73 ± 0.19 |
| Tumor/Blood | 1.13 ± 0.42 | 0.76 ± 0.45 | 1.64 ± 0.15 | 1.53 ± 0.41 | 0.88 ± 0.19 | 1.37 ± 0.15 |
| Tumor/Brain | 2.45 ± 0.92 | 1.19 ± 0.72 | 3.81 ± 0.35 | 2.89 ± 0.78 | 2.35 ± 0.51 | 2.36 ± 0.32 |
Uptake of FET-Gly ([18F]2) and FET-Ala ([18F]3) in the tumors was fast and reached maximum within 30 min. While FET-Ala ([18F]3) had long retention in 9L tumors, FET-Gly ([18F]2) exhibited a pronounced washout of 31 % at 60 min. In contrast, AcFET ([18F]4) showed slower uptake kinetics. Its uptake increased from 0.61 % ID/g at 30 min to 0.85 % ID/g at 60 min. FET-Ala ([18F]3) had the highest tumor uptake (1.18 % ID/g) among the three FET prodrugs. Except for the pancreas, the uptake of FET-Ala ([18F]3) in 9L tumors was higher than in normal organs and tissues. Tumor uptake and tumor-to-background ratios of FET-Ala ([18F]3) are comparable to those reported for FET in the same tumor model [29]. At the 30 minute time point, tumor-to-muscle, tumor-to-blood and tumor-to-brain ratios of FET-Ala ([18F]3) were 2.2, 1.6 and 3.8, respectively.
3.6. Small animal imaging studies
Dynamic small animal PET (APET) imaging studies of FET prodrugs were performed on rats bearing the 9L tumor model and compared with images done with FET ([18F]1) and FDG (Fig. 4). To assess their kinetics, regions of interest (such as tumor and muscle) were drawn to generate time-activity curves (Fig. 5). The tumors could be visualized with FET ([18F]1) and its prodrugs. There is no apparent bone uptake. Kinetic analysis showed that the tumor uptake of FET-Gly ([18F]2), FET-Ala ([18F]3), and ([18F]1) quickly reached a maximum within 20 min. ([18F]1) and FET-Ala ([18F]3) exhibited a long retention in the 9L tumors, with uptake remaining more or less constant after 20 min. In contrast, FET-Gly showed washout in the same time period. AcFET ([18F]4) had slower kinetics; its tumor uptake kept increasing until 60 min. The kinetics of FET and its derivatives is quite different from that of FDG, which had a continuously increasing tumor uptake due to the metabolic trapping of FDG in cancer cells [30]. FET ([18F]1), FET-Ala ([18F]3), and FET-Gly ([18F]2) all had a similar maximum tumor-to-muscle ratio of 5, which is higher than that of AcFET ([18F]4). These results are consistent with the biodistribution data.
Fig. 4.

Small animal PET images of FET-Gly ([18F]2, (A)), FET-Ala ([18F]3, (B)), AcFET ([18F]4, (C)), FET ([18F]1, (D)), and [18F]FDG (E) in tumor bearing rats. Color-coded PET images from summed 2 h data are shown in transverse, coronal, and sagittal views from left to right following the injection of PET tracers. The arrowhead points to the 9L tumors.
Fig. 5.
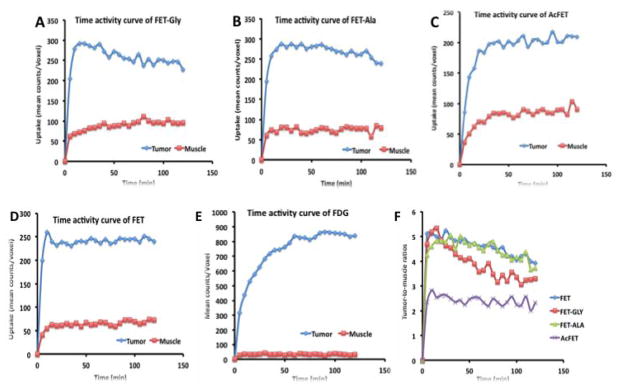
Time activity curves of FET-Gly([18F]2, (A)), FET-Ala ([18F]3, (B)), AcFET ([18F]3, (C)), FET ([18F]1, (D)), and [18F]FDG (E) in rats bearing 9L tumors. The comparison of tumorto-muscle ratios of FET and its derivatives is in the panel (F).
4. Discussion
Commonly used FDG/PET studies are designed for tumor detection based on the fact that tumor tissues often exhibit a higher glucose metabolism. Since the normal brain already actively metabolizes glucose, FDG/PET is not useful for imaging glioblastoma because of the high background activity in the brain. Therefore, alternative tracers are needed because they show high tumor uptake and can distinguish glioma from normal brain tissue. [15]. FET ([18F]1) is one of the amino acid based imaging agents that has shown promising results in brain tumor imaging studies [12, 31].
In this study, the dipeptides FET-Gly ([18F]2), FET-Ala ([18F]3), and the N-acetyl derivative AcFET ([18F]4) were evaluated as prodrugs of FET. These prodrugs could be readily hydrolyzed in vivo and release FET, ([18F]1). These prodrugs were prepared with a similar labeling procedure as used for FET [32]. Attempts have been made to synthesize L-alanyl-O-(2-[18F]fluoroethyl)-L-tyrosine and L-glycyl-O-(2-[18F]fluoroethyl)-L-tyrosine. However, under the same fluorination conditions as described before, a significant amount of isomerization occurred. The radiochemical yield of dipeptides FET-Gly ([18F]2) and FET-Ala ([18F]3) is significantly lower than that of AcFET ([18F]4), which is comparable to FET ([18F]1). This is most likely due to the instability of the peptide bond under the harsh nucleophilic fluorination conditions. Additional optimization of the radiolabeling will be needed. One widely adopted alternative strategy for labeling peptides is to use a prosthetic group, which allows efficient and site-specific labeling of peptides with 18F [33]. Indirect labeling via prosthetic groups, such as 2-[18F]fluoroethyl bromide, may improve the radiochemical yields of FET-Gly ([18F]2) and FET-Ala ([18F]3).
Hydrolysis studies of the three FET prodrugs in rat and human blood demonstrated that FET-Ala ([18F]3) had the shortest half-life, which made it most similar to FET. This is not surprising since the rate of dipeptide hydrolysis is generally far greater for the dipeptides containing an alanyl rather than a glycyl sub-unit [34]. The N-AcFET ([18F]4) was likely hydrolyzed by aminoacylase, which converts N-acyl-L-amino acids to L-amino acids. The aminoacylase-1/metallopeptidas 20 (Acy1/M20) family is of particular interest to this project. It is divided into two functional subclasses. One subclass features degradative dipeptidases and exopeptidases (EC 3.4), while the other comprises of various amidohydrolases acting on carbon–nitrogen bonds rather than peptide bonds (EC 3.5) (2, 3). [35, 36]. Mammalian Acy1, in particular, has been applied in racemic resolution and reverse hydrolysis. Although AcFET ([18F]4) could be metabolized to FET ([18F]1) in rats, it remained stable in human blood. The in vivo degradation reaction rate for releasing FET is far slower than the rates of FET-Gly ([18F]2) and FET-Ala ([18F]3). We conclude that AcFET ([18F]4) is not suitable as a prodrug for PET imaging.
Consistent with the results of the hydrolysis studies, the biodistribution and imaging studies demonstrated that FET-Ala ([18F]3) and FET ([18F]1) were comparable. Since FET-Gly ([18F]2), FET-Ala ([18F]3), and AcFET ([18F]4) cannot enter into 9L cells, their uptake in the implanted 9L tumors is the result of accumulation of hydrolyzed product FET ([18F]1). When one of the FET prodrugs is injected via intravenous injection, it enters the bloodstream where it is hydrolyzed to FET ([18F]1). Cells then take them up, likely with system L amino acid transporters, which are broadly expressed in both tumor cells and normal tissues [37]. FET prodrugs may also enter into cells in their native forms through peptide transporters or amino acid transporters that could accept N-acetyl derivatives. Proton-coupled peptide transporters (PEPTs) PEPT1 and PETP2, which belong to the proton-coupled peptide transporter SLC15A family, transport di/tripeptides across the cell membrane [38]. The expression of these transporters is mostly restricted to intestinal and renal brush-order cells [39]. The T-type amino acid transporter 1 (TAT1) is reported to tolerate N-acetyl aromatic amino acid derivatives including N-acetyl tyrosine and phenylalanine [40]. The expression of TAT1 is mostly limited to the colon, liver, and placenta [40]. These transporters could potentially affect the absorption and excretion of the FET prodrugs. The data presented in this paper suggest that FET-Ala ([18F]3) displayed the shortest half-life in blood. Therefore, it is the most efficient derivative in delivering FET ([18F]1) to the tumor tissue.
5. Conclusion
In summary, we have synthesized three new FET derivatives and evaluated their potential as prodrugs of FET. Of these ligands, FET-Ala ([18F]3) has the fastest hydrolysis rate in the blood. In vivo biodistribution and PET imaging studies of xenographed 9L tumors in rats showed that the tumor uptake and washout of FET-Ala ([18F]3) was comparable to FET ([18F]1). FET-Ala ([18F]3) demonstrated prominent and selective tumor uptake in vivo and shows promise as a FET prodrug.
Acknowledgments
This work was supported in part by grants from Stand-Up 2 Cancer (SU2C), PA Health Department, and National Institutes of Health (CA-164490).
Abbreviations
- ASC
alanine-serine-cysteine preferring amino acid transporter system
- ASCT2
system ASC transporter subtype 2
- BCH
2-amino-bicylo[2.2.1]heptane-2-carboxylic acid
- FACBC
3-[18F]fluoro-cyclobutyl-1-carboxylic acid
- FC
flash chromatography
- FDG
2-[18F]fluoro-2-deoxy-D-glucose
- FDOPA
6-[18F]fluoro-3,4-dihydroxyphenylalanine
- FET
O-(2-[18F]fluoroethyl)-L-tyrosine
- FET-Ala
O-(2-[18F]fluoroethyl)-L-tyrosyl-L-alanine
- FET-Gly
O-(2-[18F]fluoroethyl)-L-tyrosyl-L-glycine
- AcFET
N-acetyl O-(2-[18F]fluoroethyl)-L-tyrosine
- HPLC
High performance liquid chromatography
- HRMS
High-resolution mass spectrometry
- IMT
3-[123I]Iodo-alpha-methyl-L-tyrosine
- PBS
phosphate buffered saline
- PEPTs
peptide transporters
- SPE
solid-phase extraction
- TFA
trifluoroacetic acid
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Haberkorn U, Ziegler SI, Oberdorfer F, Trojan H, Haag D, Peschke P, et al. FDG uptake, tumor proliferation and expression of glycolysis associated genes in animal tumor models. Nucl Med Bio. 1994;21:827–834. doi: 10.1016/0969-8051(94)90162-7. [DOI] [PubMed] [Google Scholar]
- 2.Ganapathy V, Thangaraju M, Prasad PD. Nutrient transporters in cancer: Relevance to Warburg hypothesis and beyond. Pharmacol Thera. 2008 doi: 10.1016/j.pharmthera.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 3.Palacin M, Estevez R, Bertran J, Zorzano A. Molecular biology of mammalian plasma membrane amino acid transporters. Physiol Rev. 1998;78:969–1054. doi: 10.1152/physrev.1998.78.4.969. [DOI] [PubMed] [Google Scholar]
- 4.McConathy J, Yu W, Jarkas N, Seo W, Schuster DM, Goodman MM. Radiohalogenated nonnatural amino acids as PET and SPECT tumor imaging agents. Med Res Rev. 2011 doi: 10.1002/med.20250. published online July 26. [DOI] [PubMed] [Google Scholar]
- 5.Petrirena GJ, Goldman S, Delattre JY. Advances in PET imaging of brain tumors: a referring physician’s perspective. Curr Opin Oncol. 2011;23:617–623. doi: 10.1097/CCO.0b013e32834aa752. [DOI] [PubMed] [Google Scholar]
- 6.Heiss WD, Raab P, Lanfermann H. Multimodality assessment of brain tumors and tumor recurrence. J Nucl Med. 2011;52:1585–1600. doi: 10.2967/jnumed.110.084210. [DOI] [PubMed] [Google Scholar]
- 7.Singhal T, Narayanan TK, Jain V, Mukherjee J, Mantil J. 11C-L-methionine positron emission tomography in the clinical management of cerebral gliomas. Mol Imag Biol. 2008;10:1–18. doi: 10.1007/s11307-007-0115-2. [DOI] [PubMed] [Google Scholar]
- 8.Minn H, Kauhanen S, Seppanen M, Nuutila P. 18F-FDOPA: a multiple-target molecule. J Nucl Med. 2009;50:1915–1918. doi: 10.2967/jnumed.109.065664. [DOI] [PubMed] [Google Scholar]
- 9.Kaira K, Oriuchi N, Shimizu K, Tominaga H, Yanagitani N, Sunaga N, et al. 18F-FMT uptake seen within primary cancer on PET helps predict outcome of non-small cell lung cancer. J Nucl Med. 2009;50:1770–1776. doi: 10.2967/jnumed.109.066837. [DOI] [PubMed] [Google Scholar]
- 10.Wester H, Herz M, Weber W, Heiss P, Senekowitsch-Schmidtke R, Schwaiger M, et al. Synthesis and radiopharmacology of O-(2-[18F]fluoroethyl)-L-tyrosine for tumor imaging. J Nucl Med. 1999;40:205–212. [PubMed] [Google Scholar]
- 11.Yu W, Williams L, Camp V, Olson J, Goodman M. Synthesis and biological evaluation of anti-1-amino-2-[18F]fluoro-cyclobutyl-1-carboxylic acid (anti-2-[18F]FACBC) in rat 9L gliosarcoma. Bioorg Med Chem Lett. 2010;20:2140–2143. doi: 10.1016/j.bmcl.2010.02.048. [DOI] [PubMed] [Google Scholar]
- 12.Langen KJ, Hamacher K, Weckesser M, Floeth F, Stoffels G, Bauer D, et al. O-(2-[18F]fluoroethyl)-L-tyrosine: uptake mechanisms and clinical applications. Nucl Med Bio. 2006;33:287–294. doi: 10.1016/j.nucmedbio.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 13.Piroth MD, Pinkawa M, Holy R, Klotz J, Nussen S, Stoffels G, et al. Prognostic value of early [18F]fluoroethyltyrosine positron emission tomography after radiochemotherapy in glioblastoma multiforme. Int J Radiat Oncol Biol Phys. 2011;80:176–184. doi: 10.1016/j.ijrobp.2010.01.055. [DOI] [PubMed] [Google Scholar]
- 14.Piroth MD, Holy R, Pinkawa M, Stoffels G, Kaiser HJ, Galldiks N, et al. Prognostic impact of postoperative, pre-irradiation 18F-fluoroethyl-l-tyrosine uptake in glioblastoma patients treated with radiochemotherapy. Radiother Oncol. 2011;99:218–224. doi: 10.1016/j.radonc.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 15.Klasner BD, Krause BJ, Beer AJ, Drzezga A. PET imaging of gliomas using novel tracers: a sleeping beauty waiting to be kissed. Expert Rev Anticancer Ther. 2010;10:609–613. doi: 10.1586/era.10.37. [DOI] [PubMed] [Google Scholar]
- 16.Mueller D, Klette I, Kalb F, Baum RP. Synthesis of O-(2-[18F]fluoroethyl)-L-tyrosine based on a cartridge purification method. Nucl Med Biol. 2011;38:653–658. doi: 10.1016/j.nucmedbio.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 17.Lee TS, Ahn SH, Moon BS, Chun KS, Kang JH, Cheon GJ, et al. Comparison of 18F-FDG, 18F-FET and 18F-FLT for differentiation between tumor and inflammation in rats. Nucl Med Biol. 2009;36:681–686. doi: 10.1016/j.nucmedbio.2009.03.009. [DOI] [PubMed] [Google Scholar]
- 18.Rau FC, Weber WA, Wester HJ, Herz M, Becker I, Kruger A, et al. O-(2-[18F]Fluoroethyl)-L-tyrosine (FET): a tracer for differentiation of tumour from inflammation in murine lymph nodes. Eur J Nucl Med Mol Imaging. 2002;29:1039–1046. doi: 10.1007/s00259-002-0821-6. [DOI] [PubMed] [Google Scholar]
- 19.Pauleit D, Floeth F, Tellmann L, Hamacher K, Hautzel H, Muller HW, et al. Comparison of O-(2-18F-fluoroethyl)-L-tyrosine PET and 3-123I-iodo-alpha-methyl-L-tyrosine SPECT in brain tumors. J Nucl Med. 2004;45:374–381. [PubMed] [Google Scholar]
- 20.Stadlbauer A, Prante O, Nimsky C, Salomonowitz E, Buchfelder M, Kuwert T, et al. Metabolic imaging of cerebral gliomas: spatial correlation of changes in O-(2-18F-fluoroethyl)-L-tyrosine PET and proton magnetic resonance spectroscopic imaging. J Nucl Med. 2008;49:721–729. doi: 10.2967/jnumed.107.049213. [DOI] [PubMed] [Google Scholar]
- 21.Ishiwata K, Shinoda M, Ishii S, Nozaki T, Senda M. Synthesis and evaluation of an 18F-labeled dopa prodrug as a PET tracer for studying brain dopamine metabolism. Nucl Med Bio. 1996;23:295–301. doi: 10.1016/0969-8051(95)02083-7. [DOI] [PubMed] [Google Scholar]
- 22.Giorgioni G, Claudi F, Ruggieri S, Ricciutelli M, Palmieri GF, Di Stefano A, et al. Design, synthesis, and preliminary pharmacological evaluation of new imidazolinones as L-DOPA prodrugs. Bioorg Med Chem. 2010;18:1834–1843. doi: 10.1016/j.bmc.2010.01.041. [DOI] [PubMed] [Google Scholar]
- 23.Inoue O, Hosoi R, Momosaki S, Yamamoto K, Amitani M, Yamaguchi M, et al. Evaluation of [14C]phenylacetate as a prototype tracer for the measurement of glial metabolism in the rat brain. Nucl Med Biol. 2006;33:985–989. doi: 10.1016/j.nucmedbio.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 24.Momosaki S, Hosoi R, Sanuki T, Todoroki K, Yamaguchi M, Gee A, et al. [14C]Benzyl acetate is a potential radiotracer for the measurement of glial metabolism in the rat brain. Nucl Med Biol. 2007;34:939–944. doi: 10.1016/j.nucmedbio.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 25.Mori T, Sun L, Kobayashi M, Kiyono Y, Okazawa H, Furukawa T, et al. Preparation and evaluation of ethyl [18F]fluoroacetate as a proradiotracer of [18F]fluoroacetate for the measurement of glial metabolism by PET. Nucl Med Bio. 2009;36:155–162. doi: 10.1016/j.nucmedbio.2008.11.006. [DOI] [PubMed] [Google Scholar]
- 26.del Amo V, McGlone AP, Soriano JM, Davis AP. Two-colour screening in combinatorial chemistry: prospecting for enantioselectivity in a library of steroid-based receptors. Tetrahedron. 2009;65:6370–6381. [Google Scholar]
- 27.Billing JF, Nilsson UJ. Cyclic peptides containing a delta-sugar amino acid-synthesis and evaluation as artificial receptors. Tetrahedron. 2005;61:863–874. [Google Scholar]
- 28.Martarello L, McConathy J, Camp VM, Malveaux EJ, Simpson NE, Simpson CP, et al. Synthesis of syn- and anti-1-amino-3-[18F]fluoromethyl-cyclobutane-1-carboxylic acid (FMACBC), potential PET ligands for tumor detection. J Med Chem. 2002;45:2250–2259. doi: 10.1021/jm010242p. [DOI] [PubMed] [Google Scholar]
- 29.Moon BS, Lee TS, Lee KC, An GI, Cheon GJ, Lim SM, et al. Syntheses of F-18 labeled fluoroalkyltyrosine derivatives and their biological evaluation in rat bearing 9L tumor. Bioorg Med Chem Lett. 2007;17:200–204. doi: 10.1016/j.bmcl.2006.09.063. [DOI] [PubMed] [Google Scholar]
- 30.Pauwels EK, Ribeiro MJ, Stoot JH, McCready VR, Bourguignon M, Maziere B. FDG accumulation and tumor biology. Nucl Med Bio. 1998;25:317–322. doi: 10.1016/s0969-8051(97)00226-6. [DOI] [PubMed] [Google Scholar]
- 31.Pauleit D, Stoffels G, Bachofner A, Floeth FW, Sabel M, Herzog H, et al. Comparison of 18F-FET and 18F-FDG PET in brain tumors. Nucl Med Biol. 2009;36:779–787. doi: 10.1016/j.nucmedbio.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 32.Hamacher K, Coenen HH. Efficient routine production of the 18F-labelled amino acid O-2-18F fluoroethyl-L-tyrosine. Appl Radiat Isot. 2002;57:853–856. doi: 10.1016/s0969-8043(02)00225-7. [DOI] [PubMed] [Google Scholar]
- 33.Okarvi SM. Recent progress in fluorine-18 labelled peptide radiopharmaceuticals. Eur J Nucl Med. 2001;28:929–938. doi: 10.1007/s002590100508. [DOI] [PubMed] [Google Scholar]
- 34.Adibi SA, Paleos GA, Morse EL. Influence of molecular structure on half-life and hydrolysis of dipeptides in plasma: importance of glycine as N-terminal amino acid residue. Metabolism: clinical and experimental. 1986;35:830–836. doi: 10.1016/0026-0495(86)90224-6. [DOI] [PubMed] [Google Scholar]
- 35.Lindner HA, Alary A, Wilke M, Sulea T. Probing the acyl-binding pocket of aminoacylase-1. Biochem. 2008;47:4266–4275. doi: 10.1021/bi702156h. [DOI] [PubMed] [Google Scholar]
- 36.Lindner HA, Alary A, Boju LI, Sulea T, Menard R. Roles of dimerization domain residues in binding and catalysis by aminoacylase-1. Biochem. 2005;44:15645–15651. doi: 10.1021/bi051180y. [DOI] [PubMed] [Google Scholar]
- 37.Yanagida O, Kanai Y, Chairoungdua A, Kim DK, Segawa H, Nii T, et al. Human L-type amino acid transporter 1 (LAT1): characterization of function and expression in tumor cell lines. Biochimica et Biophysica Acta. 2001;1514:291–302. doi: 10.1016/s0005-2736(01)00384-4. [DOI] [PubMed] [Google Scholar]
- 38.Herrera-Ruiz D, Knipp GT. Current perspectives on established and putative mammalian oligopeptide transporters. J Pharm Sci. 2003;92:691–714. doi: 10.1002/jps.10303. [DOI] [PubMed] [Google Scholar]
- 39.Rubio-Aliaga I, Daniel H. Mammalian peptide transporters as targets for drug delivery. Trends in Pharm Sci. 2002;23:434–440. doi: 10.1016/s0165-6147(02)02072-2. [DOI] [PubMed] [Google Scholar]
- 40.Kim DK, Kanai Y, Chairoungdua A, Matsuo H, Cha SH, Endou H. Expression cloning of a Na+-independent aromatic amino acid transporter with structural similarity to H+/monocarboxylate transporters. J Biol Chem. 2001;276:17221–17228. doi: 10.1074/jbc.M009462200. [DOI] [PubMed] [Google Scholar]