

Summary
Gangliosides are sialic acid-containing glycosphingolipids (GSLs) that are most abundant in the nervous system. They are localized primarily in the outer leaflets of plasma membranes and participated in cell-cell recognition, adhesion, and signal transduction and are integral components of cell surface microdomains or lipid rafts along with proteins, sphingomyelin and cholesterol. Ganglioside-rich lipid rafts play an important role in signaling events affecting neural development and the pathogenesis of certain diseases. Disruption of ganglioside synthase genes in mice induces developmental defects and neural degeneration. Targeting ganglioside metabolism may represent a novel therapeutic strategy for intervention in certain diseases. In this review, we focus on recent advances on metabolic and functional studies of gangliosides in normal brain development and in certain neurological disorders.
Keywords: Glycosyltransferase (ganglioside synthase), gangliosides, knockout mouse, lipid rafts, neurodevelopment, neurological diseases
Introduction
Gangliosides
Gangliosides are sialic acid-containing glycosphingolipids (GSLs) ubiquitously distributed in tissues and body fluids, and are more abundantly expressed in the nervous system [1]. Heterogeneity and diversity of the structures in their carbohydrate chains are characteristic hallmarks of gangliosides; so far, 188 gangliosides with different carbohydrate structures have been identified in vertebrates [2]. The structural complexity of gangliosides renders them ideally suited to participate in regulation of many cellular functions, including serving to as modulators of signaling receptors, such as neurotrophic factor, neurotransmission, and interaction with regulatory proteins in the nervous system [3]. On the cell surface, gangliosides are involved in cell-cell recognition and adhesion and signal transduction within specific cell surface microdomains, termed caveolae [4], lipid rafts [5], or GSL-enriched microdomains [6], with other membrane components such as sphingomyelin and cholesterol. Accumulated evidence strongly suggests that gangliosides are co-localized in the microdomain structures with signaling molecules and adhesion molecules. In addition to cell plasma membranes, gangliosides are also known to occur in intracellular organelles, such as the nuclear membranes, and they have recently been proposed to play important roles in modulating intracellular and intranuclear calcium homeostasis and the ensuing cellular functions [7].
1.a. Biosynthesis and regulation of ganglioside expression during brain development
Figure 1 shows the structures and metabolic pathways of major GSLs and ganglio-series gangliosides in the nervous system. The latter are classified by the presence of one or more sialic acid residues linked to different galactose and/or sialic acid residues and are classified into asialo-, a-, b- and c-series gangliosides. Gangliosides having sialic acid residue(s) linked to the inner N-acetylgalactosamine residue, such as GT1aα originally reported as GTx [8], are classified as α-series gangliosides.
Figure 1.
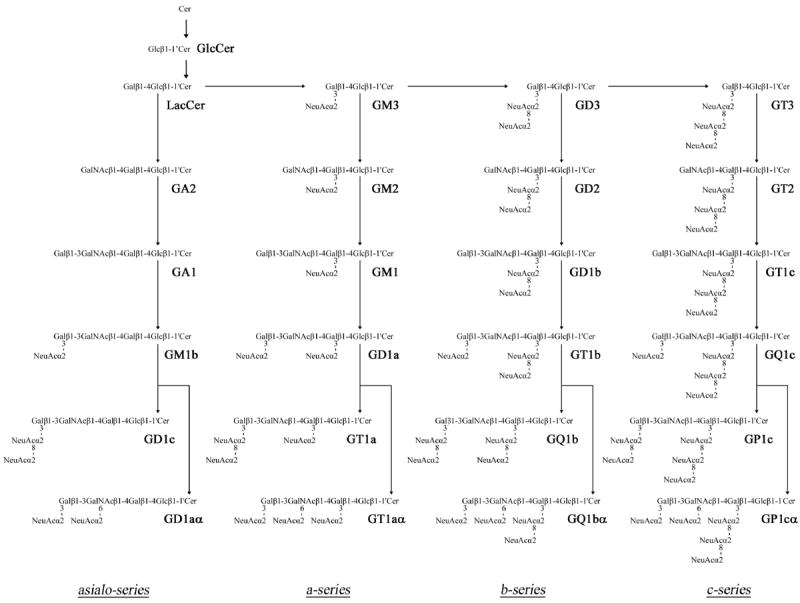
Structures and biosynthetic pathways of ganglio-series gangliosides. The nomenclature for gangliosides and the components are based on those of Svennerholm [102] and IUPAC-IUBMB Joint Commission on Biochemical Nomenclature [103], respectively. Glycosyltransferases catalyzing the synthesis of glycosphingolipids, including gangliosides, are underlined. GD1α, GT1aα, GQ1bα, and GP1cα are classified as belonging to the α-series gangliosides. (Reproduced from [52]).
Ganglioside biosynthesis commences from glucosylceramide, whose synthesis occurs in the endoplasmic reticulum; structural extension occurs primarily in the Golgi apparatus by sequential addition of carbohydrate moieties [9] to an existing acceptor glycolipid molecule. The reactions are catalyzed by a series of specific glycosyltransferases (GTs). With the exception of GM4, which is derived from galactosylceramide (GalCer), most complex ganglioside species are derived from glucosylceramide (GlcCer) and lactosylceramide (LacCer). First, a simple ganglioside, GM3, is synthesized by addition of a sialic acid to LacCer by the action of CMP-sialic acid:LacCer α2-3 sialyltransferase (GM3-synthase, GM3S, or ST-I). GD3 and GT3 are synthesized by sequential addition of sialic acids to GM3 and GD3 by the action of CMP-sialic acid:GM3 α2-8 sialyltransferase (GD3-synthase, GD3S, or ST-II) and CMP-sialic acid:GD3 α2-8 sialyltransferase (GT3-synthase, GT3S, or ST-III), respectively. GM3, GD3 and GT3 that lack N-acetylgalactosamine further serve as precursors of more complex gangliosides belonging to the a-, b-, and c-series, respectively. Elaboration of these simple gangliosides to complex gangliosides is catalyzed by UDP-GalNAc:LacCer/GM3/GD3/GT3 β1-4 N-acetylgalactosaminyltransferase (GalNAcT or GA2/GM2/GD2/GT2-synthase), UDP-Gal:GA2/GM2/GD2/GT2 β1-3 galactosyltransferase (GA1/GM1/GD1b/GT1c-synthase or GalT-II), CMP-sialic acid: GA1/GM1/GD1b/GT1c α2-3 sialyltransferase (GM1b/GD1a/GT1b/GQ1c-synthase or ST-IV), and CMP-sialic acid:GM1b/GD1a/GT1b/GQ1c α2-8 sialyltransferase (GD1α/GT1aα/GQ1bα/GP1cα-synthase or ST-V) along specific pathways (Figure 1). Asialo-series gangliosides are also synthesized from LacCer by these glycosyltransferases along a different pathway. Interestingly, the expression levels and patterns of gangliosides undergo dramatic changes during brain development [10, 11]. For instance, in human and rodent embryonic brains, the predominant gangliosides are simple hemato-series gangliosides such as GM3, GD3 and 9-OAcGD3. As the brain develops, the expression of these simple gangliosides is down-regulated with concomitant up-regulation of complex gangliosides such as GM1, GD1a, GD1b and GT1b. This change in expression levels and patterns of gangliosides can be largely attributed to developmental changes of the expression levels and patterns of ganglioside synthases [12, 13] that are spatiotemporally regulated, both at the transcriptional and post-translational levels, by multiple systems including epigenetic modifications [14, 15]. Some of the key GTs whose genes have been cloned and their promoters are functionally analyzed are presented below.
1.a.(1). GM3-synthase (GM3S, sialyltransferase I or ST-I)
ST-I catalyzes the synthesis of GM3, and its expression is regulated in a tissue-specific manner [16] during brain development [17]. While multiple transcriptional start sites of ST-I gene are demonstrated in mouse [18] and human [19], minimum proximal regions of the ST-I gene displaying the highest promoter activity have also been found in both species [18, 20, 21]. Analyses of the promoter regions showed no TATA or CCAAT boxes but multiple Sp1 and AP2 sites. Indeed, chromatin immunoprecipitation confirmed the Sp1 and AP2 binding to these sites, which have revealed an essential role to maintain a high level of promoter activity [18]. cAMP response element-binding (CREB), one of the transcription factors of the neurotrophin brain-derived neurotrophic factor (BDNF), can be pulled-down using human or mouse promoter DNA fragment of ST-I gene [21, 18], suggesting a possible co-regulatory mechanism of GM3 expression that is shared with brain development.
1.a.(2). GD3-synthase (GD3S, sialyltransferase II, ST-II)
ST-II is a key regulatory enzyme controlling the synthesis of b- and c-series gangliosides (Figure 1). Following the cloning of the promoters of the ST-II gene from rat, mouse and human brains [22-24], Sp1 sites were identified to be common among these species. Deletion of the Sp1 sites results in a dramatic loss of the promoter activity of mouse ST-II gene [23], suggesting that Sp1 plays a major role in the transcriptional regulation of this gene. Furthermore, all of the three promoters contain a GC–GT repeat sequence motif featured in the conformation of Z-DNA structure, presenting at the suppressive control region of the promoter. This unique GC-GT repeat may therefore contribute to cis-regulation of ST-II gene expression.
1.a.(3). N-Acetylgalactosaminyltransferase (GalNAcT; GA2/GM2/GD2/GT2-synthase, GA2/GM2/GD2/GT2S)
GalNAcT is a common enzyme controlling the expression of more complex ganglio-series gangliosides along the different pathways. The human gene contains three transcription start sites corresponding to three alternative transcripts [25]. Three promoters have also been defined for the respective transcription start sites. Given that some putative transcription factor-binding sites have been found at the promoter regions based on sequence comparison, the regulation as well as biological activities of these promoters of GalNAcT gene remains to be elucidated.
1.a.(4). Galactosyltranferase II (GalT-II; GA1/GM1/GD1b/GT1c-synthase; GA1/GM1/GD1b/GT1cS)
Despite the importance of GM1 ganglioside in the neural development, the gene expression of GalT-II, the enzyme responsible for catalyzing GM1 biosynthesis from GM2, is less clear. We have cloned the promoter of mouse GalT-II gene and identified a number of its transcription factors [26]. This promoter is TATA-less, has no CCAAT box, and has a single transcription start site. Twenty-seven transcription factors have been identified to bind to the consensus sites in the promoter region, whereas 4 other factors without consensus binding sites in this region have also been recruited, possibly as components of transcription factor complexes. However, the regulation of the GalT-II gene expression, particularly during neuronal differentiation, is not clear, which should be carefully elucidated.
1.a.(5). Glucosyltransferase (GlcT, GlcCer-synthase, GlcCerS)
GlcT catalyzes the first committed step in ganglioside biosynthesis, the transfer of a glucose moiety from UDP-glucose to ceramide. Studies on the promoter of mouse GlcT gene revealed that the sequence is TATA-less while GC-rich, and contains no CCAAT box [27]. Deletion of a 41-bp fragment that contains an Sp1 site dramatically decreases the promoter activity, indicating an essential role of this site in maintaining an expression level of the mouse GlcT gene.
1.a.(6). Galactosyltransferase III (GalT-III, GalCer-synthase, GalCerS)
GalT-III is another key enzyme responsible for catalyzing the biosynthesis of galactocerebroside (GalCer), the most abundant GSL in the myelin sheath. GalCer serves as the precursor for the biosynthesis of sulfatide, another myelin-specific GSL, and GM4, the simplest ganglioside that is highly enriched in CNS myelin [28]. The promoter of the human GalT-III gene is also TATA-less while GC-rich, and has no CCAAT box [29]. At least two small regions in the promoter are characterized as the regulatory elements in our laboratory [30]. Further analysis demonstrated that the proximal region contains an Sp1 site and the distal region contains a CREB site [30]. Cell type-specific activities of the promoter have been observed between the human oligodendroglioma (HOG) and human neuroblastoma (LAN-5) cell lines, and the factor CREB may account for the different levels of GalT-III promoter activity in these cell lines due to the differential DNA binding affinities of CREB [30].
1.b. Epigenetic regulation of ganglioside synthase gene expression
Temporal and spatial expression of ganglioside synthase genes is subject to complex developmental and tissue-specific regulation. Attempts have been made to characterize the upstream promoter sequences and basal transcription factors as discussed above; however, very few studies have been reported on the underlying molecular mechanisms by which these genes are regulated. Moreover, although epigenetic regulation mechanisms including histone modification, DNA methylation, chromatin remodeling, and non-coding RNA, are fundamental for controlling gene expression [31, 32], very few studies have been undertaken on the epigenetic regulatory mechanisms of GT expression for the biosynthesis of glycoproteins or glycolipids. Recently, we have demonstrated for the first time that histone acetylation in chromatins of the 5’-region of mouse GalNAcT gene can regulate mRNA expression, and this epigenetic modification is highly correlative to the stage-specific alternations of GalNAcT mRNA level in mouse brain during development [14]. A recent report by Taniguchi and his colleagues also demonstrated that neuron-specific expression of N-acetyl-glucosaminyltransferase-IX (GnT-IX) for branched O-mannose glycan synthesis, was triggered by building an open chromatin environment based on deposit of active histone marks around GnT-IX gene’s transcription start site [33]. It would be a most interesting and fertile area to investigate the regulatory mechanism of glycosyltransferase gene expression in physiological and pathological conditions by epigenetic approaches.
2. Ganglioside in neurodevelopment and function
During neurodevelopment, particularly during the early stages, dramatic consistent changes in ganglioside expression are observed [10, 11]. Generally, in the early embryonic vertebrate brain, the pattern of ganglioside expression is limited to more simple gangliosides, predominantly GM3 and GD3; in later developmental stages, however, more complex gangliosides predominate, particularly GM1, GD1a, GD1b, and GT1b [1, 10, 11]. Much research has been presented showing that qualitative and quantitative changes in ganglioside expression in the nervous system correlate with neurodevelopmental milestones, which are summarized in Figure 2 (for a review, see [1, 15]). The tight regulation of ganglioside expression during development strongly suggests that the expression of specific ganglioside species may reflect the functional roles they play at particular developmental stages. Mounting evidence supports the notion that gangliosides serve regulatory roles in cellular events, including neuritogenesis, axonogenesis, and synaptogenesis [1, 10, 15, 34-37].
Figure 2.
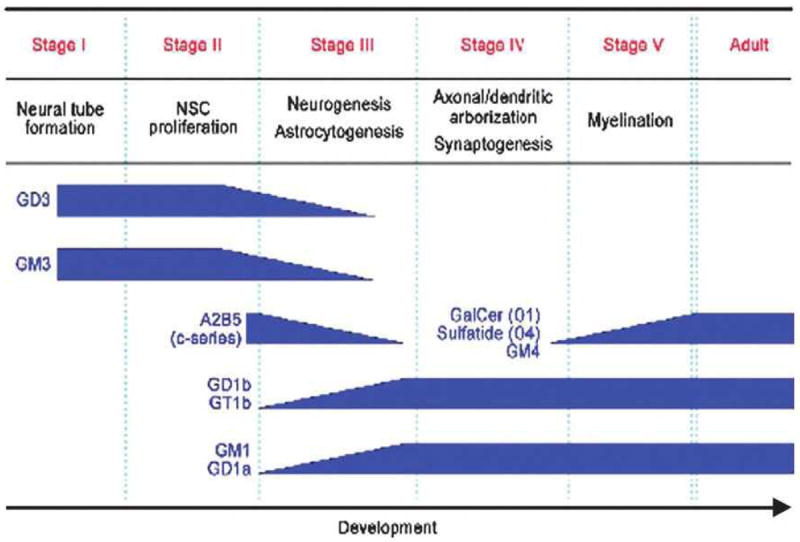
Developmental milestones and concurrent changes in GSL expression in the brain. (Reproduced from [52]).
With the advent of cloning and transgenic technology, several lines of genetically engineered mice have been established in which the expression of gangliosides and other GSLs have been altered or depleted. For example, mice lacking GalNAcT, and, therefore, GalNAc-containing gangliosides, appear to have neurological disorders such as axonal degeneration, sensory, motor and behavior deficits, and other neural dysfunctions. Simpson et al. showed that in humans an autosomal recessive infantile-onset symptomatic epilepsy syndrome in a large Old Order Amish pedigree is associated with a nonsense mutation of ST-I, a key enzyme for the synthesis of all complex gangliosides [38]. ST-I knockout mice, lacking all major a-, b- and c-series gangliosides, exhibit major developmental deficits in the central nervous system (CNS) and the peripheral nervous system (PNS) as well as susceptibility to seizures [39]. Gangliosides are involved in formation of the brain and in maintaining integrity of nervous tissues [40, 41]. Similarly, ST-II-deficient mice, lacking b- and c-series gangliosides, present subtle developmental and behavioral deficits [42]. Severe neurodegeneration develops in double knockout mice lacking ST-II and GalNAcT in which all major brain gangliosides are deficient [39]. Refractory skin injury occurs in the complex knockout mice exhibiting only GM3 ganglioside [43]. A summary of these transgenic mice and their neurological abnormalities is shown in Table 1 and a detailed description of each of these GT knockout mutant animals is seen below. It should be apparent that these animals, while rarely fetal lethal do show altered neural development and behavioral deficits [39, 43-51]. Those observations suggest that gangliosides have important biological functions in the developing nervous system. The availability of specific GT- deficient mutants allows one to dissect the biological functions of a specific ganglioside or, in most cases, a series of gangliosides during early brain development.
Table 1.
Ganglioside synthase-KO mice and their phenotypes
| Disrupted gene | Gangliosides expressed in brain* | Phenotypes of the nervous system | Reference |
|---|---|---|---|
|
| |||
| ST-I | GM1b, GD1α | (Viable) | [19, 70] |
| Complete hearing loss | |||
| Degeneration of the sensory organ of hearing in cochlea | |||
| Attention-deficit hyperactivity disorder-like behavior | |||
|
| |||
| ST-II | GM1, GD1a | (Viable) | [41, 42, 104] |
| GM3, GD1a in embryo | Impaired regeneration of the lesioned hypoglossal nerve | ||
|
| |||
| GalNAcT | GM3, GD3, 9-OAc-GD3 | (Viable) | [41, 42, 72-74, 76, 90] |
| Decreased myelination and axonal degeneration in CNS/PNS | |||
| Demyelination in PNS | |||
| Reduction in neural conduction velocity from the tibial nerve | |||
| Sensory nerve-dominant nerve degeneration and synaptic remodeling | |||
|
| |||
| ST-I/GalNAcT | (ganglio-series ganglioside deficient) | (Viable; death soon after weaning) | [48] |
| Axonal degeneration and perturbed axon-glia interaction in CNS | |||
|
| |||
| ST-II/GalNAcT | GM3 | (Viable; shortened life span) | [40-42, 77, 78] |
| Sudden death in response to lethal sound-induced seizures | |||
| Neurodegeneration by dysfunction of complement systems and inflammation | |||
| GEM/raft transfiguration, complement activation, local inflammation | |||
| Progressive dysfunction of motor coordination, marked deterioration in memory and learning | |||
| Suppressed function of muscarinic type acetylcholine receptors | |||
In wildtype mice, the predominant gangliosides are GM3 and GD3 in embryonic brains and GM1, GD1a, GD1b and GT1b in adult brains.
3. Neural stem cells (NSCs) and neural progenitor cells (NPCs)
The CNS is comprised of a variety of cells, including neurons and glial cells. These cells are generated from common NSCs during development [52-55]. At each stage of neural development, there are unique cell-surface marker glycoconjugates, including GSLs, characteristic of a variety of cell types and developmental stages [10, 55-60] (Figure 2).
NSCs appear during neural plate formation and serve as precursors to the major cell types in the early neuroectoderm and in the neural tube. As development proceeds, NSCs become progressively less abundant, and more restricted NPCs emerge. In the adult brain, NSCs are located primarily in the subventricular zone of the lateral ventricles and the subgranular layer of the dentate gyrus in the hippocampus, regions of the brain that can sustain neurogenesis even during adulthood [61, 62]. These NSCs are considered to serve as a cellular reservoir for CNS development and for replacement of cells lost due to normal cell turnover. NSCs have attracted considerable attention because of their basic biological significance and their potentially important clinical use in regenerative medicine.
3.a. NSCs and gangliosides
At present, a wide variety of glycoconjugates (glycoproteins, glycolipids, and proteoglycans) and carbohydrate antigens have been found in NSCs. These glycoconjugates, such as SSEA-1, HNK-1, PSA-NCAM, prominin-1, gp130, chondroitin sulfate proteoglycans, heparan sulfate proteoglycans, cystatin C, galectin-1, and GSLs including gangliosides and Notch, are marker molecules and also play important functional roles in NSCs [55, 60]. The signaling involved in NSC fate regulation and mediated or modulated by glycoconjugates is referred to as “glycosignaling” (Figure 3).
Figure 3.
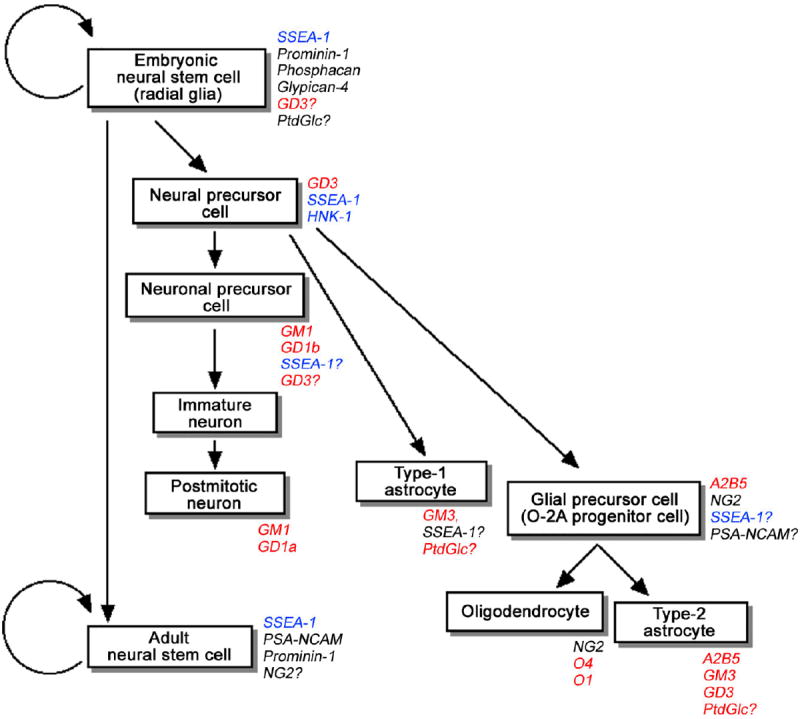
A model of mouse neural cell lineage derived from NSCs in the developing CNS. The known glycoconjugate markers are indicated in italic. Glycolipids are indicated in red. Carbohydrate antigens carried by both glycolipids and glycoproteins are indicated in blue. (Reproduced from [54])
In primary mNSCs prepared from mice at embryonic day 14 (E14.5), GD3 is the dominant GSL species expressed (>80%) [63]. In NSCs treated with methyl-β-cyclodextran, which disrupts lipid microdomains by depleting cholesterol, basic fibroblast growth factor (bFGF)-induced activation of extracellular signal-regulated kinase (ERK) MAPK and adhesion via integrins were repressed [63]. Interestingly, suppressing β1-integrin, a glycoprotein bearing the SSEA-1 carbohydrate epitope results in apoptosis [64], which could represent one of the mechanisms for determining the fate of daughter cells during early development. In addition, in mNSCs treated with PDMP, an inhibitor of GSL synthesis, GD3 disappeared and the bFGF-induced ERK MAPK activation was repressed [65]. Because gangliosides, including GD3, are robustly localized in lipid rafts and caveolae, those findings suggest a specific role of gangliosides in cell-surface GSL-enriched microdomains (GEMs) in modulating the cytokine and adhesion signal transduction pathways. In fact, GD3 has been shown to enhance adhesion signals and recruit integrins to lipid rafts in melanoma cells of neuroectodermal origin [66]. That concept is consistent with our own findings that GD3, characteristic of NSCs, is present in GEMs and participates in mediating EGF/bFGF-induced β1-integrin up-regulation to promote cell proliferation and self-renewal [64]. In GlcT-conditional knockout mice, dysfunction of the cerebellum and peripheral nerves and alteration of the Ras-MAPK pathway have been found [64] [67]). In mouse neural epithelial cells (mNECs) that are rich in NSCs, knockdown of GlcT with small hairpin RNAs inhibited activation of the Ras-MAPK pathway and cellular proliferation [68]. Those studies also support the concept that GSLs including gangliosides are involved in signal transduction in stem cells. In NSCs isolated from ST-II knockout mice, however, no significant differences were seen in proliferation rates, differentiation characteristics, viability, and activation status of signaling pathways important for cell fate modulation [60], suggesting there may be a more complex mechanism in which other GSLs compensate for the lack of GD3. Those discrepancies can best be resolved using a combination of GT-deficient mice that compensate the loss of GD3.
4. GTs in neurodevelopment and neurodegenerative disorders
As described above, dramatic changes in ganglioside expression are observed during neural development [10, 11]. How those drastic developmental changes are regulated, however, has not been fully elucidated. Recently, an analysis has been completed of the developmental changes of the expression patterns of GSLs including gangliosides and the GT genes in developing mouse brains [10]. In E12 to E14 brains, GD3 was a predominant ganglioside. After E16, the concentrations of GM3 and GD3 markedly decreased, and the concentrations of a-series gangliosides, such as GD1a, increased (Figure 5). Analysis of GT gene expression patterns revealed that GalNAcT becomes dominant as compared with the GT expression patterns of ST-II in the brain during development. As a result, more GD1a than GD3 is synthesized in late embryonic, postnatal, and adult brains. Those results suggest that the expression levels of GalNAcT and ST-II regulate the pathway shift of ganglioside expression patterns during development.
Figure 5.
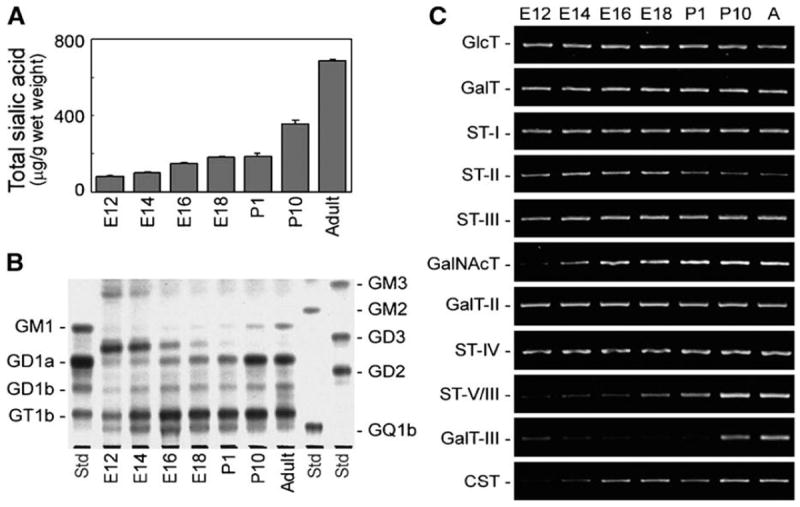
(A) Expression levels of gangliosides in the developing mouse brain. (B) Expression patterns of gangliosides in developing mouse brain analyzed by thin-layer chromatography. During development, the ganglioside expression patterns in mouse brain shifted from simple gangliosides such as GM3 and GD3 to complex gangliosides such as GM1 and GD1a. (C) Expression levels of GT genes in the developing mouse brain analyzed by RT-PCR. During development, GalNAcT (GA2/GM2/GD2/GT2-synthase) were increased, and ST-II (GD3-synthase) slightly decreased. “A” indicates adult mouse brain. (Reproduced from [66])
4.a. Disruption of glycosyltransferase (GT) genes and their knockout mice
In recent years, a number of specific GT gene knockout mutant mice have been generated. They provide powerful tools for studying the biological functions of some of the gangliosides during brain development. Most of these mutant mice survive with only varying degrees of phenotypes, ranging from severe to subtle. Nonetheless, they provide critical development cues for the functional aspects of specific ganglioside(s) in the nervous system. Table 1 lists the available mutant mice that are available.
4.a.(1). ST-I (GM3S) knockout mice
In these mutant mice, all major brain gangliosides such as GM1, GD1a, GD1b, and GT1b are lacking, which is compensated by the accumulation of asialo-series gangliosides in the brain [49]. No significant differences are found in body weights between mutant and littermate control mice. Histochemical examination of tissues from mutant mice shows no apparent abnormalities in brain, liver, adipose tissue, and muscle [48, 69]. GM3-synthase knockout mice are able to live longer than 1 year [48, 69]. Despite the lack of GM3-synthase activities, the primary embryonic fibroblasts of these mutant mice retain a decreased amount of gangliosides (21% of normal) with a completely different pattern compared with those in the wild-type control. The gangliosides in the GM3-synthase null fibroblasts consist mainly of GM1b, GalNAc-GM1b, and GD1α, with both N-acetyl and N-glycolylneuraminic acids (NeuAc and NeuGc) and diverse ceramide structures. All these gangliosides are products of the asialo-pathway of ganglioside synthesis, not normally expressed in fibroblasts [69]. Thus, the over-expression of the asialo-series gangliosides in these mice may have compensatory effects for maintaining “normal” neurologic function and life span in these mice [49]. On the other hand, subtle behavioral abnormalities have been noted in these mutant mice. For example, Niimi et al. [70] examined motor activity, cognitive and emotional behaviors, in juvenile GM3-knockout mice and found that these mutant mice showed hyperactivity in the motor activity test, Y-maze test, and elevated plus maze test, which represented phenotypes resembling attention-deficit hyperactivity disorder, thus indicating a role of GSLs in maintaining neuropsychological balance. In the Y-maze test, there is significantly less spontaneous alternation behavior in the male mutant mice than in wild-type mice, suggesting a hormonal effect.
4.a.(2). ST-II (GD3S) knockout mice
ST-II knockout mice, lacking both b- and c-series gangliosides, display no overt phenotype [42, 43]. The mutant mice appear to undergo normal development and have a normal life span with no apparent abnormal behavior [42]. Histochemical examination of mutant mice revealed no obvious changes in the nervous tissue. Moreover, no difference in Fas-mediated apoptotic reaction of lymphocytes between wild type and the mutant mice can be detected; on the other hand, there is impaired nerve regeneration of axotomized hypoglossal nerves, suggesting that b- and c-series gangliosides are more involved in neural repair rather than in the differentiation of the nerve cells and apoptotic process induced via Fas [43]. Further studies revealed that the mutant mice exhibited increased thermal and mechanical sensory responses compared to the wild-type mice [71]. No significant differences, however, were observed in the conduction velocity of the sciatic nerve and in apparent morphology in the spinal cord and sciatic nerve of the mutant and the control animals. These results suggest that b- and c-series gangliosides are critical in the development and/or maintenance of the sensory nervous system [71].
4.a.(3). GalNAcT (GM2/GD2S) knockout mice
By disrupting the GalNAcT gene that encodes the enzyme responsible for the biosynthesis of complex gangliosides, mice express only simple gangliosides such as GM3, GD3, and 9-O-acetyl GD3, which are expressed robustly in the mutant mouse brains [72]. Although the mice lack all complex ganglio-series gangliosides, they do not show any gross histological defects in their nervous systems or apparent behavioral deficits. A slight reduction in the conduction velocity from the tibial nerve to the somatosensory cortex, but not to the lumbar spine, could be demonstrated, suggesting the involvement of complex gangliosides in neural functions, such as neuronal transmission [73]. The mutant mice display decreased myelination, Wallerian axonal degeneration in both the central and peripheral nervous systems [74]. Further studies indicated that these mice develop significant progressive neuropathies, including deficits in reflexes, strength, coordination, and balance [75]. Quantitative indices of motor abilities, applied at 8 and 12 months of age, also revealed progressive gait disorders, including reduced stride length and width, increased hindpaw print length, as well as a marked reduction in ability to rearing [75]. Compared to controls, the mutant mice tend to walk in small labored movements. Twelve-month-old mice display a significant incidence of tremor and catalepsy. Evidence of sensory dysfunction, neural degeneration, and glial proliferation is apparent [74]. Morphological changes in the synaptic vesicles and modes of synaptic contacts with central terminals were detected, suggesting synaptic remodeling following nerve degeneration [76]. Moreover, Sugiura et al. [76] reported that these behavior deficits are worsened in aging mutant mice. These results suggest that complex gangliosides are essential in the maintenance of the integrity of the cytoarchitecture and function of the nervous system; a lack of these complex gangliosides results in neural degeneration in a sensory nerve-dominant manner [76].
4.a.(4). GalNAcT and ST-I double knockout mice
The double knockout mice lack both GalNAcT and ST-I genes and they lack all complex “brain-type” ganglio-series gangliosides, resulting in several early developmental deficits. Histochemical examinations of the young mutant brains revealed severe pathological changes such as prominent vacuolization in the cerebellar and spinal white matters, along with enhanced apoptosis, axonal degeneration and perturbed axon-glia interactions in the cerebral cortex [48]. Mice developed significant and progressive behavioral neuropathies starting at 2 weeks of age, including deficits in reflexes, strength, coordination, and balance. The majority of the mutant mice die within 3 weeks of birth [48].
4.a.(5). GalNAcT and ST-II double knockout mice
When both GalNAcT and ST-II genes are disrupted simultaneously, the mutant mice express primarily GM3 with no “brain-type” ganglio-series gangliosides. The GM3-only mice exhibited weight loss, progressive motor and sensory dysfunctions and deterioration in spatial learning and memory with aging [77, 78]. Additionally, the responses to treatment with oxotremorine, an agonist of muscarinic acetylcholine receptors (mAChRs), were markedly attenuated, indicating an impairment of mAChR functions in the GM3-only mice [78], while there was no clear causal association with any aforementioned neurological abnormalities. Likewise, substantial degeneration of Purkinji neurons has also been reported in the cerebellar cortex, which may result from regional complement activation and inflammatory reactions, as shown by deposits of C1q complement in the cerebella [41]. A degeneration-rescuing effect could be achieved by cross-breeding the double knockout animals with animals carrying the disrupted gene of complement C3 [77]. A striking phenotype of high susceptibility to sound-induced seizures has been noted in another derived line of the double knockout mouse with a distinct genetic background, C57BL/6 [42]. Refractory skin lesions resulting from frequent scratching developed at >6 months of age, possibly due to sensory nerve impairment [79]. These mutant mice display normal birth, but with a shortened life span, typically with sudden death of 50% of the animals by 30 weeks of age [42, 79]. In addition, these mice are extremely susceptible to induction of lethal audiogenic seizures by sound stimulus [42]. These observations reinforce the notion that gangliosides play an essential role in the proper functioning of the nervous system.
4.b. Ganglioside synthase gene knockout mutants as a tool for studying neurological diseases
In addition to providing normal developmental cues, ganglioside synthase gene knockout animals can be used as valuable tools for studying disease mechanisms. Some of the notable examples are described below.
4.b.(1). ST-I (GM3S) mutation
As indicated earlier, in humans an autosomal recessive infantile-onset symptomatic epilepsy syndrome in a large Old Order Amish pedigree is associated with a loss-of-function mutation of ST-I, which causes the synthesis of all complex gangliosides to be affected [38]. ST-I null mice represent a useful model for studying the disease mechanisms. In those mice, hearing ability is impaired at the onset of hearing development and completely lost by 17 days after birth (P17), showing a deformity in hair cells in the organ of Corti [49]. By 2 months of age, the organ of Corti is selectively and completely disappeared without any effect on balance or motor function or in the histology of vestibule [49]. These mutant mice are viable and appear to have a heightened sensitivity to insulin. The basis for the increased insulin sensitivity in the mutant mice is apparently associated with enhanced insulin receptor phosphorylation in skeletal muscle [80]. Importantly, the mutant mice are protected from high-fat diet-induced insulin resistance. These results suggest that GM3 ganglioside is a negative regulator of insulin signaling, which is important for nervous system development [81]. In addition, ST-I-deficient mice show gender-specific phenotypes resembling attention-deficit hyperactivity disorder, namely hyperactivity, reduced attention, and increased impulsive behaviors. Administration of methylphenidate hydrochloride, however, does not ameliorate hyperactivity in either male or female mutant mice [70]. In this regard, it is interesting to note that in humans of the old Order Amish pedigree, deficiency of GM3-synthase results in infantile-onset sympromatic epilepsy syndrome associated with developmental stagnation and blindness [38]. The vision loss results from CNS and optic atrophy, although retinal function appears to be normal into the teenage years [82].
4.b.(2). Alzheimer’s disease
The onset of AD, the most common form of dementia and neurodegenerative disease, has been proposed to be initiated by aggregation of Aβ caused by gangliosides [83-85]. Gangliosides GM3 and GM1 are suggested to play precipitating roles in the deposition of the Aβ as amyloid angiopathy and senile plaques, respectively, in the Alzheimer brain. Oikawa et al. [86] reported the profile of amyloid deposition in the brains of transgenic mice expressing a mutant amyloid precursor protein with a disrupted GalNAcT gene, in which GM3 accumulated in both vascular and parenchyma tissues obtained from the brains of the mutant mouse, whereas GM1 was lacking in those tissues. These mice show a significantly increased level of deposited Aβ in vascular tissues. Furthermore, formation of severe dyshoric-form amyloid angiopathy, in which amyloid extending from the blood vessel walls deeply into the surrounding parenchyma has been observed [86].
Bernardo et al. [87] prepared double-transgenic mutant mice expressing APP/PSEN1, a mouse model of Alzheimer’s diseases (AD), and triple-transgenic mice expressing APP/PSEN1 with deficient GD3-synthase, ST-II. As expected, the double-transgenic mice exhibit impaired memory tasks and accumulated Aβ deposits in the brain. Remarkably, in the triple-transgenic mice lacking ST-II, both Aβ deposits-associated neuropathology and behavioral impairments are almost completely eliminated. The APP/PSEN1/ST-II-/- triple mutant mice perform as well as the wild-type GD3S-/- mice. How the altered ganglioside biosynthesis can result in eliminating the formation of Aβ is not known. It would be of great interest in targeting ST-II for intervening the cognitive deficits, amyloid plaque formation, and neurodegeneration associated with AD.
More recently, we found an increase of Chol-1α antigens (GQ1bα and GT1aα), which are specifically expressed in cholinergic neurons [88], in the brain of AD model of transgenic mice [89]. The increase of Chol-1α gangliosides in AD mouse brains may present evidence for generation of cholinergic neurons, which should be of interest in investigating the capability of adult neurogenesis in AD brains.
4.b.(3). Parkinson’s disease
Parkinson’s disease (PD) is a progressive neurodegenerative movement disorder resulting from the loss of dopaminergic neurons in the substantia nigra. Wu et al. [90] reported that in mutant mice with disrupted GalNAcT gene, α-synuclein expression was greatly elevated in substantia nigra pars compacta of the brain. These mice show overt motor disability on aging, loss of dopaminergic neurons and aggregation of α-synuclein, resulting in Parkinson-like symptoms. Manifestation of Parkinsonism can be attenuated by administration of LIGA-20, a membrane permeable lyso derivative of GM1 that is capable of penetrating the blood-brain barrier and entering living neurons. The discovery of the possibility that GM1 is capable of attenuating α-synuclein aggregation in neurons underlies another facet of the myriad of ganglioside functions.
4.b.(4). Guillain-Barré Syndrome
Guillain-Barré syndrome (GBS) and its variants are autoimmune neuropathies that frequently occur as a result of enteritis resulting from an infectious event from agents such as Campylobacter jejuni (C. jejuni). These neuropathies are recognized as several disorders characterized by an immune-mediated attack on the peripheral nerve, particularly in the myelin sheath of sensory and motor nerves. Increased antibody titers in GBS and variants are thought to be a result of the production of antibodies to bacterial carbohydrate-containing surface antigen(s) that cross-react with the GSLs, including gangliosides, of the myelin sheath and the axons of nerve cells [91-93]. The presynaptic neuromuscular junction (NMJ) has been considered a potential target vulnerable to autoimmune attack in Guillain-Barré Syndrome (GBS) and related diseases. The NMJ is rich in gangliosides and lacks blood-nerve barrier, thereby readily allowing access to circulating autoantibodies. The NMJ is the binding site for a wide variety of bacterial toxins [94, 95]. These observations support the long-held hypothesis that gangliosides play a major functional role in synaptic transmission. GalNAcT/ST-II double knockout mice develop significant and progressive behavioral neuropathies, including deficits in reflexes, strength, coordination, and balance [77]. Treatment of NMJs with botulinum neurotoxin type A readily induced a drastic reduction of nerve stimulation-evoked acetylcholine release (quantal content) in the wild-type mice. In contrast, the quantal content and miniature endplate potentials of the NMJs of the double knockout animals are not affected by treatment with botulinum neurotoxin [96], supporting the notion that complex gangliosides are involved in synaptic transmission. In addition, Zitman et al. [97] determined the synaptic transmission at the NMJs in ST-II knockout mouse and GalNAcT/ST-II double knockout mouse and found no major synaptic deficits in either null-mutant. However, some degree of rundown of acetylcholine release at high intensity (40 Hz) was present at the NMJs of the double knockout mice and a temperature-specific increase in acetylcholine release at 35°C was observed in the NMJs of ST-II knockout mice, compared to that in the wild type. These kinetic changes might reflect a change in postsynaptic acetylcholine receptor behavior. These data indicate that it is highly unlikely that transmission failure at NMJs contributes to the progressive motor defects of GalNAcT null-mutants and that, despite the presence of some kinetic changes of synaptic signals, neuromuscular transmission remains successful in ST-II null-mutant mice. The overall conclusion is that gangliosides may modulate temperature- and use-dependent fine-tuning of transmitter release, but are largely dispensable players in transmitter release. The mutant mice lacking GalNAcT have disrupted paranodal junctions, altered lipid rafts, mislocalization and dysfunction of ion channels, and motor nerve conduction slowing. These results suggest that gangliosides play important roles in stabilizing neuron-glia interactions at the paranodal junctions [98].
4.b.(5). Complement-dependent neurodegeneration
Furukawa et al. [3] recently reported the significance of complement regulatory proteins, such as CD55 and CD59, in protection of host organs and tissues from complement attack in degenerative diseases. They prepared triple knockout mice with disrupted complement regulating protein genes and GalNAcT genes. Generation of the triple knockout mice lacking complement activity and gangliosides provide a tool for studying the role of complement activation and inflammation, which causes neurodegeneration. This study indicates that to maintain the integrity of lipid rafts, normal composition of gangliosides is essential, since they play important roles in regulation of the complement system and suppression of inflammation.
2.b.(6). Huntington’s disease
Huntington’s disease (HD) is a member of this family of Type II trinucleotide repeat neurodegenerative disorders. Muutation in HD is an unstable expanded polyglutamine repeat tract, which is expressed at protein level. Patients of HD suffer from progressive motor, cognitive and psychiatric dysfunction. A number of genetic models of HD have been developed, which allow for investigation of the early phases of disease process, at several different levels of cell function. In addition, these models are being used to investigate the potential of a variety of therapeutic agents for clinical use. Recently, Desplats et al. [99] reported changes of gangliosides in the forebrains of R6/1 transgenic mice expressing exon 1 of the human HD gene, which carries 115 CAG repeats driven by the human huntingtin promoter. The concentration of total gangliosides did not change between the R6/1 transgenic and WT mice. The most prominent feature, however, was a reduction in GM1 content (61%) in the transgenic mice, which was compensated by an elevation of b-series gangliosides. In human brain with HD, the GD3 content was significantly elevated (162%), but not that of other gangliosides, when compared to aged-matched controls. Correlations were found between a decrease in gene expression of GalNAcT and a decrease in the downstream products GD1a, GD1b, and GT1b. Further studies [100] demonstrated that the concentration of total ganglioside was significantly lower (30%) in the cerebellum of R6/1 transgenic mice than in the WT mice. Moreover, in R6/1 mice, the Purkinje cell-enriched ganglioside LD1 (NeuAc-NeuAcα2-3Galβ1-4GlcNAcβ1-3Galβ1-4Glcβ1-1Cer) and the granule cell-enriched GD1a were significantly lower than those in the wild-type control. In contrast, the total gangliosides in the cerebellum of patients with HD were slightly elevated (24%). However, in HD striatum, the concentration of total gangliosides decreased as compared to normal subjects [101], which may be due to abnormal expression of the GT genes involved in the synthesis of gangliosides [99]. These data demonstrate an association of gangliosides in the pathogenesis of HD.
Concluding remarks
Many biosynthetic and catalytic enzymes responsible for ganglioside metabolism have been characterized, and glycogenes encoding these enzymes cloned and studied. These efforts have formed a firm foundation for elucidating the biological functions of specific gangliosides. Future research should be focused on their functions not only as structural components of biomembranes, but also their biological roles in mediating cell-cell recognition and adhesion, and cell migration, as well as in signal transduction. Recent studies on the disruption of the expression of ganglioside-synthase genes are bearing fruit in developing an understanding of these important glycolipids in normal brain development and their role in certain diseases. These findings further underlie the importance of gangliosides in maintaining membrane integrity and in regulation of brain development.
Figure 4.
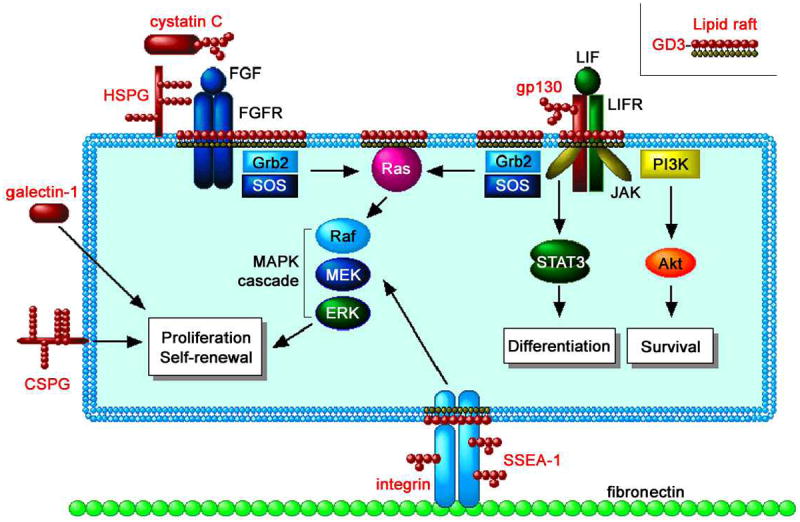
A wide variety of glycoconjugates such as heparin sulfate proteoglycan (HSPG), chondrotin sulfate proteoglycan (CSPG), N-glycosylated cystatin C, and galectins-1 are involved in signaling pathways inducing proliferation and self-renewal of NSCs. The Ras-MAPK pathway mediating the proliferation signal is modulated in the lipid rafts, cell-surface microdomains rich in GSLs (e.g., GD3). Modification of GSL expression represses the Ras-MAPK pathway but not the JAK-STAT pathway (inducing astrocyte differentiation) and the PI3K-Akt pathway (inducing survival), suggesting a specific role of GSLs in the Ras-MAPK pathway. The glycoconjugate antigens and related molecules are indicated in red. (Reproduced from [54])
Acknowledgments
This study was supported by USPHS grants (NS11853-36 and NS26994-21) and a grant from the Children’s Medical Research Foundation, Chicago, IL, to RKY. RKY also grateful acknowledges the contributions from many of his past and present collaborators for the work performed in his laboratory. It is a great pleasure to contribute to this special issue of “Neurochemical Research” honoring Dr. Robert W. Ledeen whose pioneering contributions to the field of ganglioside research have greatly enriched the field. RKY is also deeply indebted to him for his initiation and guidance into the ganglioside field during the early phase of his career development at the Albert Einstein College of Medicine, Bronx, NY.
Abbreviations
- GSL
glycosphingolipid
- CNS
central nervous system
- PNS
peripheral nervous system
- GT
glycosyltransferase
- TNFα
Tumor necrosis factor α
- NMJ
neuromuscular junction
- Tag
transgenic mouse
- WT
wild-type mouse
References
- 1.Yu RK, Nakatani Y, Yanagisawa M. The role of glycosphingolipid metabolism in the developing brain. J Lipid Res. 2009;50(Suppl):S440–445. doi: 10.1194/jlr.R800028-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yu RK, Yanagisawa M, Ariga T. Glycosphingolipid structures. In: Kamerling JP, editor. Comprehensive Glycoscience. Elsevier; Oxford, UK: 2007. pp. 73–122. [Google Scholar]
- 3.Furukawa K, Ohmi Y, Ohkawa Y, Tokuda N, Kondo Y, Tajima O. Regulatory mechanisms of nervous systems with glycosphingolipids. Neurochem Res. 2011;36:1578–1586. doi: 10.1007/s11064-011-0494-2. [DOI] [PubMed] [Google Scholar]
- 4.Anderson RG. The caveolae membrane system. Annu Rev Biochem. 1998;67:199–225. doi: 10.1146/annurev.biochem.67.1.199. [DOI] [PubMed] [Google Scholar]
- 5.Simons K, Ehehalt R. Cholesterol, lipid rafts, and disease. J Clin Invest. 2002;110:597–603. doi: 10.1172/JCI16390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hakomori S, Handa K, Iwabuchi K, Yamamura S, Prinetti A. New insights in glycosphingolipid function: “glycosignaling domain,” a cell surface assembly of glycosphingolipids with signal transducer molecules, involved in cell adhesion coupled with signaling. Glycobiology. 1998;8:xi–xix. doi: 10.1093/oxfordjournals.glycob.a018822. [DOI] [PubMed] [Google Scholar]
- 7.Ledeen RW, Wu G. Nuclear sphingolipids: metabolism and signaling. J Lipid Res. 2008;49:1176–1186. doi: 10.1194/jlr.R800009-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakamura K, Inagaki F, Tamai Y. A novel ganglioside in dogfish brain. Occurrence of a trisialoganglioside with a sialic acid linked to N-acetylgalactosamine. J Biol Chem. 1988;263:9896–9900. [PubMed] [Google Scholar]
- 9.Maccioni HJ. Glycosylation of glycolipids in the Golgi complex. J Neurochem. 2007;103(Suppl 1):81–90. doi: 10.1111/j.1471-4159.2007.04717.x. [DOI] [PubMed] [Google Scholar]
- 10.Ngamukote S, Yanagisawa M, Ariga T, Ando S, Yu RK. Developmental changes of glycosphingolipids and expression of glycogenes in mouse brains. J Neurochem. 2007;103:2327–2341. doi: 10.1111/j.1471-4159.2007.04910.x. [DOI] [PubMed] [Google Scholar]
- 11.Yu RK, Macala LJ, Taki T, Weinfield HM, Yu FS. Developmental changes in ganglioside composition and synthesis in embryonic rat brain. J Neurochem. 1988;50:1825–1829. doi: 10.1111/j.1471-4159.1988.tb02484.x. [DOI] [PubMed] [Google Scholar]
- 12.Ishii A, Ikeda T, Hitoshi S, et al. Developmental changes in the expression of glycogenes and the content of N-glycans in the mouse cerebral cortex. Glycobiology. 2007;17:261–276. doi: 10.1093/glycob/cwl076. [DOI] [PubMed] [Google Scholar]
- 13.Yu RK, Ariga T, Yanagisawa M, Zeng G. Gangliosides in the nervous system: Biosynthesis and degradation. In: Fraser-Reid B, Tatsuka K, Thiem J, editors. Glycoscience. Springer-Verlag, Berlin-Heiderberg; Germany: 2008. pp. 1671–1695. [Google Scholar]
- 14.Suzuki Y, Yanagisawa M, Ariga T, Yu RK. Histone acetylation-mediated glycosyltransferase gene regulation in mouse brain during development. J Neurochem. 2011;116:874–880. doi: 10.1111/j.1471-4159.2010.07042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu RK, Bieberich E, Xia T, Zeng G. Regulation of ganglioside biosynthesis in the nervous system. J Lipid Res. 2004;45:783–793. doi: 10.1194/jlr.R300020-JLR200. [DOI] [PubMed] [Google Scholar]
- 16.Fukumoto S, Miyazaki H, Goto G, Urano T, Furukawa K. Expression cloning of mouse cDNA of CMP-NeuAc:Lactosylceramide alpha2,3-sialyltransferase, an enzyme that initiates the synthesis of gangliosides. J Biol Chem. 1999;274:9271–9276. doi: 10.1074/jbc.274.14.9271. [DOI] [PubMed] [Google Scholar]
- 17.Kono M, Takashima S, Liu H, et al. Molecular cloning and functional expression of a fifth-type alpha 2,3-sialyltransferase (mST3Gal V: GM3 synthase) Biochem Biophys Res Commun. 1998;253:170–175. doi: 10.1006/bbrc.1998.9768. [DOI] [PubMed] [Google Scholar]
- 18.Xia T, Zeng G, Gao L, Yu RK. Sp1 and AP2 enhance promoter activity of the mouse GM3-synthase gene. Gene. 2005;351:109–118. doi: 10.1016/j.gene.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 19.Chung T-W, Choi H-J, Lee Y-C, Kim C-H. Molecular mechanism for transcriptional activation of ganglioside GM3 synthase and its function in differentiation of HL-60 cells. Glycobiology. 2005;15:233–244. doi: 10.1093/glycob/cwh156. [DOI] [PubMed] [Google Scholar]
- 20.Kim S-W, Lee S-H, Kim K-S, Kim C-H, Choo Y-K, Lee Y-C. Isolation and characterization of the promoter region of the human GM3 synthase gene. Biochim Biophys Acta. 2002;1578:84–89. doi: 10.1016/s0167-4781(02)00505-5. [DOI] [PubMed] [Google Scholar]
- 21.Zeng G, Gao L, Xia T, Tencomnao T, Yu RK. Characterization of the 5’-flanking fragment of the human GM3-synthase gene. Biochim Biophys Acta. 2003;1625:30–35. doi: 10.1016/s0167-4781(02)00573-0. [DOI] [PubMed] [Google Scholar]
- 22.Zeng G, Gao L, Yu RK. Isolation and functional analysis of the promoter of the rat CMP-NeuAc:GM3 alpha2,8 sialyltransferase gene 1. Biochim Biophys Acta. 1998;1397:126–130. doi: 10.1016/s0167-4781(98)00030-x. [DOI] [PubMed] [Google Scholar]
- 23.Takashima S, Kono M, Kurosawa N, et al. Genomic organization and transcriptional regulation of the mouse GD3 synthase gene (ST8Sia I): comparison of genomic organization of the mouse sialyltransferase genes. J Biochem (Tokyo) 2000;128:1033–1043. doi: 10.1093/oxfordjournals.jbchem.a022831. [DOI] [PubMed] [Google Scholar]
- 24.Furukawa K, Horie M, Okutomi K, Sugano S. Isolation and functional analysis of the melanoma specific promoter region of human GD3 synthase gene. Biochim Biophys Acta. 2003;1627:71–78. doi: 10.1016/s0167-4781(03)00076-9. [DOI] [PubMed] [Google Scholar]
- 25.Furukawa K, Soejima H, Niikawa N, Shiku H. Genomic organization and chromosomal assignment of the human beta1, 4-N-acetylgalactosaminyltransferase gene. Identification of multiple transcription units. J Biol Chem. 1996;271:20836–20844. doi: 10.1074/jbc.271.34.20836. [DOI] [PubMed] [Google Scholar]
- 26.Xia T, Gao L, Yu RK, Zeng G. Characterization of the promoter and the transcription factors for the mouse UDP-Gal:betaGlcNAc beta1,3-galactosyltransferase gene. Gene. 2003;309:117–123. doi: 10.1016/s0378-1119(03)00496-7. [DOI] [PubMed] [Google Scholar]
- 27.Ichikawa S, Ozawa K, Hirabayashi Y. Molecular cloning and characterization of the mouse ceramide glucosyltransferase gene. Biochem Biophys Res Commun. 1998;253:707–711. doi: 10.1006/bbrc.1998.9855. [DOI] [PubMed] [Google Scholar]
- 28.Yu RK, Lee SH. In vitro biosynthesis of sialosylgalactosylceramide (G7) by mouse brain microsomes. J Biol Chem. 1976;251:198–203. [PubMed] [Google Scholar]
- 29.Tencomnao T, Yu RK, Kapitonov D. Characterization of the human UDP-galactose:ceramide galactosyltransferase gene promoter. Biochim Biophys Acta. 2001;1517:416–423. doi: 10.1016/s0167-4781(00)00283-9. [DOI] [PubMed] [Google Scholar]
- 30.Tencomnao T, Kapitonov D, Bieberich E, Yu RK. Transcriptional regulation of the human UDP-galactose:ceramide galactosyltransferase (hCGT) gene expression: functional role of GC-box and CRE. Glycoconj J. 2004;20:339–351. doi: 10.1023/B:GLYC.0000033630.58533.16. [DOI] [PubMed] [Google Scholar]
- 31.Goldberg AD, Allis CD, Bernstein E. Epigenetics: a landscape takes shape. Cell. 2007;128:635–638. doi: 10.1016/j.cell.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 32.Mohn F, Schübeler D. Genetics and epigenetics: stability and plasticity during cellular differentiation. Trends Genet. 2009;25:129–136. doi: 10.1016/j.tig.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 33.Kizuka Y, Kitazume S, Yoshida M, Taniguchi N. Brain-specific expression of N-acetylglucosaminyltransferase IX (GnT-IX) is regulated by epigenetic histone modifications. J Biol Chem. 2011;286:31875–31884. doi: 10.1074/jbc.M111.251173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bieberich E, MacKinnon S, Silva J, Yu RK. Regulation of apoptosis during neuronal differentiation by ceramide and b-series complex gangliosides. J Biol Chem. 2001;276:44396–44404. doi: 10.1074/jbc.M107239200. [DOI] [PubMed] [Google Scholar]
- 35.Fang Y, Wu G, Xie X, Lu ZH, Ledeen RW. Endogenous GM1 ganglioside of the plasma membrane promotes neuritogenesis by two mechanisms. Neurochem Res. 2000;25:931–940. doi: 10.1023/a:1007596223484. [DOI] [PubMed] [Google Scholar]
- 36.Wu G, Fang Y, Lu ZH, Ledeen RW. Induction of axon-like and dendrite-like processes in neuroblastoma cells. J Neurocytol. 1998;27:1–14. doi: 10.1023/a:1006910001869. [DOI] [PubMed] [Google Scholar]
- 37.Wu G, Lu ZH, Xie X, Li L, Ledeen RW. Mutant NG108-15 cells (NG-CR72) deficient in GM1 synthase respond aberrantly to axonogenic stimuli and are vulnerable to calcium-induced apoptosis: they are rescued with LIGA-20. J Neurochem. 2001;76:690–702. doi: 10.1046/j.1471-4159.2001.00036.x. [DOI] [PubMed] [Google Scholar]
- 38.Simpson MA, Cross H, Proukakis C, et al. Infantile-onset symptomatic epilepsy syndrome caused by a homozygous loss-of-function mutation of GM3 synthase. Nat Genet. 2004;36:1225–1229. doi: 10.1038/ng1460. [DOI] [PubMed] [Google Scholar]
- 39.Proia RL. Glycosphingolipid functions: insights from engineered mouse models. Philos Trans R Soc Lond B Biol Sci. 2003;358:879–883. doi: 10.1098/rstb.2003.1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ohmi Y, Tajima O, Ohkawa Y, Mori A, Sugiura Y, Furukawa K. Gangliosides play pivotal roles in the regulation of complement systems and in the maintenance of integrity in nerve tissues. Proc Natl Acad Sci U S A. 2009;106:22405–22410. doi: 10.1073/pnas.0912336106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ohmi Y, Tajima O, Ohkawa Y, Yamauchi Y, Sugiura Y, Furukawa K. Gangliosides are essential in the protection of inflammation and neurodegeneration via maintenance of lipid rafts: elucidation by a series of ganglioside-deficient mutant mice. J Neurochem. 2011;116:926–935. doi: 10.1111/j.1471-4159.2010.07067.x. [DOI] [PubMed] [Google Scholar]
- 42.Kawai H, Allende ML, Wada R, et al. Mice expressing only monosialoganglioside GM3 exhibit lethal audiogenic seizures. J Biol Chem. 2001;276:6885–6888. doi: 10.1074/jbc.C000847200. [DOI] [PubMed] [Google Scholar]
- 43.Okada M, Itoh Mi M, Haraguchi M, et al. b-series Ganglioside deficiency exhibits no definite changes in the neurogenesis and the sensitivity to Fas-mediated apoptosis but impairs regeneration of the lesioned hypoglossal nerve. J Biol Chem. 2002;277:1633–1636. doi: 10.1074/jbc.C100395200. [DOI] [PubMed] [Google Scholar]
- 44.Dupree JL, Coetzee T, Suzuki K, Popko B. Myelin abnormalities in mice deficient in galactocerebroside and sulfatide. J Neurocytol. 1998;27:649–659. doi: 10.1023/a:1006908013972. [DOI] [PubMed] [Google Scholar]
- 45.Ezoe T, Vanier MT, Oya Y, et al. Biochemistry and neuropathology of mice doubly deficient in synthesis and degradation of galactosylceramide. J Neurosci Res. 2000;59:170–178. doi: 10.1002/(sici)1097-4547(20000115)59:2<170::aid-jnr3>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 46.Furukawa K, Takamiya K, Okada M, Inoue M, Fukumoto S, Furukawa K. Novel functions of complex carbohydrates elucidated by the mutant mice of glycosyltransferase genes. Biochim Biophys Acta. 2001;1525:1–12. doi: 10.1016/s0304-4165(00)00185-9. [DOI] [PubMed] [Google Scholar]
- 47.Takamiya K, Yamamoto A, Furukawa K, et al. Mice with disrupted GM2/GD2 synthase gene lack complex gangliosides but exhibit only subtle defects in their nervous system. Proc Natl Acad Sci U S A. 1996;93:10662–10667. doi: 10.1073/pnas.93.20.10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yamashita T, Wu YP, Sandhoff R, et al. Interruption of ganglioside synthesis produces central nervous system degeneration and altered axon-glial interactions. Proc Natl Acad Sci U S A. 2005;102:2725–2730. doi: 10.1073/pnas.0407785102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yoshikawa M, Go S, Takasaki K, et al. Mice lacking ganglioside GM3 synthase exhibit complete hearing loss due to selective degeneration of the organ of Corti. Proc Natl Acad Sci U S A. 2009;106:9483–9488. doi: 10.1073/pnas.0903279106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yu RK, Nakatani Y, Yanagisawa M. The role of glycosphingolipid metabolism in the developing brain. J Lipid Res. 2009;50(Suppl):S440–445. doi: 10.1194/jlr.R800028-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu RK, Tsai YT, Ariga T, Yanagisawa M. Structures, biosynthesis, and functions of gangliosides-an overview. Journal of oleo science. 2011;60:537–544. doi: 10.5650/jos.60.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McKay R. Stem cells in the central nervous system. Science. 1997;276:66–71. doi: 10.1126/science.276.5309.66. [DOI] [PubMed] [Google Scholar]
- 53.Temple S, Alvarez-Buylla A. Stem cells in the adult mammalian central nervous system. Curr Opin Neurobiol. 1999;9:135–141. doi: 10.1016/s0959-4388(99)80017-8. [DOI] [PubMed] [Google Scholar]
- 54.Weiss S, Reynolds BA, Vescovi AL, Morshead C, Craig CG, van der Kooy D. Is there a neural stem cell in the mammalian forebrain? Trends Neurosci. 1996;19:387–393. doi: 10.1016/s0166-2236(96)10035-7. [DOI] [PubMed] [Google Scholar]
- 55.Yanagisawa M, Yu RK. The expression and functions of glycoconjugates in neural stem cells. Glycobiology. 2007;17:57R–74R. doi: 10.1093/glycob/cwm018. [DOI] [PubMed] [Google Scholar]
- 56.Liour SS, Dinkins MB, Su CY, Yu RK. Spatiotemporal expression of GM1 in murine medial pallial neural progenitor cells. J Comp Neurol. 2005;491:330–338. doi: 10.1002/cne.20696. [DOI] [PubMed] [Google Scholar]
- 57.Liour SS, Kraemer SA, Dinkins MB, Su CY, Yanagisawa M, Yu RK. Further characterization of embryonic stem cell-derived radial glial cells. Glia. 2006;53:43–56. doi: 10.1002/glia.20257. [DOI] [PubMed] [Google Scholar]
- 58.Yanagisawa M, Taga T, Nakamura K, Ariga T, Yu RK. Characterization of glycoconjugate antigens in mouse embryonic neural precursor cells. J Neurochem. 2005;95:1311–1320. doi: 10.1111/j.1471-4159.2005.03452.x. [DOI] [PubMed] [Google Scholar]
- 59.Yang CR, Liour SS, Dasgupta S, Yu RK. Inhibition of neuronal migration by JONES antibody is independent of 9-O-acetyl GD3 in GD3-synthase knockout mice. J Neurosci Res. 2007;85:1381–1390. doi: 10.1002/jnr.21264. [DOI] [PubMed] [Google Scholar]
- 60.Yu RK, Yanagisawa M. Glycosignaling in neural stem cells: involvement of glycoconjugates in signal transduction modulating the neural stem cell fate. J Neurochem. 2007;103(Suppl 1):39–46. doi: 10.1111/j.1471-4159.2007.04710.x. [DOI] [PubMed] [Google Scholar]
- 61.Doetsch F, Caille I, Lim DA, Garcia-Verdugo JM, Alvarez-Buylla A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell. 1999;97:703–716. doi: 10.1016/s0092-8674(00)80783-7. [DOI] [PubMed] [Google Scholar]
- 62.Seri B, Garcia-Verdugo JM, McEwen BS, Alvarez-Buylla A. Astrocytes give rise to new neurons in the adult mammalian hippocampus. J Neurosci. 2001;21:7153–7160. doi: 10.1523/JNEUROSCI.21-18-07153.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yanagisawa M, Nakamura K, Taga T. Roles of lipid rafts in integrin-dependent adhesion and gp130 signalling pathway in mouse embryonic neural precursor cells. Genes Cells. 2004;9:801–809. doi: 10.1111/j.1365-2443.2004.00764.x. [DOI] [PubMed] [Google Scholar]
- 64.Suzuki Y, Yanagisawa M, Yagi H, Nakatani Y, Yu RK. Involvement of beta1-integrin up-regulation in basic fibroblast growth factor- and epidermal growth factor-induced proliferation of mouse neuroepithelial cells. J Biol Chem. 2010;285:18443–18451. doi: 10.1074/jbc.M110.114645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yanagisawa M, Nakamura K, Taga T. Glycosphingolipid synthesis inhibitor represses cytokine-induced activation of the Ras-MAPK pathway in embryonic neural precursor cells. J Biochem. 2005;138:285–291. doi: 10.1093/jb/mvi129. [DOI] [PubMed] [Google Scholar]
- 66.Ohkawa Y, Miyazaki S, Hamamura K, et al. Ganglioside GD3 enhances adhesion signals and augments malignant properties of melanoma cells by recruiting integrins to glycolipid-enriched microdomains. J Biol Chem. 2010;285:27213–27223. doi: 10.1074/jbc.M109.087791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jennemann R, Sandhoff R, Wang S, et al. Cell-specific deletion of glucosylceramide synthase in brain leads to severe neural defects after birth. Proc Natl Acad Sci U S A. 2005;102:12459–12464. doi: 10.1073/pnas.0500893102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jung JU, Ko K, Lee DH, Ko K, Chang KT, Choo YK. The roles of glycosphingolipids in the proliferation and neural differentiation of mouse embryonic stem cells. Exp Mol Med. 2009;41:935–945. doi: 10.3858/emm.2009.41.12.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shevchuk NA, Hathout Y, Epifano O, et al. Alteration of ganglioside synthesis by GM3 synthase knockout in murine embryonic fibroblasts. Biochim Biophys Acta. 2007;1771:1226–1234. doi: 10.1016/j.bbalip.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 70.Niimi K, Nishioka C, Miyamoto T, et al. Impairment of neuropsychological behaviors in ganglioside GM3-knockout mice. Biochem Biophys Res Commun. 2011;406:524–528. doi: 10.1016/j.bbrc.2011.02.071. [DOI] [PubMed] [Google Scholar]
- 71.Handa Y, Ozaki N, Honda T, et al. GD3 synthase gene knockout mice exhibit thermal hyperalgesia and mechanical allodynia but decreased response to formalin-induced prolonged noxious stimulation. Pain. 2005;117:271–279. doi: 10.1016/j.pain.2005.06.016. [DOI] [PubMed] [Google Scholar]
- 72.Furukawa K, Aixinjueluo W, Kasama T, et al. Disruption of GM2/GD2 synthase gene resulted in overt expression of 9-O-acetyl GD3 irrespective of Tis21. J Neurochem. 2008;105:1057–1066. doi: 10.1111/j.1471-4159.2008.05232.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Takamiya K, Yamamoto A, Furukawa K, et al. Mice with disrupted GM2/GD2 synthase gene lack complex gangliosides but exhibit only subtle defects in their nervous system. Proc Natl Acad Sci U S A. 1996;93:10662–10667. doi: 10.1073/pnas.93.20.10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sheikh KA, Sun J, Liu Y, et al. Mice lacking complex gangliosides develop Wallerian degeneration and myelination defects. Proc Natl Acad Sci U S A. 1999;96:7532–7537. doi: 10.1073/pnas.96.13.7532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chiavegatto S, Sun J, Nelson RJ, Schnaar RL. A functional role for complex gangliosides: motor deficits in GM2/GD2 synthase knockout mice. Exp Neurol. 2000;166:227–234. doi: 10.1006/exnr.2000.7504. [DOI] [PubMed] [Google Scholar]
- 76.Sugiura Y, Furukawa K, Tajima O, Mii S, Honda T. Sensory nerve-dominant nerve degeneration and remodeling in the mutant mice lacking complex gangliosides. Neuroscience. 2005;135:1167–1178. doi: 10.1016/j.neuroscience.2005.07.035. [DOI] [PubMed] [Google Scholar]
- 77.Tajima O, Egashira N, Ohmi Y, et al. Reduced motor and sensory functions and emotional response in GM3-only mice: emergence from early stage of life and exacerbation with aging. Behav Brain Res. 2009;198:74–82. doi: 10.1016/j.bbr.2008.10.024. [DOI] [PubMed] [Google Scholar]
- 78.Tajima O, Egashira N, Ohmi Y, et al. Dysfunction of muscarinic acetylcholine receptors as a substantial basis for progressive neurological deterioration in GM3-only mice. Behav Brain Res. 2010;206:101–108. doi: 10.1016/j.bbr.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 79.Inoue M, Fujii Y, Furukawa K, et al. Refractory skin injury in complex knock-out mice expressing only the GM3 ganglioside. J Biol Chem. 2002;277:29881–29888. doi: 10.1074/jbc.M201631200. [DOI] [PubMed] [Google Scholar]
- 80.Yamashita T, Hashiramoto A, Haluzik M, et al. Enhanced insulin sensitivity in mice lacking ganglioside GM3. Proc Natl Acad Sci U S A. 2003;100:3445–3449. doi: 10.1073/pnas.0635898100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.de la Monte SM, Wands JR. Review of insulin and insulin-like growth factor expression, signaling, and malfunction in the central nervous system: relevance to Alzheimer’s disease. Journal of Alzheimer’s disease : JAD. 2005;7:45–61. doi: 10.3233/jad-2005-7106. [DOI] [PubMed] [Google Scholar]
- 82.Farukhi F, Dakkouri C, Wang H, Wiztnitzer M, Traboulsi EI. Etiology of vision loss in ganglioside GM3 synthase deficiency. Ophthalmic Genet. 2006;27:89–91. doi: 10.1080/13816810600862626. [DOI] [PubMed] [Google Scholar]
- 83.Yanagisawa K. Pathological significance of ganglioside clusters in Alzheimer’s disease. J Neurochem. 2011;116:806–812. doi: 10.1111/j.1471-4159.2010.07006.x. [DOI] [PubMed] [Google Scholar]
- 84.Ariga T, McDonald MP, Yu RK. Role of ganglioside metabolism in the pathogenesis of Alzheimer’s disease--a review. J Lipid Res. 2008;49:1157–1175. doi: 10.1194/jlr.R800007-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ariga T, Wakade C, Yu RK. The pathological roles of ganglioside metabolism in Alzheimer’s disease: effects of gangliosides on neurogenesis. Int J Alzheimers Dis. 2011 doi: 10.4061/2011/193618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Oikawa N, Yamaguchi H, Ogino K, et al. Gangliosides determine the amyloid pathology of Alzheimer’s disease. Neuroreport. 2009;20:1043–1046. doi: 10.1097/WNR.0b013e32832e4b9d. [DOI] [PubMed] [Google Scholar]
- 87.Bernardo A, Harrison FE, McCord M, et al. Elimination of GD3 synthase improves memory and reduces amyloid-beta plaque load in transgenic mice. Neurobiol Aging. 2009;30:1777–1791. doi: 10.1016/j.neurobiolaging.2007.12.022. [DOI] [PubMed] [Google Scholar]
- 88.Ando S, Hirabayashi Y, Kon K, Inagaki F, Tate S, Whittaker VP. A trisialoganglioside containing a sialyl alpha 2-6 N-acetylgalactosamine residue is a cholinergic-specific antigen, Chol-1 alpha. J Biochem. 1992;111:287–290. doi: 10.1093/oxfordjournals.jbchem.a123751. [DOI] [PubMed] [Google Scholar]
- 89.Ariga T, Yanagisawa M, Wakade C, et al. Ganglioside metabolism in a transgenic mouse model of Alzheimer’s disease: expression of Chol-1alpha antigens in the brain. ASN Neuro. 2010;2:e00044. doi: 10.1042/AN20100021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wu G, Lu ZH, Kulkarni N, Amin R, Ledeen RW. Mice lacking major brain gangliosides develop parkinsonism. Neurochem Res. 2011;36:1706–1714. doi: 10.1007/s11064-011-0437-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kaida K, Ariga T, Yu RK. Antiganglioside antibodies and their pathophysiological effects on Guillain-Barré syndrome and related disorders–a review. Glycobiology. 2009;19:676–692. doi: 10.1093/glycob/cwp027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yu RK, Usuki S, Ariga T. Ganglioside molecular mimicry and its pathological roles in Guillain-Barré syndrome and related diseases. Infection and immunity. 2006;74:6517–6527. doi: 10.1128/IAI.00967-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yu RK, Ariga T, Usuki S, Kaida K. Pathological roles of ganglioside mimicry in Guillain-Barré syndrome and related neuropathies. Advances in experimental medicine and biology. 2011;705:349–365. doi: 10.1007/978-1-4419-7877-6_17. [DOI] [PubMed] [Google Scholar]
- 94.Willison HJ, Yuki N. Peripheral neuropathies and anti-glycolipid antibodies. Brain. 2002;125:2591–2625. doi: 10.1093/brain/awf272. [DOI] [PubMed] [Google Scholar]
- 95.Ariga T, Yu RK. Antiglycolipid antibodies in Guillain-Barre syndrome and related diseases: review of clinical features and antibody specificities. J Neurosci Res. 2005;80:1–17. doi: 10.1002/jnr.20395. [DOI] [PubMed] [Google Scholar]
- 96.Bullens RW, O’Hanlon GM, Wagner E, et al. Roles of complex gangliosides at the neuromuscular junction. Ann N Y Acad Sci. 2003;998:401–403. doi: 10.1196/annals.1254.051. [DOI] [PubMed] [Google Scholar]
- 97.Zitman FM, Todorov B, Jacobs BC, et al. Neuromuscular synaptic function in mice lacking major subsets of gangliosides. Neuroscience. 2008;156:885–897. doi: 10.1016/j.neuroscience.2008.08.034. [DOI] [PubMed] [Google Scholar]
- 98.Susuki K, Baba H, Tohyama K, et al. Gangliosides contribute to stability of paranodal junctions and ion channel clusters in myelinated nerve fibers. Glia. 2007;55:746–757. doi: 10.1002/glia.20503. [DOI] [PubMed] [Google Scholar]
- 99.Desplats PA, Denny CA, Kass KE, et al. Glycolipid and ganglioside metabolism imbalances in Huntington’s disease. Neurobiol Dis. 2007;27:265–277. doi: 10.1016/j.nbd.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Denny CA, Desplats PA, Thomas EA, Seyfried TN. Cerebellar lipid differences between R6/1 transgenic mice and humans with Huntington’s disease. J Neurochem. 2010;115:748–758. doi: 10.1111/j.1471-4159.2010.06964.x. [DOI] [PubMed] [Google Scholar]
- 101.Higatsberger MR, Sperk G, Bernheimer H, Shannak KS, Hornykiewicz O. Striatal ganglioside levels in the rat following kainic acid lesions: comparison with Huntington’s disease. Exp Brain Res. 1981;44:93–96. doi: 10.1007/BF00238752. [DOI] [PubMed] [Google Scholar]
- 102.Svennerholm L. Chromatographic separation of human brain gangliosides. J Neurochem. 1963;10:613–623. doi: 10.1111/j.1471-4159.1963.tb08933.x. [DOI] [PubMed] [Google Scholar]
- 103.IUPAC-IUBMB Joint Commission on Biochemical Nomenclature. Nomenclature of glycolipids. Pure Appl Chem. 1997;69:2475–2487. [Google Scholar]
- 104.Okada M, Itoh Mi M, Haraguchi M, et al. b-series Ganglioside deficiency exhibits no definite changes in the neurogenesis and the sensitivity to Fas-mediated apoptosis but impairs regeneration of the lesioned hypoglossal nerve. J Biol Chem. 2002;277:1633–1636. doi: 10.1074/jbc.C100395200. [DOI] [PubMed] [Google Scholar]