

Abstract
The long bones of the vertebrate body are built by the initial formation of a cartilage template that is later replaced by mineralized bone. The proliferation and maturation of the skeletal precursor cells (chondrocytes) within the cartilage template and their replacement by bone is a highly coordinated process which, if misregulated, can lead to a number of defects including dwarfism and other skeletal deformities. This is exemplified by the fact that abnormal bone development is one of the most common types of human birth defects. Yet, many of the factors that initiate and regulate chondrocyte maturation are not known. We identified a recessive dwarf mouse mutant (pug) from an N-ethyl-N-nitrosourea (ENU) mutagenesis screen. pug mutant skeletal elements are patterned normally during development, but display a ~20% length reduction compared to wild-type embryos. We show that the pug mutation does not lead to changes in chondrocyte proliferation but instead promotes premature maturation and early ossification, which ultimately leads to disproportionate dwarfism. Using sequence capture and high-throughput sequencing, we identified a missense mutation in the Xylosyltransferase 1 (Xylt1) gene in pug mutants. Xylosyltransferases catalyze the initial step in glycosaminoglycan (GAG) chain addition to proteoglycan core proteins, and these modifications are essential for normal proteoglycan function. We show that the pug mutation disrupts Xylt1 activity and subcellular localization, leading to a reduction in GAG chains in pug mutants. The pug mutant serves as a novel model for mammalian dwarfism and identifies a key role for proteoglycan modification in the initiation of chondrocyte maturation.
Keywords: forward genetics, Xylt1, dwarfism, chondrocytes, skeletal development, proteoglycans, CSPGs, HSPGs, glycosaminoglycans
INTRODUCTION
One of the defining features of vertebrates is their internal skeleton. While the global organization of the skeleton is conserved across vertebrates, changes in the number, size, or shape of specific skeletal elements correspond to newly acquired or modified structures induced through evolutionary pressure. Within a species, however, the organization and morphology of skeletal elements are much more constrained; suggesting that tight regulation of skeletal development is essential for an animal’s reproductive fitness. Most skeletal elements in the appendicular and axial skeleton of the vertebrate body are formed through the process of endochondral ossification that involves the formation of a cartilage anlage, within which chondrocytes undergo a maturation process before being replaced by mature bone. This process begins with the condensation and aggregation of mesenchymal cells, which then differentiate into chondrocytes (Shimizu et al., 2007). As the cartilage anlage grows, the chondrocytes become arranged into morphologically distinct zones of resting, proliferative, prehypertrophic, and hypertrophic chondrocytes, called growth plates (Goldring et al., 2006). Progression through the growth plates occurs as chondrocytes mature, resulting in the most mature cells located near the center of the skeletal element. While bone is laid down in the center of the anlage by invading osteoblasts, the element continues to grow on either end within the growth plates via highly regulated proliferation of a subset of the chondrocyte pool. This process, if misregulated, leads to skeletal abnormalities that can affect the size, shape and strength of skeletal elements (Baldridge et al., 2010).
Multiple signaling pathways regulate chondrogenesis, and tight regulation of their signaling output is critical for normal skeletal development. For example, achondroplasia, the most common form of human dwarfism, is associated with excess Fibroblast growth factor (Fgf) signaling due to activating mutations in Fgfr3. This results in the rhizomelic shortening of limb bones, or a more severe shortening of proximal limb elements compared to distal elements (Deng et al., 1996; Ornitz, 2005; Rousseau et al., 1994; Segev et al., 2000). Signaling molecules such as Indian Hedgehog (Ihh) and Parathyroid hormone related protein (PTHrP) (Retting et al., 2009; Shimizu et al., 2007) also regulate chondrogenesis and maturation. A loss of either PTHrP or its receptor, PTH1R, results in reduced skeletal element size and premature maturation, while a loss of Ihh results in reduced skeletal elements size due to delayed maturation and a disorganization of the growth plate (Karaplis et al., 1994; Lanske et al., 1996; Vortkamp et al., 1996). Though these signaling pathways each have specific roles, they do not act in isolation; crosstalk between the major pathways helps to maintain the proper rate and coordination of chondrocyte maturation to create an appropriately sized and patterned skeleton (Amizuka et al., 2004; Kronenberg, 2003; Minina et al., 2002).
Normal endochondral ossification also depends upon a distinct and complex extracellular matrix (ECM) comprising cartilage-specific collagens, elastin, and proteoglycans. The ECM functions both as a structural component for the skeleton, especially at the joints, but also as a regulator of signaling molecule diffusion and function within the growth plate. Signaling factors must traverse and interact with the extracellular matrix in order to induce their effects. Ihh, expressed in prehypertrophic chondrocytes, signals both short-range to activate its downstream targets such as Patched1 (Ptch1) and Gli1, but also long-range to regulate the expression of PTHrP at the heads of the developing skeletal elements (Gritli-Linde et al., 2001; Karaplis et al., 1994; Karp et al., 2000; Kobayashi et al., 2005; Mak et al., 2008; St-Jacques et al., 1999). This long-range signaling is critical to modulate maturation of the resting pool of chondrocytes (Kronenberg, 2003; Lanske et al., 1996; Vortkamp et al., 1996). Runx2 also functions downstream of Ihh in order to regulate maturation from prehypertrophic to hypertrophic chondrocytes, and is required for bone formation (Enomoto et al., 2000; Enomoto-Iwamoto et al., 2001; Takarada et al., 2013; Takeda et al., 2001). Similarly, during endochondral ossification, Fgf7, 8, 9, 17, and 18 are expressed primarily in the perichondrium, or the fibrous layer of cells surrounding the condensed chondrocytes, and signal to Fgfr3 expressed on proliferative and resting chondrocytes (Hung et al., 2007; Liu et al., 2002; Ornitz, 2005). Despite a large body of work on human bone diseases and animal models, the relationship between skeletal development and defects in the ECM are understudied.
To identify factors that regulate skeletal element length, we took a forward genetics approach and studied a recessive mouse mutant, pug that was identified from a N-ethyl-N-nitrosourea mutagenesis screen. Homozygous pug animals display a dwarfism phenotype, with shortened long bones compared to their wild type or heterozygous littermates. However, this is not due to changes in the proliferation of chondrocytes, but rather to their early maturation. The premature progression of pug chondrocytes through the maturation process occurs prior to changes in Ihh and Fgf signaling, suggesting chondrocyte maturation initiates independently of these signaling pathways. We further show that the underlying genetic defect in pug mutants is a missense mutation in the Xylosyltransferase 1 (Xylt1) gene. Xylosyltransferases catalyze a rate-limiting step in the addition of glycosaminoglycan (GAG) side chains to proteoglycans (Cuellar et al., 2007; Esko et al., 1985; Kearns et al., 1993; Ponighaus et al., 2007; Prante et al., 2006), which is critical for proteoglycan function within the extracellular matrix. We show that Xylt1 is specifically expressed in chondrocytes during embryonic development and, as in other studies, Xylt1 protein localizes within the cis-Golgi where it functions as a xylosyltransferase (Nuwayhid et al., 1986; Schön et al., 2006a). We find that pug is a hypomorphic allele of Xylt1 that results both in reduced Xylt1 activity and altered Xylt1 subcellular localization. We further show that GAG chain levels are reduced in pug mutants and these changes likely underlie the dwarfism phenotype. The defect in proteoglycan processing and subsequent premature maturation in pug mutants highlights a key role for Xylt1 and glycosaminoglycans in the regulation of chondrocyte maturation during skeletal development.
MATERIALS & METHODS
Mouse Strains
The pug mutant was identified as part of a mutagenesis screen (described in (Liem et al., 2009)). Briefly, C57BL6/J males were mutagenized with N-ethyl-N-nitrosourea (ENU) and crossed to FVB/NJ females. F1 males were further mated to FVB/NJ females to generate G2 females. Embryos from F1 × G2 crosses were analyzed for recessive phenotypes. The pug mutant was identified at embryonic day (e) 18.5 based on shorter limbs compared to littermates. PTHrP-LacZ mice were crossed to pug animals and genotyped as described previously for LacZ (Chen et al., 2006).
Mapping of the pug Mutation
Using a whole genome single nucleotide polymorphism (SNP) panel (Moran et al., 2006), pug was mapped to Chromosome 7 and the pug interval was subsequently narrowed to between SNP markers rs31909733 (122.29 Mb) and rs6261444 (130.1 Mb) via meiotic recombination mapping. The pug mutation has been crossed >10 generations onto the FVB/NJ background, which removed more than 99.9% of the original mutagenized C57BL/6J background, supporting the idea that the pug phenotype is monogenic. Mutant characterization was carried out at various stages of crossing into the FVB/NJ background.
Sequence Capture
A Nimblegen mouse Sequence Capture 385K array was designed to contain oligos complementary to the exons within the pug genomic locus (NCBI37/mm9 Chr7: 122,297,973–130,145,967Mb), minus repetitive sequences. Genomic DNA from a pug homozygote was isolated and then sheared by sonication, and adaptors were ligated to the resulting fragments. The adaptor-ligated templates were fractionated by agarose gel electrophoresis and fragments of the desired size were excised. Extracted DNA was amplified by ligation-mediated PCR, purified, and hybridized to the Sequence Capture array. The array was washed, and bound DNA was eluted, purified, and amplified by ligation-mediated PCR (similar to methods employed in (Choi et al., 2009)). The capture and sequencing experiments were performed at the W.M. Keck Foundation for Biotechnology Resources at Yale. This array also contained sequences from Chromosomes 4 & 11, unrelated to the pug locus. For details about these sequences, please contact the authors.
Sequencing and Mutation Analysis
Captured libraries were sequenced on an Illumina Genome Analyzer II as single-end, 75-bp reads using previously described methods (Choi et al., 2009). Illumina reads were first trimmed based on their quality scores to remove low-quality regions using the program Btrim (Kong, 2011). A cutoff of 20 for average quality scores within a moving window of size 5-bp was used. Minimum acceptable read length was 25-bp. Other parameters of Btrim were set to defaults. The pre-processed reads were then aligned to the mouse genome reference sequence (mm9) using the BWA mapping program (Li and Durbin, 2009). The mapping results were converted into SAMtools pileup format using SAMtools programs (Li et al., 2009). PCR duplicates were removed using the rmdup command from SAMtools. Single nucleotide variations (SNVs) were called using SAMtools’ pileup command. Further filtering was performed using in-house scripts to exclude those SNV calls that had less than three reads or a SNP score less than 20. Annotation was added based on the UCSC RefSeq gene model (http://genome.ucsc.edu/ (Pruitt et al., 2009)).
Stage Matching Early Embryos
To ensure that control and pug mutant embryos from e13 litters were matched in terms of embryonic development, we paired embryos based on crown-rump length, measured using an eyepiece micrometer. Using this comparative method, we observed no difference in the distribution of crown-rump length between pug and control embryos, indicating that overall embryo size was unchanged in the pug mutants at these early stages. We defined early e13 embryos as having a crown-rump length of 7.9mm – 9.3mm while e13.5 embryos were defined as those with a crown-rump length of 9.35mm – 10.3mm.
Expression Analysis
In situ hybridization and immunofluorescence analysis were performed using standard methods (Nagy et al., 2002). Antibodies were obtained from various sources: XYLT1 (1:25–50; Sigma, Prestige Antibodies, St Louis, MO), GM130 (1:500; BD Biosciences, Franklin Lakes, NJ), phospho-histone H3 (1:500; Millipore, Temecula, CA) and phospho-44/42 MAPK (i.e ERK1/2; 1:100; Cell Signaling, Danvers, MA). OCT and paraffin embedded samples underwent heat-induced epitope retrieval in 10mM sodium citrate in the 2100-Retriever (E.M.S., Hatfield, PA) following manufacturer’s specifications. Quantifications of phospho-44/42 MAPK and phospho-histone H3 were performed using standard plug-ins in ImageJ.
Skeletal Staining
Embryonic skeletons were prepared and stained with Alcian Blue and Alizarin Red, then cleared using standard methods (Nagy et al., 2002). Adult skeletons were processed according to standard procedures (Selby, 1987). Briefly, animals were skinned, eviscerated and washed in several changes of potassium hydroxide solution over several days. Following staining with Alizarin Red, adult tissue was cleared in 2:1:2 mix of glycerin:benzyl alcohol: 70% ethanol.
Histological Analysis and Staining
Specimens were collected and processed for paraffin embedding using standard methods (Nagy et al., 2002). Sections (7 microns) were cut on a Leitz Leica 1512 microtome and processed with either Gill’s No. 2 hematoxylin followed by 0.5% eosin, or 0.001% Fast Green (FCF) solution followed by 0.1% Safranin O solution.
Micro Computer Tomography (μCT)
Micro-CT analyses were performed through the Yale Core Center for Musculoskeletal Disorders, following standard procedures (Kawano et al., 2013). Briefly, adult animals were euthanized and the femurs were isolated, stripped of soft tissue and stored in 70% ethanol. The bones were scanned using a Scanco μCT-35 instrument (Scanco, Bruttisellen, Switzerland) as previously described (Itokowa et al., 2011). Both 2- and 3-D μCT data included bone volume to total volume fraction (BV/TV), trabecular number (Tb.N), thickness (Tb.Th), space (Tb.Sp), connectivity density (Conn.D), structural model index (SMI), and degree of anisotrophy (DA). Cortical thickness averaged for both cortices (Ct.Th), bone volume to total volume fraction (Ct.BV/TV), endosteal radius (endo.radius), periosteal radius (peri.radius), and polar moment of inertia (pMOI) were also quantified.
in vivo Xylosyltransferase Activity Assay
Proximal hindlimb cartilage dissected from P0 pug and wild type limbs was homogenized in 12mM MgCl2, 4mM MnCl2 50mM KCl and 50mM 4-morpholineethanesulfonic acid (MES) buffer, pH6.5. Xylosyltransferase activity was determined by the incorporation of [14C]-D-xylose into either a synthetic bikunin peptide acceptor or chicken aggrecan as previously described (Kearns et al., 1991), with minor modifications. Reaction mixture contained: 12mM MgCl2, 4mM MnCl2, 50mM KCl, 50mM MES, pH 6.5, 7.5mM KF, 0.2 mg/ml bikunin peptide (QEEEGSGGGQLV) or 1 mg/ml recombinant aggrecan CS region, 12mM UDP-xylose and 0.5mCi/ml [14C]-UDP-xylose and 60 mg of protein extract per assay. After incubation at 37°C for 1h, for bikunin peptide reaction products were absorbed onto a AG50W-X2 column, washed with 0.01 N HCl and eluted with 2M NH4OH. For aggrecan protein acceptor, radioactive products were TCA/PTA precipitated. Incorporated radioactivity was then measure by liquid scintillation counting.
Cell Culture Xylosyltransferase Assay
Chinese hamster ovary (CHO) and CHO-K1-pgsA-745 (abbreviated CHO-745) cell lines were maintained in Ham’s F-12 medium containing 10% FBS, 100 U/ml penicillin G, and 100 μg/ml streptomycin sulfate. CHO-745, deficient in the xylosyltransferase activity required for the synthesis of both heparan sulfate and chondroitin sulfate proteoglycans, was kindly provided by J. Esko, University of California, San Diego (Esko et al., 1985). Triplicate dishes at 70% confluence were transfected with a construct engineered to express wild-type Xylt1 or a mutant form of Xylt1 bearing the pug mutation, using Mirus-LT1 as the transfection reagent and following the manufacturer’s instructions. Forty-eight hours after transfection cells were collected in 12mM MgCl2, 4mM MnCl2, 50mM KCl and 50mM 4-morpholineethanesulfonic acid (MES) buffer, pH6.5 and xylosyltransferase activity determined as described above using the bikunin peptide as acceptor.
Biosynthetic Radiolabeling of Proteoglycans with Sulfate
Cartilage dissected from P0 wild-type or pug limbs was incubated for 24 hours with 200μCi/ml [35S]H2SO4 in F12 media and then homogenized in 5 M guanidine, 100 mM E-aminocaprionic acid, 10 mM EDTA, 5 mM benzamidine, 5 mM phenylmethyl-sulfonylfluoride, and 10 mM N-ethylmaleimide. Proteoglycans were purified by cesium chloride density gradient centrifugation as described previously (Domowicz et al., 2000), extensively dialyzed against 100 mM ammonium acetate (pH 7.0) and digested with chondroitinase ABC (0.5 U/ml) at 37°C for 4h in 5 mM EDTA, 100 mM Tris, pH 7.4. Digested and undigested proteoglycan samples were TCA/PTA precipitated and radioactivity measured by liquid scintillation counting. Counts were normalized for total protein in the original homogenate.
RESULTS
Forward genetics uncovers a chondrodysplastic mouse mutant
Despite recent advances in skeletal biology, the genetic mechanisms that balance chondrocyte proliferation and maturation are still unclear. To uncover new factors that regulate these processes, we took advantage of an unbiased ENU-induced mutagenesis screen in mice. We focused on the strictly recessive pug mutant that was first detected due to reduced limb length at embryonic day (e) 18.5. We discovered that this difference in limb length could be traced back to early stages suggesting that the pug mutant could provide crucial insight into skeletal development. At e14.5 we observed an ~18% reduction in stylopod (humerus, femur) element length, and ~14% reduction in zeugopod (radius/ulna, tibia/fibula) length in pug mutants (Figure 1A). By e16.5, both the pug stylopod and zeugopod are ~18% shorter than controls and this disparity remains until late embryonic stages (Figure 1A,D,E). While the crown-rump length of pug embryos showed no difference compared to controls (Figure 1B,C), reduced skeletal element length was observed in other bones (ex. mandible Figure 1B,C) that suggested a general defect in endochondral ossification.
Figure 1. pug animals display reduced skeletal element length.

A, graph showing pug skeletal element lengths compared to control littermates at various ages. Values are normalized such that control element lengths are set to 100%. Embryonic pug skeletons display a 15–20% reduction in skeletal element length beginning at e14.5 compared to their littermates. As adults, the severity of skeletal length reduction increases to ~40% in proximal stylopod elements (ex. humerus), but remains at ~20% in more distal zeugopod (ex. ulna) elements. B–E, e14.5 and e16.5 skeletons; red = bone marked by alizarin red staining; blue = cartilage marked by alcian blue staining. e14.5 pug mutant limb elements are shorter than controls (D, bracket). At e16.5, pug mutants show a similar crown-rump length and ribcage size compared to controls (B,C), but shorter limb elements (E, bracket), as well as a shorter mandible (yellow arrows in B,C). The decrease in element length is not accompanied by changes in skeletal patterning or joint morphology during embryonic stages. F–H, adult pug animals (postnatal day (P)119) show an overall reduction in body size, in addition to shortened snouts, limbs, and tails compared to control littermates. I–M Adult skeletons (P119) stained with alizarin red. Adult skeletons reveal shortening of the skull (yellow arrows in I,K mark the mandible length) and limbs with more pronounced shortening of proximal limb elements (L, M) compared to littermate controls. Adult pug animals also display a smaller ribcage and broadening of the ribs (arrowheads in K and insets). Scale bar in B = 0.25cm, scale bars in D = 0.25mm, scale bars in E = 0.5mm, scale bars in I–M = 0.5cm.
Skeletal defects in pug mutants become progressively worse with age
Bone development is not complete at birth. In the mouse, elongation and growth of the axial and appendicular skeleton continues throughout postnatal development until twelve weeks after birth. To test whether the pug phenotype merely represented a delay in embryonic skeletal development, we generated and analyzed pug homozygous adults. Homozygous pug mutants are semi-viable, comprising only 7.4% (n=20/270) of animals weaned from crosses between heterozygotes instead of the expected 25%. Though the cause of pre-wean age mortality was not directly investigated, compression of the lung cavity due to defects in the ribcage (see below) could cause respiratory distress. Alternatively, the small size of pug homozygotes could place them at a competitive disadvantage for nursing. Homozygous pug animals that survived to adulthood are fertile, appeared to have a normal life expectancy, and continued to show a dwarfed appearance, including a shortened snout, shorter overall body size, and short limbs (Figure 1F–M). Interestingly, adult pug animals showed increased severity of the skeletal phenotype compared to embryos. At embryonic stages, no obvious differences were observed in the morphology of the ribs (Figure 1B,C), while pug adults displayed a much smaller ribcage, with short, thick ribs (Figure 1F,G, arrowheads). In addition, pug adults showed rhizomelic or disproportionate shortening of the limbs. While zeugopod elements were ~20% shorter than controls, a similar reduction as observed in embryos, stylopod elements were reduced by ~40% (Figure 1A,L–M). In humans, rhizomelic shortening of the limbs is a key feature of multiple skeletal dysplasias including achondroplasia, Ellis-van Creveld syndrome, and thanatophoric dysplasia (Azouz et al., 1998; Baldridge et al., 2010; Peraita-Ezcurra et al., 2012), suggesting that the pug mutant is a new animal model to understand the molecular and cellular mechanisms underlying these defects.
Adult pug bones display structural defects
The persistent shortening of adult pug limb elements demonstrated that the embryonic phenotype is not due to a delay in skeletal development. In addition, the adult phenotype suggested that there might also be underlying structural defects in the bones. To test this, micro Computer Tomography (μCT) was performed on adult animals after the cessation of postnatal growth. μCT demonstrated an increase in the ratio of trabecular bone volume over total volume in pug adult femurs, while the full bone scans revealed significant changes in trabecular architecture in pug adults (Figure 2A–C). Trabecular bone volume, thickness, space, and connective density were all increased in pug, while trabecular number was reduced, indicating a tighter packing of trabecular bone in the epiphysis (Figure 2B). This increase in trabecular bone volume in the epiphysis in pug adult femurs corresponds to a reduction in trabecular bone within the diaphysis, resulting in a significant alteration in trabecular bone distribution within the whole element (Figure 2A′). Cortical bone forms by direct differentiation of the perichondrium into bone forming cells. Strikingly, no major changes in cortical bone structure were observed (Figure 2A″, 2B), suggesting that the primary defect in pug mutants is due to changes in endochondral ossification. Though cortical bone volume and density were unchanged, the periosteal and endosteal radius of pug femurs was slightly decreased, potentially leading to a weakening of long bones in pug adults. No evidence of altered joint morphology or other morphological defects were observed. In an attempt to define the primary defect in pug mutants, we further examined embryonic skeletal development.
Figure 2. Adult pug homozygotes display defects in trabecular architecture.

A-A″ uCT scans of pug and control femurs revealed defects in trabecular bone content within the diaphysis in pug mutants. Specifically, there is a greater density of trabecular bone at the distal head of the bone in pug and a reduction in trabecular bone along the length of the bone (A, lines mark the length of trabecular bone region). Within the region containing trabecular bone, in pug mutants, the organization appears altered compared to controls (A′), with some regions showing higher density and others displaying lower density. In contrast, pug mutant cortical bone appears relatively normal (A″). B, graph showing several measurements of trabecular and cortical bone. Values are normalized such that control measurements are set to 1. pug femurs displayed an increase in trabecular volume, thickness, space and connective density, but a reduction in trabecular number (B). No significant changes were observed in cortical bone density or structure in pug femurs while small, but significant decreases were observed in endosteal and periosteal radii (B). C, graph showing several additional measurements of trabecular and cortical bone. Values are not normalized. While the degree of anisotrophy did not change in pug mutants, the polar moment of inertia was significantly decreased. We also observed changes in the structural model index that indicated a shift from rod-like trabecular bone towards plate-like bone in pug animals (C). Tb.BV/TV – trabecular bone volume/total volume; Tb.N – trabecular number; Tb.Th – trabecular thickness; Tb.Sp – trabecular space; Conn.D – connective density; Ct.BV/TV – cortical bone volume/trabecular volume; Ct.Th – cortical thickness; endo.radius – endosteal radius; peri.radius – periosteal radius; SMI – structural model index; DA – degree of anisotrophy; pMOI(mm4) – polar moment of inertia. ** p < 0.005, *** p < 0.0005.Scale bar in A = 1mm, scale bars in A′ and A″ = 100um.
Chondrocyte identity is specified normally in pug mutants
To assess embryonic skeletal development in pug mutants, we focused on the limbs and examined the morphology and molecular profiles of the developing chondrocytes. Histological comparison of chondrocyte domains at e16.5 revealed that chondrocytes in all stages of maturation were present, including resting, columnar, prehypertrophic and hypertrophic, but the sizes of their domains were reduced in pug compared to wild type (Figure 3A–B). Analysis of the resting chondrocyte domain revealed a tighter packing of resting chondrocytes at the heads of pug skeletal elements compared to controls (Figure 3C) suggesting a potential defect in chondrocyte size or in the ECM surrounding the resting chondrocytes.
Figure 3. The maturing chondrocyte domains are reduced in pug skeletal elements.

A–B‴ hematoxylin and eosin stained paraffin-embedded sections of the proximal humerus heads at e16.5. Histological analysis revealed a reduction in the sizes of all states of maturing chondrocytes. Specifically, insets in A′ and B′ show an increased density of resting chondrocytes in pug compared to controls (quantified in C). Column formation in proliferative chondrocytes in A″ and B″ show little difference between pug and controls, though cells appear more tightly packed. Changes to both the height (D) of the hypertrophic chondrocyte domain and number of hypertrophic chondrocytes along the axis of growth (E) can be seen in pug (A‴,B‴). F, Serial sections of OCT-embedded e16.5 distal humerus heads, at the junction with the radius/ulna. The humeral heads are outlined by dotted lines. Within the skeletal elements, pug mutant chondrocytes maintain their molecular signature by appropriately expressing Sox9 and Col2a in resting, columnar and prehypertrophic regions and ColX in hypertrophic regions (B). RC – resting chondrocytes, CC – columnar chondrocytes, PC – prehypertrophic chondrocytes, HC – hypertrophic chondrocytes, B – bone. Scale bars in A–B = 100um, scale bars in A′–B‴ = 50um, scale bars in F = 100um. *** p<0.000001, * p<0.05
We further observed that the domain of columnar chondrocytes was reduced in pug, and that the cells appeared to be more tightly packed. However, the overall organization of chondrocytes within columns was comparable to controls, indicating that chondrocytes are able to progress through early maturation stages (Figure 3A″, B″). In later maturation stages, we observed a decrease in the height of the prehypertrophic and hypertrophic domains in pug mutants (Figure 3A‴,B‴,D) however this did not appear to be due to tighter packing of chondrocytes but rather to fewer cells along the axis of growth (Figure 3E), indicating an alteration in pug chondrocyte maturation.
We examined several markers of chondrocyte maturation to test whether maturation was proceeding normally in pug mutants. Sox9, a general chondrogenesis marker expressed in resting, columnar and prehypertrophic chondrocytes (Zhao et al., 1997) is expressed normally in pug chondrocytes (Figure 3F). Col2a1a, the cartilage specific form of Collagen 2, is also broadly expressed in chondrocytes and is upregulated in prehypertrophic chondrocytes (Cheah et al., 1991; Zhao et al., 1997) while ColX, is specifically upregulated in hypertrophic chondrocytes (Gibson and Flint, 1985). We largely observed a similar pattern of expression of these markers in pug and wild type animals suggesting that although domains of maturing chondrocytes are smaller in pug, they maintain normal identity and express the appropriate markers for their maturation status (Figure 3F). The upregulation of Col2a1a in prehypertrophic chondrocytes was not as obvious in pug mutants compared to controls suggesting that the transition from columnar to prehypertrophic chondrocytes may not be completely normal in pug mutants, however prehypertrophic chondrocytes appear morphologically normal. Together, these data show that all types of chondrocyte are present and express the typical complement of markers in pug embryos.
Chondrocyte proliferation is unchanged in pug mutants
The reduced sizes of the pug skeletal elements and chondrocyte domains suggested that the primary defect could lie in chondrocyte proliferation or maturation. Decreased cell division in either the resting or columnar chondrocyte pools could reduce the terminal size of a skeletal element. To test this, we calculated a mitotic index using phosphorylated histone H3 staining. If proliferation defects cause the pug phenotype, we would expect to see a reduction in the mitotic index; however, we observed no significant change in proliferation rate in pug (1.74% (+/− 0.4)) compared to wild type (1.66% (+/; 0.5)) at e13.5 (Figure 4A,B). To test whether altered proliferation contributed to the more pronounced pug phenotype at later stages, we analyzed BrdU incorporation and pHH3 staining at e16.5 and e18.5 but did not observe any significant difference compared to controls (data not shown). Given that pug skeletal elements are already significantly reduced by e14.5, changes in proliferation do not appear to cause the initial reduced size of pug skeletal elements nor do they exacerbate the phenotype during later embryonic development.
Figure 4. Proliferation is unchanged and does not account for reduced bone length in pug mutants.
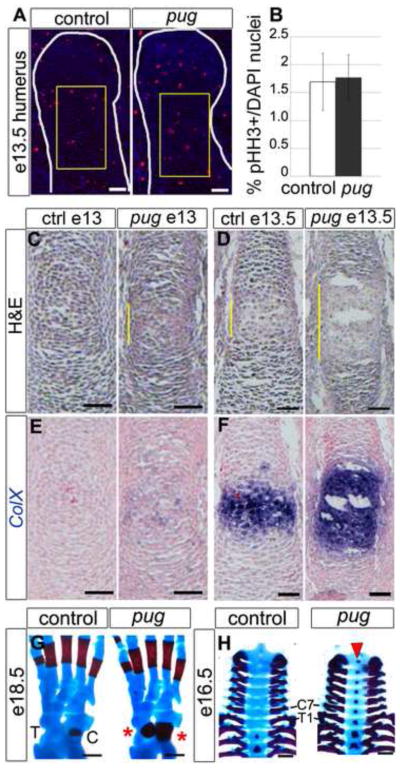
A, sections of humeral heads at e13.5 (outlined in white), showing phospho-histone H3 (pHH3) staining in red, DAPI in blue. B, graph showing the mitotic indices of control and pug chondrocytes based on pHH3 staining. During early chondrocyte organization at e13.5, proliferation is unchanged between pug and controls. Representative regions used for mitotic index quantification are marked by yellow boxes in A. C,D, hematoxylin and eosin-stained sections of humeri at e13.0 (C) and e13.5 (D). At e13.0 and e13.5, pug humeri display evidence of premature maturation in the center of the element, as shown by cell morphology (C–D, yellow bars). E,F, serial sections of C,D marked by ColX in situ hybridization (blue) and counterstained with nuclear fast red (in pink). These elements also display a premature (E) and broadened (F) domain of ColX expression in pug mutants, indicating maturation into hypertrophic chondrocytes. G, dorsal view of e18.5 hindlimbs, anterior is to the right, showing early ossification (red) of the talus and expanded ossification of the calcaneus ( red asterisks). H, ventral view of cervical and thoracic vertebrae showing premature ossification in the cervical region and T1 at e16.5 (red arrowhead). In G,H, red = bone marked by alizarin red staining; blue = cartilage marked by alcian blue staining. C – calcaneus; T – talus; C7 – seventh cervical vertebra; T1 – first thoracic vertebra. Scale bars in A–F = 50um, scale bars in G = 250um, scale bars in H = 500um.
Chondrocytes mature more rapidly in pug mutants
To determine if changes in maturation could cause dwarfism in pug homozygotes, we analyzed chondrocytes at multiple stages of bone development. In early e13 embryos, just after the cartilage template has formed, wild-type skeletal elements primarily contained resting chondrocytes. In contrast, pug skeletal elements already contained distinct prehypertrophic and hypertrophic-like chondrocytes at the center of the element, which had initiated ColX expression, a hypertrophic chondrocyte marker (Figure 4C,E). By e13.5, pug skeletal elements contained a broader domain of hypertrophic-like chondrocytes, based on morphology and ColX expression, compared to wild-type elements (Figure 4D,F). The overall size of the skeletal elements was comparable between pug and wild type at e13.5 (see Figure 5A–D), demonstrating that premature chondrocyte maturation preceded changes in element length. This suggests that premature initiation of chondrocyte maturation is the primary defect in pug mutants, leading to smaller skeletal elements. We observed other examples of premature maturation at later stages in pug embryos including premature ossification of the talus in the hindlimb at e18.5 (Figure 4G) and cervical vertebrae at e16.5, (Figure 4H). These data suggest that the gene affected in pug mutants normally curbs chondrocyte maturation in skeletal elements that form through endochondral ossification.
Figure 5. Short-range Ihh signaling increases after the onset of premature maturation in pug skeletal elements.

A–H′, J-J′, L–N′, sections through humeri at the stages indicated showing in situ hybridizations for probes as indicated (in purple), and counterstained with nuclear fast red (in pink). At e13, when premature chondrocyte maturation has begun in pug, Ihh expression is expanded (A,A′), but the expression of the direct targets Ptch1 and Gli1 are similar between size-matched wild type and pug animals (B–C′), as is the expression of Runx2 (D-D′). Within 12 hours, the broad domain of Ihh in pug animals (E-E′) is matched by increased short-range Ihh signaling as reflected by expanded Ptch1 and Gli1 expression (F–G′), and a broader domain of Runx2 expression (H-H′). I,I′, K-K′, wholemount e13.5 (I-I′) and e14.5 (K-K′) limbs of pug mutants and controls carrying one copy of PTHrP-LacZ, stained for β–galactosidase activity (in blue). No obvious changes were observed in long-range signaling, indicated by similar patterns of PTHrP-LacZ (I-I′, K-K′). Closer analysis of PTHrP-LacZ by in situ hybridization shows no reduction in PTHrP expression in pug (J-J′). Increased Ihh signaling is maintained throughout embryonic development (M–N′, marked by Ptch1 and Gli1) even though the domain of Ihh expressing cells decreases (L-L′). Scale bars in A–H′, J-J′, L–N′ = 100um, scale bars in I-I′ = 500um, scale bars in K-K′ = 250um.
The pug mutation results in increased Ihh signaling
Several signaling pathways including Ihh tightly regulate the timing and rate of chondrocyte maturation. Ihh is expressed in prehypertrophic chondrocytes, and activates short-range downstream targets such as Gli1, Ptch1, and Runx2 to regulate maturation and promote proliferation (Gritli-Linde et al., 2001; Karp et al., 2000; Mak et al., 2008; St-Jacques et al., 1999; Enomoto et al., 2000; Enomoto-Iwamoto et al., 2001; Takarada et al., 2013; Takeda et al., 2001). To test if alterations in Ihh signaling could underlie the maturation defect in pug mutants, we examined the expression of these genes at early stages of skeletal element formation. In young e13 embryos we did not observe major changes in the direct targets of Ihh signaling (Ptch1 and Gli1) within chondrocytes, despite a broadened domain of Ihh expression in pug mutants (Figure 5A–C′). Additionally, no changes were observed in Runx2 expression in pug where is it expressed throughout the center of the element and in the surrounding mesenchymal cells (Figure 5D-D′). This suggests that the premature maturation of pug chondrocytes occurs independently of changes in Ihh signaling. By e13.5, we observed increased Ptch1 expression in pug chondrocytes and perichondrial cells compared to controls (Figure 5F-F′), as well as increased Gli1 expression within pug chondrocytes, flanking the Ihh domain (Figure 5G-G′). Runx2 is also expressed in a broader domain at e13.5 in pug mutants, which likely reflects the expanded domain of maturing chondrocytes marked by Ihh expression (Figure 5E-E′, H-H′). The observed increase in the levels of short-range Ihh targets persists at later stages, including e16.5 (Figure 5L–N′). The apparent increase in short-range Ihh signaling early is likely a secondary effect of premature maturation, which causes a broader domain of cells to express Ihh.
The pug mutation does not inhibit PTHrP
Ihh also signals long range to PTHrP, which protects the proliferating pool of chondrocytes at the heads of bones by inhibiting chondrocyte maturation (Karaplis et al., 1994; Lanske et al., 1996; Vortkamp et al., 1996). Reduction or loss of PTHrP expression in resting chondrocytes can lead to dwarfism. To determine if this caused the pug phenotype, we used a PTHrP-lacZ reporter mouse line to follow PTHrP expression (Chen et al., 2006). However, we did not observe any noticeable changes in PTHrP-lacZ expression at early (e13.5) or late (e14.5–e18.5) embryonic stages (Figure 5I–K′ and not shown) in pug mutants. While some variability was evident in the staining, we did not observe a decrease in PTHrP expression either by whole mount analysis or section in situs (Figure J-J′). These data suggest that long-range Ihh signaling is not reduced by the pug mutation and that premature maturation is not due to loss of PTHrP expression.
The pug mutation does not affect Fgf signaling through ERK1/2 in chondrocytes
Fgf signaling also plays a key role in regulating chondrocyte maturation, and activating mutations in Fgfr3 cause achondroplasia, (Amizuka et al., 2004; Deng et al., 1996; Rousseau et al., 1994) suggesting that changes in Fgf signaling might contribute to the pug phenotype. Instead, we observed similar expression of Fgfr3 in young e13 pug mutants and controls (Figure 6A,B). Subsequently, Fgfr3 expression became increased in the center of pug skeletal elements by e13.5 compared to controls (Figure 6C,D), similar to the increase in short-range Ihh signaling at this timepoint. This increased expression of Fgfr3 was maintained throughout embryonic development (Figure 6E,F). However, the increased Fgfr3 levels in pug mutants do not cause increased Fgf signaling through ERK1/2. We observed similar levels of phosphorylated ERK1/2 (pERK+) nuclei in pug mutants and controls at e13.5 (Figure 6G,H). In control e13.0 resting chondrocytes, 1.91% (+/− 0.19%) of nuclei were pERK+ compared to 1.93% (+/− 0.025%) in pug resting chondrocytes, while control columnar chondrocytes displayed 1.99% (+/− 0.56%) pERK+ nuclei compared to 1.90% (+/− 0.26%) in pug columnar chondrocytes. These data suggest that the changes in Fgfr3 expression may reflect changes in chondrocyte maturation but do not cause the premature initiation of maturation in pug embryos. Together, these data show that premature chondrocyte maturation in pug mutants is not due to changes in early Ihh or Fgf signaling, two key pathways that regulate maturation.
Figure 6. Fgfr3 expression increases after the onset of premature maturation but does not lead to increased signaling through ERK1/2.

A–F, sections through humeri at the stages indicated showing in situ hybridizations for Fgfr3 (in purple), and counterstained with nuclear fast red (in pink). At e13, the expression of Fgfr3 appeared similar between control and pug animals (A–B), and levels increased at the center of pug elements by e13.5 (C–D). As embryonic development proceeds, this increase in Fgfr3 expression is maintained (E–F). G,H, sections through e13.5 humeri stained for phosphorylated Erk1/2 (in brown). Increased Fgfr3 expression at e13.5 does not result in an increase in Fgf signaling, as evidenced by similar pERK positive cells in controls and pug animals in both the resting and columnar chondrocyte pools (G–H, red arrowheads). Scale bars in A–F = 100um, scale bars in G–H = 20um.
The pug mutation affects the Xylosyltransferase 1 (Xylt1) gene
To determine the molecular lesion responsible for the pug phenotype, the locus was narrowed to a 7.83 MB interval on Chromosome 7 using meiotic recombination mapping. We then used Sequence Capture and Illumina sequencing to identify the causative mutation (see Methods for details). This approach identified an exonic T to A transversion (c.T2794A) near the 3′ end of the Xylosyltransferase 1 (Xylt1) gene. We confirmed that this mutation segregated with the pug phenotype using Sanger sequencing (Figure 7A). The pug mutation results in a missense mutation in the Xylt1 protein (W932R) affecting a residue that is highly conserved from humans to C. elegans (Figure 7B,C). The initial step in the addition of GAG chains to all proteoglycans requires the transfer of xylose to specific serine residues on the core protein (Muller et al., 2006; Roch et al., 2010; Schön et al., 2006a; Stoolmiller et al., 1972). In vertebrates, this process is catalyzed by two xylosyltransferases, Xylt1 and Xylt2. These proteins share considerable identity in the xylosyltransferase domain (>70%) and appear to have similar enzymatic activity (Condac et al., 2007; Cuellar et al., 2007; Ponighaus et al., 2007; Roch et al., 2010).
Figure 7. The pug mutation affects the Xylosyltransferase 1 gene, which is specifically expressed in chondrocytes.

A, sequencing traces from wild-type, heterozygous and homozygous pug mutant samples showing a T->A transversion mutation in pug mutants and carriers. The pug mutation causes a missense (W932R) in the Xylt1 protein. B, schematic of the mouse Xylt1 protein highlighting the transmembrane domain, core 2/I-branching and Xylosyltransferase domains. The pug mutation occurs in near the C-terminus of Xylt1 (red line), outside of any known protein domains. C. Alignment of the C-terminus of Xylt1 orthologs across multiple animal lineages. The mutated tryptophan residue (W932 in mouse) is highly conserved. D, sections through wild-type forelimb skeletal elements at several stages, showing in situ hybridization patterns for Xylt1 transcripts (purple) and counterstained with nuclear fast red (in pink). Xylt1 transcripts are specifically found in developing skeletal chondrocytes, with strong expression from e13.5 through birth. E–H, sections through e16.5 humeri, showing close-ups of resting chondrocytes. Xylt1 normally localizes to the cis-Golgi (E–F), however this localization is lost in the pug mutant (G–H). DAPI marks nuclei (blue) and GM130 marks the cis-Golgi (red). In cell types that did not show any evidence of Xylt1 transcripts by in situ hybridization, the Xylt1 antibody appeared to localize to discrete punctae within the cell similar to the pattern observed in pug mutants (H). Scale bars in D (e13.5 and e16.5) = 250um and (e18.5) = 100um, scale bars in E–H = 10um.
Xylt1 is specifically expressed in chondrocytes of developing skeletal elements
To better understand when and where Xylt1 may be acting during mouse skeletal development, we first examined the expression pattern of the Xylt1 transcript in mouse embryos. In the developing skeleton, Xylt1 mRNA is strongly expressed in all chondrocytes, beginning before e13.5 (Figure 7D and data not shown). As chondrocytes within the skeletal element mature, they maintain Xylt1 expression until they have hypertrophied but Xylt1 transcripts were not observed in mature bone (Figure 7D and data not shown). Xylt1 transcripts were also not observed in non-skeletal tissue types, suggesting that during mid to late embryonic development, Xylt1 may function exclusively in the axial and appendicular skeleton. Xylt1 and Xylt2 are expressed in a highly overlapping pattern in the adult mouse (Condac et al., 2007). However, we found no evidence of Xylt2 expression during embryonic development in control embryos (not shown), consistent with gene expression database reports for Xylt2 (Diez-Roux et al., 2011; Visel et al., 2004). We did not observe Xylt2 expression in pug mutants at embryonic stages indicating that Xylt2 is not upregulated to compensate for the loss of Xylt1 function in pug mutant chondrocytes.
The pug mutation disrupts Xylt1 protein localization
The pug mutation affects a residue 21 amino acids from the C-terminus of Xylt1 (Figure 7C). Polyphen and SIFT analyses (Adzhubei et al., 2010; Kumar et al., 2009) suggest that the substitution of an Arginine for a Tryptophan at this position is likely deleterious to the protein; however, this site is not within any of the known functional domains in Xylt1. In order to determine how the W932R missense mutation affects Xylt1 protein, we first examined the subcellular localization of the protein within chondrocytes. In previous studies, Xylt1 has been shown to act in the Golgi where it catalyzes the xylosylation of proteoglycan core proteins, (Cardin and Weintraub, 1989; Muller et al., 2006; Schön et al., 2006a; Stoolmiller et al., 1972). Consistent with this, we observed colocalization of Xylt1 protein with the cis-Golgi marker GM130 in wild-type chondrocytes (Figure 7E,F). Strikingly, in pug chondrocytes Xylt1 was not observed in the cis-Golgi (Figure 7G,H) suggesting that the missense mutation affects localization of the Xylt1 protein and thus alters its normal function within the Golgi.
The pug mutation causes reduced xylosyltransferase activity
The reduced levels of Xylt1 in the cis-Golgi in pug mutants could be the primary defect as xylosyltransferases are thought to function primarily in this organelle. However, to test whether the W932R mutation also affected activity of the Xylt1 protein, we measured the ability of Xylt1 to transfer xylose onto proteoglycan core proteins both in vivo and in cell culture. Using proximal hindlimb cartilage from postnatal day 0 (P0) neonates, we observed only ~20% xylosyltransferase activity in pug mutants compared to controls (Figure 8A). This dramatic reduction in xylosyltransferase activity was found using proteins with either single (bikunin) or multiple (aggrecan core protein) acceptor sites. As mentioned previously, we did not detect Xylt2 transcripts in control or pug mutant animals. To determine if the remaining xylosyltransferase activity in pug cartilage could be due to functional Xylt2 that was below our level of detection, we examined Xylt1 activity in cultured cells. Specifically, we expressed wild-type and pug alleles of Xylt1 in both wild type and xylosyltransferase deficient CHO-745 cells (Esko et al., 1985). Consistent with our observations in cartilage, Xylt1 protein carrying the pug mutation exhibited reduced activity comparable to 10–20% of the wild-type Xylt1 activity (Figure 8B). These data indicate that pug animals carry a hypomorphic mutation and that Xylt1W932R retains a low level of xylosyltransferase activity. The similar reduction in Xylt1W932R activity in vivo and in cell culture suggests that Xylt2 does not compensate for reduced Xylt1 activity in pug mutant chondrocytes.
Figure 8. The pug mutation strongly reduces xylosyltransferase activity in chondrocytes, curtailing proteoglycan processing.

A,B graphs showing xylosyltransferase activity in cell lysates from isolated chondrocytes (A) or transfected CHO cells (B). Values are normalized such that control activity is set to 100%. In xylosyltransferase assays using whole cartilage from P0 embryos, pug cartilage showed an 80% reduction in activity compared to control cartilage using both bikunin and chick aggrecan as acceptor proteins (A). In cell culture, the pug allele of Xylt1 also showed ~80% reduction in activity compared to a wild type Xylt1 construct when overexpressed in chinese hamster ovary (CHO) cells or in CHO cells deficient in xylosyltransferase activity (CHO 745) (B). C,C′, graphs showing percentage of radio-labeled, newly-synthesized GAG chains per mg of limb cartilage protein, after 24 hr incubation in [35S] sulfate-containing media. C′ is an expanded view of chondroitinase-treated samples shown in C. The overall incorporation of sulfate onto newly-synthesized GAG chains is reduced by ~40% in pug cartilage compared to controls. Fractionating the total sulfate incorporated counts in CsCl density gradients shows a 50% reduction in the modifications on high-density proteoglycans, and a subsequent increase in low-density proteoglycans likely due to incompletely processed large molecular weight proteoglycans (C). Digestion with chondroitinase (ChABC) removes CS-GAG chains from the analysis and revealed a reduction in the sulfation of non-CS-GAGs in pug samples (C′). D, sections of humeri at stages indicated, stained for Safranin O (orange-brown), which marks GAG chains. Reduced GAG chains in pug cartilage compared to controls could be observed in vivo by diminished Safranin O staining at several developmental stages (D) Staining outside of skeletal elements at 13.5 reflect the early extracellular matrix comprised of versican and other proteglycans. Scale bars in D = 100um.
The pug mutation reduces xylosyltransferase-dependent GAG chain modifications on proteoglycans
Two major classes of proteoglycans, heparan sulfate proteoglycans (HSPGs) and chondroitin sulfate proteoglycans (CSPGs), are highly expressed in chondrocyte extracellular matrix and have been shown to play a key role in regulating chondrocyte maturation (Cortes et al., 2009; Koziel et al., 2004). Xylosyltransferases are essential for normal proteoglycan processing and function (Baeg et al., 2001; Cortes et al., 2009; Nybakken and Perrimon, 2002; Venkatesan et al., 2012; Yan and Lin, 2009), thus the pug mutation should negatively affect normal proteoglycan processing within chondrocytes. To test this, we analyzed sulfate incorporation in isolated P0 cartilage as a measure of proteoglycan processing (Domowicz et al., 2000). pug cartilage showed a 34% reduction in total proteoglycan sulfation compared to control cartilage (Figure 8C). We then separated the proteoglycan samples into low-density and high-density fractions on dissociative CsCl gradients. We observed that the strongest decrease in sulfate incorporation occurred in the high-density proteoglycan fraction in pug mutants, which in cartilage is composed primarily of aggrecan, a CSPG normally modified by hundreds of GAG chains. Digestion with chondroitinase revealed that the proteoglycans that are partially processed in pug are primarily members of the CSPG class (Figure 8C′). However, the undigested portion of proteoglycans, including HSPGs, also displayed reduced sulfation in pug, suggesting that the pug mutation affects the ability of Xylt1 to add GAG chains to multiple classes of proteoglycans. To test whether the reduced xylosyltransferase activity observed in our assays causes GAG chain defects in vivo, we stained wild type and pug skeletal elements with Safranin O, a marker of sulfated GAG chains (Rosenberg, 1971). We observed a strong reduction in Safranin O staining in pug mutant skeletal elements compared to wild type at both early and late stages in skeletal development (Figure 8D), indicating that the decrease in Xylt1 activity translates into a reduction in proteoglycan processing in cell culture and in vivo.
DISCUSSION
The vertebrate skeleton is laid down and undergoes significant patterning and differentiation during embryogenesis. Defects in skeletal development can affect the size and morphology of the bones, but also the structure and strength of these elements throughout the life of the organism. Though the major signaling pathways that regulate endochondral ossification have been well characterized, the interplay between these signaling factors and the extracellular matrix within the developing skeleton is just beginning to be appreciated. Xylt1, as a major regulator of proteoglycan processing in chondrocyte maturation, maintains the integrity of the extracellular matrix. Defects in xylosyltransferase function in the cartilage affect not only the formation of a proper proteoglycan network, but also affect signaling within the developing growth plate to prematurely initiate chondrocyte maturation and subsequently alter the structure of adult bone.
Xylt1 is essential for normal maturation of skeletal chondrocytes
Proteoglycans, particularly HSPGs and CSPGs, play critical roles in endochondral skeletal development. The proper function of proteoglycans depends upon the polysaccharide moieties, the glycosaminoglycan side chains that are added to the core protein. Mutations in HSPG or CSPG core proteins or in the enzymes that modify their GAG chains affect chondrocyte maturation, proliferation and ossification (Cortes et al., 2009; Domowicz et al., 2009; Hilton et al., 2005; Ratzka et al., 2008; Watanabe et al., 1994; Watanabe et al., 2010; Wilson et al., 2012). Xylosyltransferases play the crucial first step in the initiation of GAG chain synthesis on HSPGs and CSPGs by adding a xylose to specific serine residues on the core proteins (Cuellar et al., 2007; Esko et al., 1985; Muller et al., 2006; Ponighaus et al., 2007; Prante et al., 2006). Following xylosylation, additional sugar moieties are added resulting in a tetrasaccharide linker, followed by varying lengths of disaccharide units (condroitin, heparan, dermatan or keratan sulfate) (Schwartz, 2000). We show that a hypomorphic Xylt1 mutation in pug animals strongly reduces xylosyltransferase activity in the developing skeleton, and leads to reduced GAG chains in skeletal elements and premature chondrocyte maturation. This has a profound effect on the endochondral skeleton and leads to disproportionate dwarfism in pug homozygous mice.
Decreased xylosyltransferase activity in pug mutants leads to signaling defects
Previous studies in both vertebrates and invertebrates have uncovered important roles for GAG chains in regulating morphogen gradient formation through the direct binding of signaling molecules (Cortes et al., 2009; Yan and Lin, 2009). HSPGs have been implicated as major regulators of Fgf diffusion within the ECM, both in flies and in mammalian systems, and more recently in regulating Ihh diffusion within the ECM (Baeg et al., 2001; Cardin and Weintraub, 1989; Kalinina et al., 2009; Koziel et al., 2004; Nybakken and Perrimon, 2002). In addition, recent work indicates that proper CSPG processing plays an important role in allowing Ihh to diffuse through the extracellular matrix surrounding chondrocytes (Cortes et al., 2009). Ihh and Fgfs normally act to prevent chondrocyte maturation, and as the pug mutation affects both HSPGs and CSPGs, one prediction would be that both of these signaling pathways should be affected in pug mutants, and thus would affect maturation rate. Indeed, we observed an upregulation of short-range Ihh signaling as well as an increase in Fgfr3 levels. In the case of Fgfr3, no downstream changes in MAPK signaling were observed, and long-range Ihh signaling through PTHrP, which normally inhibits chondrocyte maturation, was not affected in pug mutants. This indicates that changes in Ihh and Fgf signaling likely do not drive the premature maturation of pug chondrocytes. Interestingly, CSPGs and HSPGs appear to have opposing functions in regulating the diffusion of Ihh through the ECM, with CSPGs acting to expand the domain of Ihh diffusion, and HSPGs acting to restrict the domain of Ihh diffusion. As our pug mutant appears to affect both CSPGs and HSPGs, we observed an alteration in the level of signaling propagated within Ihh-receiving cells, indicating an increase in the efficacy of the Ihh signal in a processed proteoglycan-deficient growth plate.
Strikingly, we found that the maturation of chondrocytes occurs approximately half a day earlier in pug mutants than in wild type and prior to changes in Fgf and Ihh signaling pathways. However, after premature maturation is initiated, Ihh target genes become upregulated in the pug growth plate and surrounding perichondrium. This short-range Ihh signaling drives maturation and ossification of perichondrial bone (Hammond and Schulte-Mercer, 2009; St-Jacques et al., 1999) and the increase that we observed in pug mutants may make the dwarfism phenotype more severe by driving premature ossification of the perichondrium.
Though the pug mutation does not lead to complete loss of GAG chain formation in the growth plate, the reduction in proteoglycan processing is sufficient to disrupt chondrocyte maturation. These data reveal that proteoglycans not only regulate the diffusion of key signaling molecules but also provide maturation cues to chondrocytes, independent of Ihh and Fgf signaling. While the mechanisms that underlie the decision to initiate maturation are still unclear, the premature maturation phenotype observed in pug in the absence of changes in signaling points to an extracellular matrix-associated cue that defines and determines the normal timing of chondrocyte maturation.
Xylt1 function is conserved across vertebrates
A recent study described the characterization of two zebrafish mutant lines carrying point mutations in the ortholog of Xylt1 (Eames et al., 2011). Similar to our findings, the zebrafish Xylt1 gene is expressed largely in developing chondrocytes, and the fish Xylt1 mutants displayed strong similarities to pug mutants including premature maturation and ossification of endochondral skeletal elements. Together, these data indicate that the role of Xylt1 during skeletal development is largely conserved across vertebrate lineages.
Interestingly, there are distinct differences between the zebrafish and mouse Xylt1 mutants. Fish homozygous for Xylt1 mutations do not show the dwarfism we observe in pug mutants, although they do show a shortened craniofacial phenotype, which is also present in pug mutants. In addition, in contrast to pug mutants that show reduced postnatal survival, homozygous Xylt1 zebrafish mutants are completely viable. Finally, the fish study noted a decrease in CSPG disaccharides but not HSPGs in their Xylt1 mutants, while our sulfate incorporation assay suggests that both CSPGs and HSPGs are affected by reduced Xylt1 function. One explanation for the differences between the zebrafish and mouse Xylt1 mutant phenotypes is that they are due to the specific nature of each mutation, which could be hypomorphic to different degrees. Future studies on the xylosyltransferase activity of the mutant zebrafish Xylt1 alleles would help determine whether these phenotypic differences are due to varying levels of activity of the mutant isoforms or represent real differences in the requirement for Xylt1 in mouse vs. fish development.
Xylosyltransferases and human health
Polymorphisms in XYLT1 and XYLT2 have been associated with modifying the severity of pseudoxanthoma elasticum, a multisystem disorder in humans involving mineralization of elastic fibers (Schön et al., 2006c). Xylosyltransferase activity has also been used as a biomarker for several other diseases including diabetic nephropathy, male infertility and osteoarthritis (Gotting et al., 2002; Götting et al., 2007; Götting et al., 1999; Schön et al., 2006b). Most recently, Schreml et al., identified a homozygous point mutation in XYLT1 (c.C1441T) in two siblings exhibiting short stature and other skeletal anomalies and developmental defects (Schreml et al., 2013). Our studies show that the pug mutant shares many of the phenotypes described in the human patients carrying homozygous mutations in XYLT1, including skeletal dysplasias, short stature, broadening of the ribs, craniofacial defects, and progressive bone growth defects. Interestingly, the human phenotypes appear milder than those observed in pug homozygotes. These differences could be due to the fact that XYLT1 protein levels are not reduced in patient cells, nor was xylosyltransferase activity reduced in contrast to our findings in pug mutants. The pug mutant mouse shows a more severe skeletal growth defect than short stature and shares defining characteristics with human dwarfism including rhizomelic shortening of long bones and thoracic kyphosis (Azouz et al., 1998). This suggests that XYLT1 is a potential candidate for additional cases of human dwarfism or short stature syndrome where the underlying genetic defect has yet to be determined. Further characterization of the pug mutant has the potential to reveal targets for effective treatment as well as the future diagnosis of human dwarfism cases resulting from primary defects to the skeleton.
Xylt1 and Xylt2 display specificity in expression and functional requirements
Xylt1 and Xylt2 are expressed in a highly overlapping pattern in adult mice (Condac et al., 2007); however, the phenotypes of the loss of function mutants reveal an elegant specificity for xylosyltransferases during development and in adults. The Xylt2 knockout mouse displays no developmental defects, but develops liver cysts postnatally (Condac et al., 2007). The liver is one of the few tissues in the adult mouse that specifically expresses Xylt2, but not Xylt1. In contrast, Xylt1 is primarily expressed in developing skeletal elements and the defect in pug mutants is restricted to chondrocytes. The restriction of the pug phenotype to cartilage elements and the Xylt2 phenotype to the liver suggests a redundancy in xylosyltransferase function in other tissue types, both during development and adult growth. However, the restricted expression pattern of Xylt1 during development, coupled with the lack of Xylt2 expression during these stages argue against redundancy but instead suggests that other proteins might act as xylosyltransferases to initiate GAG chain synthesis during embryonic development. Analysis of pug-Xylt2 double mutants could reveal redundancies in xylosyltransferase function during development if Xylt1 and Xylt2 are expressed broadly, but at levels we could not detect in our analyses. Alternatively, the double mutants could indicate a requirement for previously unidentified xylosyltransferases in the development of embryonic tissues.
HIGHLIGHTS.
The pug mutation causes disproportionate dwarfism in mice.
Dwarfism in pug mutants follows premature chondrocyte maturation.
pug mutants have a missense mutation in the Xylosyltransferase 1 gene.
The pug mutation disrupts Xylt1 protein localization and enzymatic activity.
The pug allele of Xylt1 fails to produce normal levels of glycosaminoglycans.
Acknowledgments
We would like to thank Kasey Christopher, Sunjin Lee, Mustafa Khokha, Arthur Horwich, Michael Hurwitz, Karl Insogna and Caren Gundberg for helpful discussions. We also thank Steven Reilly, Benhua Sun, Judy Henry and Stephanie Andrade for technical assistance. We also thank Arthur Broadus for providing PTHrP-lacZ mice.
FUNDING
This work was supported by pilot project funding through a NIH grant to the Yale Core Center for Musculoskeletal Disorders (1P30DK090744), NIH grants no. R01NS044385 to Kathryn Anderson, R01HD017332 to Nancy Schwartz, NIH T32GM007499 (Emily K. Mis), and institutional startup funds from the Yale Genetics Department (S.D. Weatherbee).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nature methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amizuka N, Davidson D, Liu H, Valverde-Franco G, Chai S, Maeda T, Ozawa H, Hammond V, Ornitz DM, Goltzman D, Henderson JE. Signalling by fibroblast growth factor receptor 3 and parathyroid hormone-related peptide coordinate cartilage and bone development. Bone. 2004;34:13–25. doi: 10.1016/j.bone.2003.08.009. [DOI] [PubMed] [Google Scholar]
- Azouz EM, Teebi AS, Eydoux P, Chen MF, Fassier F. Bone dysplasias: an introduction. Can Assoc Radiol J. 1998;49:105–109. [PubMed] [Google Scholar]
- Baeg GH, Lin X, Khare N, Baumgartner S, Perrimon N. Heparan sulfate proteoglycans are critical for the organization of the extracellular distribution of Wingless. Development. 2001;128:87–94. doi: 10.1242/dev.128.1.87. [DOI] [PubMed] [Google Scholar]
- Baldridge D, Shchelochkov O, Kelley B, Lee B. Signaling pathways in human skeletal dysplasias. Annu Rev Genomics Hum Genet. 2010;11:189–217. doi: 10.1146/annurev-genom-082908-150158. [DOI] [PubMed] [Google Scholar]
- Cardin AD, Weintraub HJ. Molecular modeling of protein-glycosaminoglycan interactions. Arteriosclerosis. 1989;9:21–32. doi: 10.1161/01.atv.9.1.21. [DOI] [PubMed] [Google Scholar]
- Cheah KS, Lau ET, Au PK, Tam PP. Expression of the mouse alpha 1(II) collagen gene is not restricted to cartilage during development. Development. 1991;111:945–953. doi: 10.1242/dev.111.4.945. [DOI] [PubMed] [Google Scholar]
- Chen X, Macica CM, Dreyer BE, Hammond VE, Hens JR, Philbrick WM, Broadus AE. Initial characterization of PTH-related protein gene-driven lacZ expression in the mouse. J Bone Miner Res. 2006;21:113–123. doi: 10.1359/JBMR.051005. [DOI] [PubMed] [Google Scholar]
- Choi M, Scholl UI, Ji W, Liu T, Tikhonova IR, Zumbo P, Nayir A, Bakkaloglu A, Ozen S, Sanjad S, Nelson-Williams C, Farhi A, Mane S, Lifton RP. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:19096–19101. doi: 10.1073/pnas.0910672106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condac E, Silasi-Mansat R, Kosanke S, Schoeb T, Towner R, Lupu F, Cummings RD, Hinsdale ME. Polycystic disease caused by deficiency in xylosyltransferase 2, an initiating enzyme of glycosaminoglycan biosynthesis. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:9416–9421. doi: 10.1073/pnas.0700908104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes M, Baria AT, Schwartz NB. Sulfation of chondroitin sulfate proteoglycans is necessary for proper Indian hedgehog signaling in the developing growth plate. Development. 2009;136:1697–1706. doi: 10.1242/dev.030742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuellar K, Chuong H, Hubbell SM, Hinsdale ME. Biosynthesis of chondroitin and heparan sulfate in chinese hamster ovary cells depends on xylosyltransferase II. The Journal of biological chemistry. 2007;282:5195–5200. doi: 10.1074/jbc.M611048200. [DOI] [PubMed] [Google Scholar]
- Deng C, Wynshaw-Boris A, Zhou F, Kuo A, Leder P. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell. 1996;84:911–921. doi: 10.1016/s0092-8674(00)81069-7. [DOI] [PubMed] [Google Scholar]
- Diez-Roux G, Banfi S, Sultan M, Geffers L, Anand S, Rozado D, Magen A, Canidio E, Pagani M, Peluso I, Lin-Marq N, Koch M, Bilio M, Cantiello I, Verde R, De Masi C, Bianchi SA, Cicchini J, Perroud E, Mehmeti S, Dagand E, Schrinner S, Nürnberger A, Schmidt K, Metz K, Zwingmann C, Brieske N, Springer C, Hernandez AM, Herzog S, Grabbe F, Sieverding C, Fischer B, Schrader K, Brockmeyer M, Dettmer S, Helbig C, Alunni V, Battaini MA, Mura C, Henrichsen CN, Garcia-Lopez R, Echevarria D, Puelles E, Garcia-Calero E, Kruse S, Uhr M, Kauck C, Feng G, Milyaev N, Ong CK, Kumar L, Lam M, Semple CA, Gyenesei A, Mundlos S, Radelof U, Lehrach H, Sarmientos P, Reymond A, Davidson DR, Dollé P, Antonarakis SE, Yaspo ML, Martinez S, Baldock RA, Eichele G, Ballabio A. A high-resolution anatomical atlas of the transcriptome in the mouse embryo. PLoS Biol. 2011;9:e1000582. doi: 10.1371/journal.pbio.1000582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domowicz M, Mangoura D, Schwartz NB. Cell specific-chondroitin sulfate proteoglycan expression during CNS morphogenesis in the chick embryo. Int J Dev Neurosci. 2000;18:629–641. doi: 10.1016/s0736-5748(00)00039-3. [DOI] [PubMed] [Google Scholar]
- Domowicz MS, Cortes M, Henry JG, Schwartz NB. Aggrecan modulation of growth plate morphogenesis. Developmental biology. 2009;329:242–257. doi: 10.1016/j.ydbio.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eames BF, Yan YL, Swartz ME, Levic DS, Knapik EW, Postlethwait JH, Kimmel CB. Mutations in fam20b and xylt1 reveal that cartilage matrix controls timing of endochondral ossification by inhibiting chondrocyte maturation. PLoS Genet. 2011;7:e1002246. doi: 10.1371/journal.pgen.1002246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto H, Enomoto-Iwamoto M, Iwamoto M, Nomura S, Himeno M, Kitamura Y, Kishimoto T, Komori T. Cbfa1 is a positive regulatory factor in chondrocyte maturation. The Journal of biological chemistry. 2000;275:8695–8702. doi: 10.1074/jbc.275.12.8695. [DOI] [PubMed] [Google Scholar]
- Enomoto-Iwamoto M, Enomoto H, Komori T, Iwamoto M. Participation of Cbfa1 in regulation of chondrocyte maturation. Osteoarthritis and cartilage/OARS, Osteoarthritis Research Society. 2001;9(Suppl A):S76–84. doi: 10.1053/joca.2001.0448. [DOI] [PubMed] [Google Scholar]
- Esko JD, Stewart TE, Taylor WH. Animal cell mutants defective in glycosaminoglycan biosynthesis. Proceedings of the National Academy of Sciences of the United States of America. 1985;82:3197–3201. doi: 10.1073/pnas.82.10.3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson GJ, Flint MH. Type X collagen synthesis by chick sternal cartilage and its relationship to endochondral development. The Journal of cell biology. 1985;101:277–284. doi: 10.1083/jcb.101.1.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldring MB, Tsuchimochi K, Ijiri K. The control of chondrogenesis. J Cell Biochem. 2006;97:33–44. doi: 10.1002/jcb.20652. [DOI] [PubMed] [Google Scholar]
- Gotting C, Kuhn J, Brinkmann T, Kleesiek K. Xylosyltransferase activity in seminal plasma of infertile men. Clinica chimica acta; international journal of clinical chemistry. 2002;317:199–202. doi: 10.1016/s0009-8981(01)00793-8. [DOI] [PubMed] [Google Scholar]
- Götting C, Kuhn J, Kleesiek K. Human xylosyltransferases in health and disease. Cell Mol Life Sci. 2007;64:1498–1517. doi: 10.1007/s00018-007-7069-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götting C, Sollberg S, Kuhn J, Weilke C, Huerkamp C, Brinkmann T, Krieg T, Kleesiek K. Serum xylosyltransferase: a new biochemical marker of the sclerotic process in systemic sclerosis. J Invest Dermatol. 1999;112:919–924. doi: 10.1046/j.1523-1747.1999.00590.x. [DOI] [PubMed] [Google Scholar]
- Gritli-Linde A, Lewis P, McMahon AP, Linde A. The whereabouts of a morphogen: direct evidence for short- and graded long-range activity of hedgehog signaling peptides. Developmental biology. 2001;236:364–386. doi: 10.1006/dbio.2001.0336. [DOI] [PubMed] [Google Scholar]
- Hammond CL, Schulte-Merker S. Two populations of endochondral osteoblasts with differential sensitivity to Hedgehog signalling. Development. 2009;136:3991–4000. doi: 10.1242/dev.042150. [DOI] [PubMed] [Google Scholar]
- Hilton MJ, Gutierrez L, Martinez DA, Wells DE. EXT1 regulates chondrocyte proliferation and differentiation during endochondral bone development. Bone. 2005;36:379–386. doi: 10.1016/j.bone.2004.09.025. [DOI] [PubMed] [Google Scholar]
- Hung IH, Yu K, Lavine KJ, Ornitz DM. FGF9 regulates early hypertrophic chondrocyte differentiation and skeletal vascularization in the developing stylopod. Developmental biology. 2007;307:300–313. doi: 10.1016/j.ydbio.2007.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itokowa T, Zhu ML, Troiano N, Bian J, Kawano T, Insogna K. Osteoclasts lacking Rac2 have defective chemotaxis and resorptive activity. Calcif Tissue Int. 2011;88:75–86. doi: 10.1007/s00223-010-9435-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalinina J, Byron SA, Makarenkova HP, Olsen SK, Eliseenkova AV, Larochelle WJ, Dhanabal M, Blais S, Ornitz DM, Day LA, Neubert TA, Pollock PM, Mohammadi M. Homodimerization controls the fibroblast growth factor 9 subfamily’s receptor binding and heparan sulfate-dependent diffusion in the extracellular matrix. Mol Cell Biol. 2009;29:4663–4678. doi: 10.1128/MCB.01780-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaplis AC, Luz A, Glowacki J, Bronson RT, Tybulewicz VL, Kronenberg HM, Mulligan RC. Lethal skeletal dysplasia from targeted disruption of the parathyroid hormone-related peptide gene. Genes & development. 1994;8:277–289. doi: 10.1101/gad.8.3.277. [DOI] [PubMed] [Google Scholar]
- Karp SJ, Schipani E, St-Jacques B, Hunzelman J, Kronenberg H, McMahon AP. Indian hedgehog coordinates endochondral bone growth and morphogenesis via parathyroid hormone related-protein-dependent and -independent pathways. Development. 2000;127:543–548. doi: 10.1242/dev.127.3.543. [DOI] [PubMed] [Google Scholar]
- Kawano T, Zhu M, Troiano N, Horowitz M, Bian J, Gundberg C, Kolodziejczak K, Insogna K. LIM kinase 1 deficient mice have reduced bone mass. Bone. 2013;52:70–82. doi: 10.1016/j.bone.2012.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearns AE, Campbell SC, Westley J, Schwartz NB. Initiation of chondroitin sulfate biosynthesis: a kinetic analysis of UDP-D-xylose: core protein beta-D-xylosyltransferase. Biochemistry. 1991;30:7477–7483. doi: 10.1021/bi00244a016. [DOI] [PubMed] [Google Scholar]
- Kearns AE, Vertel BM, Schwartz NB. Topography of glycosylation and UDP-xylose production. The Journal of biological chemistry. 1993;268:11097–11104. [PubMed] [Google Scholar]
- Kobayashi T, Soegiarto DW, Yang Y, Lanske B, Schipani E, McMahon AP, Kronenberg HM. Indian hedgehog stimulates periarticular chondrocyte differentiation to regulate growth plate length independently of PTHrP. J Clin Invest. 2005;115:1734–1742. doi: 10.1172/JCI24397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong Y. Btrim: a fast, lightweight adapter and quality trimming program for next-generation sequencing technologies. Genomics. 2011;98:152–153. doi: 10.1016/j.ygeno.2011.05.009. [DOI] [PubMed] [Google Scholar]
- Koziel L, Kunath M, Kelly OG, Vortkamp A. Ext1-dependent heparan sulfate regulates the range of Ihh signaling during endochondral ossification. Dev Cell. 2004;6:801–813. doi: 10.1016/j.devcel.2004.05.009. [DOI] [PubMed] [Google Scholar]
- Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003;423:332–336. doi: 10.1038/nature01657. [DOI] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nature protocols. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- Lanske B, Karaplis AC, Lee K, Luz A, Vortkamp A, Pirro A, Karperien M, Defize LH, Ho C, Mulligan RC, Abou-Samra AB, Jüppner H, Segre GV, Kronenberg HM. PTH/PTHrP receptor in early development and Indian hedgehog-regulated bone growth. Science. 1996;273:663–666. doi: 10.1126/science.273.5275.663. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics (Oxford, England) 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The Sequence Alignment/Map format and SAMtools. Bioinformatics (Oxford, England) 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liem KF, Jr, He M, Ocbina PJ, Anderson KV. Mouse Kif7/Costal2 is a cilia-associated protein that regulates Sonic hedgehog signaling. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:13377–13382. doi: 10.1073/pnas.0906944106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Xu J, Colvin JS, Ornitz DM. Coordination of chondrogenesis and osteogenesis by fibroblast growth factor 18. Genes & development. 2002;16:859–869. doi: 10.1101/gad.965602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak KK, Kronenberg HM, Chuang PT, Mackem S, Yang Y. Indian hedgehog signals independently of PTHrP to promote chondrocyte hypertrophy. Development. 2008;135:1947–1956. doi: 10.1242/dev.018044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minina E, Kreschel C, Naski MC, Ornitz DM, Vortkamp A. Interaction of FGF, Ihh/Pthlh, and BMP signaling integrates chondrocyte proliferation and hypertrophic differentiation. Dev Cell. 2002;3:439–449. doi: 10.1016/s1534-5807(02)00261-7. [DOI] [PubMed] [Google Scholar]
- Moran JL, Bolton AD, Tran PV, Brown A, Dwyer ND, Manning DK, Bjork BC, Li C, Montgomery K, Siepka SM, Vitaterna MH, Takahashi JS, Wiltshire T, Kwiatkowski DJ, Kucherlapati R, Beier DR. Utilization of a whole genome SNP panel for efficient genetic mapping in the mouse. Genome research. 2006;16:436–440. doi: 10.1101/gr.4563306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller S, Disse J, Schöttler M, Schön S, Prante C, Brinkmann T, Kuhn J, Kleesiek K, Götting C. Human xylosyltransferase I and N-terminal truncated forms: functional characterization of the core enzyme. The Biochemical journal. 2006;394:163–171. doi: 10.1042/BJ20051606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy A, Gertsenstain M, Vintersten K, Behringer RR. Manipulating the Mouse Embryo: A Laboratory Manual. 3. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2002. [Google Scholar]
- Nuwayhid N, Glaser JH, Johnson JC, Conrad HE, Hauser SC, Hirschberg CB. Xylosylation and glucuronosylation reactions in rat liver Golgi apparatus and endoplasmic reticulum. The Journal of biological chemistry. 1986;261:12936–12941. [PubMed] [Google Scholar]
- Nybakken K, Perrimon N. Heparan sulfate proteoglycan modulation of developmental signaling in Drosophila. Biochim Biophys Acta. 2002;1573:280–291. doi: 10.1016/s0304-4165(02)00395-1. [DOI] [PubMed] [Google Scholar]
- Ornitz DM. FGF signaling in the developing endochondral skeleton. Cytokine Growth Factor Rev. 2005;16:205–213. doi: 10.1016/j.cytogfr.2005.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peraita-Ezcurra M, Martínez-García M, Ruiz-Pérez VL, Sánchez-Gutiérrez ME, Fenollar-Cortés M, Vélez-Monsalve C, Ramos-Corrales C, Pastor I, Santonja C, Trujillo-Tiebas MJ. Ellis-van Creveld syndrome in a fetus with rhizomelia and polydactyly. Report of a case diagnosed by genetic analysis, and correlation with pathological andradiologic findings. Gene. 2012;499:223–225. doi: 10.1016/j.gene.2012.02.030. [DOI] [PubMed] [Google Scholar]
- Ponighaus C, Ambrosius M, Casanova JC, Prante C, Kuhn J, Esko JD, Kleesiek K, Gotting C. Human xylosyltransferase II is involved in the biosynthesis of the uniform tetrasaccharide linkage region in chondroitin sulfate and heparan sulfate proteoglycans. The Journal of biological chemistry. 2007;282:5201–5206. doi: 10.1074/jbc.M611665200. [DOI] [PubMed] [Google Scholar]
- Prante C, Bieback K, Funke C, Schon S, Kern S, Kuhn J, Gastens M, Kleesiek K, Gotting C. The formation of extracellular matrix during chondrogenic differentiation of mesenchymal stem cells correlates with increased levels of xylosyltransferase I. Stem Cells. 2006;24:2252–2261. doi: 10.1634/stemcells.2005-0508. [DOI] [PubMed] [Google Scholar]
- Pruitt KD, Harrow J, Harte RA, Wallin C, Diekhans M, Maglott DR, Searle S, Farrell CM, Loveland JE, Ruef BJ, Hart E, Suner MM, Landrum MJ, Aken B, Ayling S, Baertsch R, Fernandez-Banet J, Cherry JL, Curwen V, Dicuccio M, Kellis M, Lee J, Lin MF, Schuster M, Shkeda A, Amid C, Brown G, Dukhanina O, Frankish A, Hart J, Maidak BL, Mudge J, Murphy MR, Murphy T, Rajan J, Rajput B, Riddick LD, Snow C, Steward C, Webb D, Weber JA, Wilming L, Wu W, Birney E, Haussler D, Hubbard T, Ostell J, Durbin R, Lipman D. The consensus coding sequence (CCDS) project: Identifying a common protein-coding gene set for the human and mouse genomes. Genome research. 2009;19:1316–1323. doi: 10.1101/gr.080531.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratzka A, Kalus I, Moser M, Dierks T, Mundlos S, Vortkamp A. Redundant function of the heparan sulfate 6-O-endosulfatases Sulf1 and Sulf2 during skeletal development. Developmental dynamics : an official publication of the American Association of Anatomists. 2008;237:339–353. doi: 10.1002/dvdy.21423. [DOI] [PubMed] [Google Scholar]
- Retting KN, Song B, Yoon BS, Lyons KM. BMP canonical Smad signaling through Smad1 and Smad5 is required for endochondral bone formation. Development. 2009;136:1093–1104. doi: 10.1242/dev.029926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roch C, Kuhn J, Kleesiek K, Götting C. Differences in gene expression of human xylosyltransferases and determination of acceptor specificities for various proteoglycans. Biochem Biophys Res Commun. 2010;391:685–691. doi: 10.1016/j.bbrc.2009.11.121. [DOI] [PubMed] [Google Scholar]
- Rosenberg L. Chemical basis for the histological use of safranin O in the study of articular cartilage. The Journal of bone and joint surgery. American volume. 1971;53:69–82. [PubMed] [Google Scholar]
- Rousseau F, Bonaventure J, Legeai-Mallet L, Pelet A, Rozet JM, Maroteaux P, Le Merrer M, Munnich A. Mutations in the gene encoding fibroblast growth factor receptor-3 in achondroplasia. Nature. 1994;371:252–254. doi: 10.1038/371252a0. [DOI] [PubMed] [Google Scholar]
- Schön S, Prante C, Bahr C, Kuhn J, Kleesiek K, Götting C. Cloning and recombinant expression of active full-length xylosyltransferase I (XT-I) and characterization of subcellular localization of XT-I and XT-II. The Journal of biological chemistry. 2006a;281:14224–14231. doi: 10.1074/jbc.M510690200. [DOI] [PubMed] [Google Scholar]
- Schön S, Prante C, Bahr C, Tarnow L, Kuhn J, Kleesiek K, Götting C. The xylosyltransferase I gene polymorphism c.343G>T (p.A125S) is a risk factor for diabetic nephropathy in type 1 diabetes. Diabetes Care. 2006b;29:2295–2299. doi: 10.2337/dc06-0344. [DOI] [PubMed] [Google Scholar]
- Schön S, Schulz V, Prante C, Hendig D, Szliska C, Kuhn J, Kleesiek K, Götting C. Polymorphisms in the xylosyltransferase genes cause higher serum XT-I activity in patients with pseudoxanthoma elasticum (PXE) and are involved in a severe disease course. J Med Genet. 2006c;43:745–749. doi: 10.1136/jmg.2006.040972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreml J, Durmaz B, Cogulu O, Keupp K, Beleggia F, Pohl E, Milz E, Coker M, Ucar SK, Nurnberg G, Nurnberg P, Kuhn J, Ozkinay F. The missing “link”: an autosomal recessive short stature syndrome caused by a hypofunctional XYLT1 mutation. Human genetics. 2013 doi: 10.1007/s00439-013-1351-y. [DOI] [PubMed] [Google Scholar]
- Schwartz N. Biosynthesis and regulation of expression of proteoglycans. Front Biosci. 2000;5:D649–655. doi: 10.2741/a540. [DOI] [PubMed] [Google Scholar]
- Segev O, Chumakov I, Nevo Z, Givol D, Madar-Shapiro L, Sheinin Y, Weinreb M, Yayon A. Restrained chondrocyte proliferation and maturation with abnormal growth plate vascularization and ossification in human FGFR-3(G380R) transgenic mice. Hum Mol Genet. 2000;9:249–258. doi: 10.1093/hmg/9.2.249. [DOI] [PubMed] [Google Scholar]
- Selby PB. A rapid method for preparing high quality alizarin stained skeletons of adult mice. Stain Technol. 1987;62:143–146. doi: 10.3109/10520298709107984. [DOI] [PubMed] [Google Scholar]
- Shimizu H, Yokoyama S, Asahara H. Growth and differentiation of the developing limb bud from the perspective of chondrogenesis. Dev Growth Differ. 2007;49:449–454. doi: 10.1111/j.1440-169X.2007.00945.x. [DOI] [PubMed] [Google Scholar]
- St-Jacques B, Hammerschmidt M, McMahon AP. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes & development. 1999;13:2072–2086. doi: 10.1101/gad.13.16.2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoolmiller AC, Horwitz AL, Dorfman A. Biosynthesis of the chondroitin sulfate proteoglycan. Purification and properties of xylosyltransferase. The Journal of biological chemistry. 1972;247:3525–3532. [PubMed] [Google Scholar]
- Takarada T, Hinoi E, Nakazato R, Ochi H, Xu C, Tsuchikane A, Takeda S, Karsenty G, Abe T, Kiyonari H, Yoneda Y. An analysis of skeletal development in osteoblast-specific and chondrocyte-specific runt-related transcription factor-2 (Runx2) knockout mice. J Bone Miner Res. 2013;28:2064–2069. doi: 10.1002/jbmr.1945. [DOI] [PubMed] [Google Scholar]
- Takeda S, Bonnamy JP, Owen MJ, Ducy P, Karsenty G. Continuous expression of Cbfa1 in nonhypertrophic chondrocytes uncovers its ability to induce hypertrophic chondrocyte differentiation and partially rescues Cbfa1-deficient mice. Genes & development. 2001;15:467–481. doi: 10.1101/gad.845101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesan N, Barre L, Bourhim M, Magdalou J, Mainard D, Netter P, Fournel-Gigleux S, Ouzzine M. Xylosyltransferase-I regulates glycosaminoglycan synthesis during the pathogenic process of human osteoarthritis. PloS one. 2012;7:e34020. doi: 10.1371/journal.pone.0034020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Thaller C, Eichele G. GenePaint.org: an atlas of gene expression patterns in the mouse embryo. Nucleic acids research. 2004;32:D552–556. doi: 10.1093/nar/gkh029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vortkamp A, Lee K, Lanske B, Segre GV, Kronenberg HM, Tabin CJ. Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science. 1996;273:613–622. doi: 10.1126/science.273.5275.613. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Kimata K, Line S, Strong D, Gao LY, Kozak CA, Yamada Y. Mouse cartilage matrix deficiency (cmd) caused by a 7 bp deletion in the aggrecan gene. Nature genetics. 1994;7:154–157. doi: 10.1038/ng0694-154. [DOI] [PubMed] [Google Scholar]
- Watanabe Y, Takeuchi K, Higa Onaga S, Sato M, Tsujita M, Abe M, Natsume R, Li M, Furuichi T, Saeki M, Izumikawa T, Hasegawa A, Yokoyama M, Ikegawa S, Sakimura K, Amizuka N, Kitagawa H, Igarashi M. Chondroitin sulfate N-acetylgalactosaminyltransferase-1 is required for normal cartilage development. The Biochemical journal. 2010;432:47–55. doi: 10.1042/BJ20100847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson DG, Phamluong K, Lin WY, Barck K, Carano RA, Diehl L, Peterson AS, Martin F, Solloway MJ. Chondroitin sulfate synthase 1 (Chsy1) is required for bone development and digit patterning. Developmental biology. 2012;363:413–425. doi: 10.1016/j.ydbio.2012.01.005. [DOI] [PubMed] [Google Scholar]
- Yan D, Lin X. Shaping morphogen gradients by proteoglycans. Cold Spring Harb Perspect Biol. 2009;1:a002493. doi: 10.1101/cshperspect.a002493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Eberspaecher H, Lefebvre V, De Crombrugghe B. Parallel expression of Sox9 and Col2a1 in cells undergoing chondrogenesis. Developmental dynamics : an official publication of the American Association of Anatomists. 1997;209:377–386. doi: 10.1002/(SICI)1097-0177(199708)209:4<377::AID-AJA5>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]