

Abstract
Aims
Autonomic dysfunction is a feature of chronic heart failure (HF). This study tested the hypothesis that chronic open-loop electrical vagus nerve stimulation (VNS) improves LV structure and function in canines with chronic HF.
Methods and results
Twenty-six canines with HF (EF ∼35%) produced by intracoronary microembolizations were implanted with a bipolar cuff electrode around the right cervical vagus nerve and connected to an implantable pulse generator. The canines were enrolled in Control (n = 7) vs. VNS therapy (n = 7) or a crossover study, with crossovers occurring at 3 months (C × VNS, n = 6; VNS × C, n = 6). After 6 months of VNS, LVEF and LV end-systolic volume (ESV) were significantly improved compared with Control (ΔEF Control –4.6 ± 0.9% vs. VNS 6.0 ± 1.6%, P < 0.001) and (ΔESV Control 8.3 ± 1.8 mL vs. VNS –3.0 ± 2.3 mL, P = 0.002. Plasma and tissue biomarkers were also improved. In the crossover study, VNS also resulted in a significant improvement in EF and ESV compared with Control (ΔEF Control –2.3 ± 0.65% vs. VNS 6.7 ± 1.1 mL, P < 0.001 and ΔESV Control 3.2 ± 1.2 mL vs. VNS –4.0 ± 0.9 mL, P < 0.001). Initiation of therapy in the Control group at 3 months resulted in a significant improvement in EF (Control –4.7 ± 1.4% vs. VNS 3.7 ± 0.74%, P < 0.001) and ESV (Control 1.5 ± 1.2 mL vs. NS –5.5 ± 1.6 mL, P = 0.003) by 6 months.
Conclusions
In canines with HF, long-term, open-looped low levels of VNS therapy improves LV systolic function, prevents progressive LV enlargement, and improves biomarkers of HF when compared with control animals that did not receive therapy.
Keywords: Vagal, Autonomic nervous system, Parasympathetic, Neurostimulation, Heart failure
Introduction
Dysfunction of the autonomic nervous system is involved in the development and progression of chronic heart failure.1,2 In heart failure, this dysfunction is characterized by excessive sympathetic drive accompanied by parasympathetic withdrawal. The mechanisms responsible for sustained sympathetic activation and parasympathetic withdrawal are not fully understood, but are in part due to a compensatory response to the failing myocardium and a depressed baroreflex.3–5 Extensive support exists for the use of pharmacological agents to suppress sympathetic activity with beta-adrenergic receptor blockers, and has been shown to reduce mortality and morbidity in patients with heart failure.6 However, suppression of the sympathetic nervous system addresses only half of the autonomic imbalance. Clinical evidence suggests that diminished cardiac vagal activity and subsequently elevated heart rates (HRs) are predictors of mortality in heart failure.7,8 Vagal stimulation in a rodent model of heart failure reduces mortality,9 while vagal stimulation in a canine heart failure model attenuates cardiac remodelling and helps maintain cardiac function.10 In addition, a non-randomized clinical trial exploring vagal stimulation in 32 NYHA class II–IV heart failure patients has been conducted. It was found that preferential stimulation of vagal efferent fibres delivered in a closed-loop manner timed to the cardiac cycle improves quality of life, exercise capacity, and LVEF.11
The present study was designed to test the hypothesis that open-loop cervical vagus nerve stimulation (VNS) of both efferent and afferent vagal fibres can significantly improve LV function and prevent LV dilation in a canine model of heart failure. In addition, it was hypothesized that prolonged VNS would be required for continued benefit and that withdrawal of VNS therapy would lead to a resumption of progressive LV dysfunction and maladaptive remodelling.
Methods
This study was approved by the Henry Ford Health System Institutional Animal Care and Use Committee and conformed to the ‘Position of the American Heart Association on Research Animal Use’.12
Experimental heart failure model
The canine model of chronic post-ischaemic heart failure used in this study was previously described in detail.13 Briefly, LV dysfunction is produced by multiple, sequential coronary microembolizations with polystyrene microspheres (70–102 μm in diameter; Polysciences, Warrington, PA, USA), which results in loss of viable myocardium, an increase in ventricular volumes, and a reduction in LVEF. In the present study, two sequential investigations enrolled 26 healthy, mongrel canines weighing between 17 and 24 kg. Embolizations were performed 1–2 weeks apart and were discontinued when EF, determined angiographically, was ≤35%. All procedures were performed by cardiac catheterization under general anaesthesia and sterile conditions. Anaesthesia was induced using a combination of i.v. injections of hydromorphone (0.22 mg/kg) and diazepam (0.2–0.6 mg/kg), and maintained throughout the procedure with 1–2% isoflurane. The canines were intubated and ventilated with room air supplemented with oxygen. Once the target EF was reached, the VNS system was surgically implanted in all canines.
Vagus nerve stimulation and study design
A bipolar cuff electrode was implanted around the right cervical vagus nerve and connected to a custom implantable pulse generator (Boston Scientific Corporation, St. Paul, MN, USA). A period of ∼30 days was allowed for recovery from the surgical implantation of the stimulation system before animals were enrolled in the study. Fourteen canines were enrolled in a 6-month longitudinal study and randomly assigned to no therapy (Control, n =7) or active VNS therapy (VNS, n =7). In addition, 12 canines were randomly assigned to no therapy crossed-over to VNS therapy (C ×VNS, n =6) or VNS therapy crossed-over to no therapy (VNS ×C, n =6). All four groups of canines were followed for 6 months, with crossovers occurring at 3 months. Bipolar, afferent and efferent, open-loop VNS was continuously delivered at a pre-determined frequency (20 Hz), pulse width (300 μs), and duty cycle (10 s of every minute). Pulse voltage was adjusted to a level which did not cause any major side effects such as chronic coughing, and ranged from 0.6 to 1.9 V. These values were sufficiently low to not cause any acute reductions in HR. Following randomization, the VNS group received therapy titrations twice a week for 1 month (eight total titrations). Prior to data collection, VNS therapy was turned off for 5–10 min and remained off for the duration of the cardiac catheterizations and haemodynamic evaluations. Haemodynamic, ventriculographic, echocardiographic, Doppler, and ECG measurements were made at baseline, prior to any microembolizations, and were repeated at the PRE time point once the canines were in confirmed heart failure, and again at 3 and 6 months after randomization. Animals enrolled in the 6-month longitudinal study had blood samples obtained at baseline, PRE, and 3 and 6 months post-randomization. Samples were stored at –80°C for subsequent analysis. Blood samples were obtained only at 3 and 6 months for the crossover study. No animals received background pharmacological therapy during this investigation.
Cardiovascular assessment
All haemodynamic measurements were made during left and right heart catheterizations in anaesthetized canines. Aortic and LV pressures were measured with catheter tip micromanometers (Millar Instruments, Houston, TX, USA). Left ventriculograms were performed during cardiac catheterization after completion of the haemodynamic measurements using Siemens Coroskop X-ray Systems (Munich, Germany). Ventriculograms were performed with the dog placed in the right lateral decubitus position and were recorded on 35 mm digital media at 30 frames/s during an injection of 20 mL of contrast material (ISOVU-300, Bracco Diagnostics, Inc., Princeton, NJ, USA). Correction for image magnification was made using a radiopaque grid placed at the level of the left ventricle. The LV end-systolic (ESV) and end-diastolic (EDV) volumes were calculated from angiographic silhouettes using the area–length method.14 The LVEF was calculated as described previously.13 Cardiac output (CO) was calculated as the product of HR and stroke volume (SV). Ventriculograms were evaluated by a blinded sonographer. Echocardiographic and Doppler studies were performed using a General Electric VIVID-7 ultrasound system with a 3.5 MHz transducer and recorded on a VHS recorder for off-line analysis. Echocardiographic measurements were made with the dog placed in the right lateral decubitus position and recorded on a Panasonic 6300 VHS recorder for off-line analysis. The LV major and minor semi-axes were measured and used for calculation of LV end-diastolic circumferential wall stress (EDWS). Wall stress was calculated as follows:
where P is LV end-diastolic pressure (LVEDP), a is the LV major semi-axis, b is the LV minor semi-axis, and h is the LV wall thickness. Mitral inflow velocity was measured by pulsed-wave Doppler echocardiography. The velocity waveforms were used to calculate the peak mitral inflow velocity in early diastole (PE), peak mitral inflow velocity during left atrial contraction (PA), the ratio of PE to PA, and the deceleration time of early mitral inflow velocity (DT). Echocardiograms were evaluated blinded by a sonographer.
All canines in the 6-month longitudinal study underwent 24 h ambulatory ECG Holter monitoring just prior to randomization (PRE), immediately after randomization (0 month), at 3 and 6 months, and immediately after the 6 month endpoint where therapy was turned off for the VNS group to allow for comparison without VNS following chronic treatment. Animals assigned to the crossover study underwent 24 h ambulatory ECG Holter monitoring prior to randomization and at 3 and 6 months.
Gross pathology and left ventricular tissue collection
At the end of 6 months of follow-up data collection, and while under general anaesthesia, the animal chest and abdomen were opened and animals were examined for evidence of pleural effusion, pericardial effusion, and ascites. After examination, the heart was rapidly removed and placed in ice-cold Tris buffer (pH 7.4). The left and right ventricles were then separated. Three 2 mm thick transverse slices were obtained from the left ventricle; one slice from the basal third, one from the middle third, and one from the apical third. Transmural blocks were also obtained and rapidly frozen in isopentane cooled to –160°C by liquid nitrogen and stored at –70°C for biochemical examination.
Protein expression in left ventricular tissue
Freshly frozen LV tissue was used to assess levels of the following proteins using western blotting: tumour necrosis factor-α (TNF-α) (EMD Millipore, Billerica, MA, USA), interleukin-6 (IL-6) (Santa Cruz Biotechnology, Santa Cruz, CA, USA), active caspase-3 (Sigma-Aldrich, Corp., St. Louis, MO, USA), B-cell lymphoma 2 (BCL-2) (BD Biosciences, San Jose, CA, USA), connexin-43 (CNX-43) (ABCAM, Cambridge, MA, USA), endothelial nitric oxide synthase (eNOS), inducible nitric oxide synthase (iNOS), and neuronal nitric oxide synthase (nNOS) (LifeSpan Biosciences, Inc., Seattle, WA, USA), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Fitzgerald Industries, International, Sudbury, MA, USA). Protein levels were measured in LV homogenate by western blots as described previously.15,16 Primary antibodies specific to each protein were diluted based on the supplier's instructions. In all instances, the antibody was present in excess over the antigen, and the density of each protein band was in the linear scale. Band intensities were quantified in densitometric units. The left ventricle from six healthy canines was also analysed.
Protein levels in plasma
Plasma samples were used to assess levels of the following proteins using enzyme-linked immunosorbent assay (ELISA): TNF-α (Assay Designs, Inc., Ann Arbor, MI, USA), IL-6 (Assay Designs, Inc.), NT-proBNP (ALPCO, Salem, NH, USA), pro atrial natriuretic peptide (proANP) (ALPCO), and C-reactive protein (CRP) (ALPCO). The level of proANP was determined in a plasma double antibody sandwich ELISA and that of all other proteins was determined based on competitive ELISA performed according to instructions provided by the supplier.
Data analysis
Within-group comparisons were made between measurements obtained at PRE, 3 months, and 6 months using repeated measures analysis of variance (ANOVA) with α set at 0.05. If the overall ANOVA was significant, then pairwise comparisons between PRE and 3 months and 6 months were performed using the Students–Newman–Keuls test. To assess treatment effect, the change (Δ) in each measure from PRE to 3 months or 6 months was calculated for each of the two study arms in the longitudinal study. To determine whether significant differences in Δ were present between the control group and the VNS treatment groups, a t-statistic for two means was used, with P ≤ 0.05 considered significant. For all pairwise comparisons, a probability value <0.05 was considered significant. All data are reported as the mean change ±SEM.
Results
Six-month longitudinal study results
Haemodynamic, ventriculographic, echocardiographic, Doppler, and electrocardiogram measurements
There were no significant differences at Baseline and PRE for any measure between control canines and canines randomized to VNS therapy (Table 1). Following 6 months of therapy, there was a significant difference (P < 0.05) in the ΔCO (Control 0.07 ±0.05 L/min vs. VNS 0.44 ±0.8 L/min; P =0.04), ΔDT (Control –9 ±2 ms vs. VNS 12 ±4 ms; P < 0.001), and ΔSV (Control –1 ±1 mL vs. VNS 4 ±1 mL; P < 0.001) when compared with the control group.
Table 1.
Haemodynamic, ventriculographic, echocardiographic, and Doppler measures in canines randomized to the 6-month longitudinal study
| Measurement | Arm | Baseline | PRE | 3 months | 6 months |
|---|---|---|---|---|---|
| HR (b.p.m.) | Control | 81 ± 3 | 78 ± 2 | 83 ± 2 | 86 ± 3 |
| VNS | 83 ± 3 | 81 ± 2 | 82 ± 3 | 85 ± 3 | |
| mAoP (mmHg) | Control | 74 ± 5 | 68 ± 3 | 73 ± 2 | 74 ± 1 |
| VNS | 78 ± 2 | 72 ± 3 | 72 ± 3 | 80 ± 4 | |
| LVEDP (mmHg) | Control | 10 ± 0.5 | 12 ± 0.8 | 14 ± 1.0 | 14 ± 1.1 |
| VNS | 10 ± 0.5 | 14 ± 0.9 | 12 ± 0.8* | 12 ± 0.5 | |
| SV (mL)a | Control | 27 ± 1.5 | 19 ± 0.9 | 19 ± 0.9 | 19 ± 0.9 |
| VNS | 26 ± 0.9 | 19 ± 0.7 | 21 ± 0.7* | 23 ± 1.1† | |
| CO (L/min)a | Control | 2.20 ± 0.17 | 1.51 ± 0.07 | 1.56 ± 0.08 | 1.58 ± 0.03 |
| VNS | 2.17 ± 0.13 | 1.55 ± 0.07 | 1.76 ± 0.11* | 1.99 ± 0.10† | |
| PE/PA | Control | 3.3 ± 0.1 | 2.2 ± 0.2 | 2.0 ± 0.1 | 2.0 ± 0.1 |
| VNS | 3.2 ± 0.1 | 2.0 ± 0.1 | 2.3 ± 0.1* | 2.2 ± 0.1 | |
| DT (ms) | Control | 110 ± 3 | 80 ± 4 | 78 ± 5 | 72 ± 5 |
| VNS | 114 ± 2 | 76 ± 3 | 83 ± 5* | 88 ± 5† | |
| EDWS (gm/cm2) | Control | 32 ± 3 | 51 ± 4 | 62 ± 6 | 60 ± 7 |
| VNS | 37 ± 3 | 68 ± 8 | 59 ± 7* | 56 ± 4† |
Values are given as the mean ± SEM.
CO, cardiac output; DT, deceleration time of early mitral inflow velocity; EDP, LV end-diastolic pressure; EDWS, end-diastolic circumferential wall stress; HR, heart rate; mAoP, mean aortic pressure; PE/PA, ratio of peak mitral inflow velocity in early diastole (PE) to peak mitral inflow velocity during left atrial contraction (PA); SV, stroke volume; VNS, vagus nerve stimulation.
aVentriculography-derived data.
*P < 0.05 VNS vs. control Δ from PRE to 3 months; †P < 0.05 VNS vs. control Δ from 3 to 6 months.
Figure 1 presents the LVEDV, LVESV, and LVEF ventriculography data for the canines enrolled in the 6-month chronic study. There was a significant difference in the ΔESV (Control 8 ±2 mL vs. VNS –3 ±2 mL, P =0.002) and ΔEF (Control –3 ±1% vs. VNS 6 ±1%, P < 0.001) after 6 months of treatment compared with the control group. LV ΔEDV differences between control and VNS were not statistically significant (Control 7 ±2 mL vs. VNS 1 ±2 mL, P =0.055).
Figure 1.
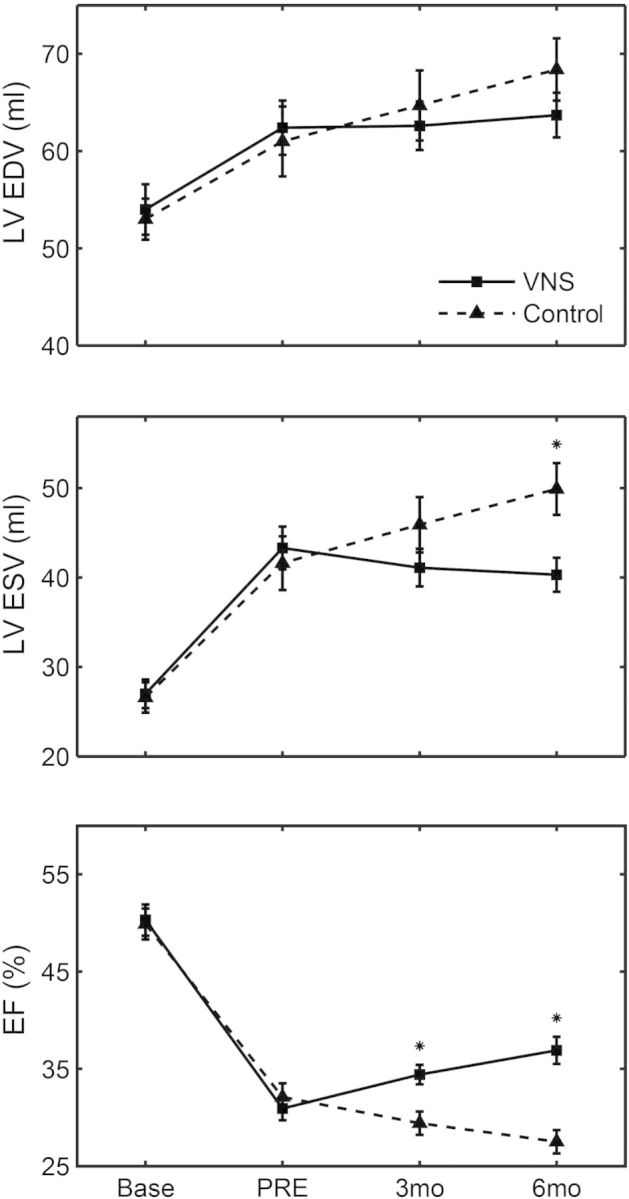
Six-month longitudinal LV end-diastolic volume (LVEDV), LV end-systolic volume (LVESV), and LVEF data obtained by ventriculography at pre-treatment (PRE), 3 months, and 6 months. Solid line, vagus nerve stimulation (VNS); dashed line, control. *P < 0.05 vs. control.
Holter data
The acute impact of vagal therapy on HR was investigated at two time points. It was first studied immediately prior to and after vagal therapy initiation, and secondly, prior to and after vagal therapy termination at 6 months. No difference in HR was seen with therapy activation or deactivation (Table 2). When looking at chronic HR levels, there was a significant reduction in mean HR at 3 months and in the mean as well as the minimum HR at 6 months in the VNS treatment group when compared with the pre-therapy time point (Table 2). The minimum HR was also decreased in the control group over the 6 months. The control group showed a significant increase in the maximum HR over 6 months and there was a significantly higher maximum HR in the control group at 6 months when compared with the VNS group.
Table 2.
Ambulatory Holter data for the 6-month longitudinal study
| Measurement | Arm | PRE OFF | 0 months OFF/ONa | 3 months OFF/ONa | 6 months OFF/ONa | 6 months OFF |
|---|---|---|---|---|---|---|
| HRmax | Control | 187 ± 13 | 190 ± 8 | 185 ± 7 | 202 ± 4* | 208 ± 10 |
| VNS | 185 ± 13 | 186 ± 9 | 179 ± 9 | 186 ± 7† | 178 ± 8† | |
| HRmin | Control | 52 ± 3 | 53 ± 2 | 49 ± 2 | 46 ± 2* | 47 ± 3 |
| VNS | 58 ± 4 | 58 ± 4 | 52 ± 4 | 51 ± 2* | 51 ± 3 | |
| HRmean | Control | 90 ± 3 | 87 ± 3 | 84 ± 2 | 83 ± 2 | 84 ± 2 |
| VNS | 100 ± 4 | 95 ± 3 | 89 ± 4* | 87 ± 3* | 86 ± 4 |
PRE, 24 h immediately before randomization, 0, 24 h immediately after randomization.
HR, heart rate; VNS, vagus nerve stimulation.
aVNS group is ON, control group is OFF. 6 months OFF was only compared with the 6 months OFF/ON and a between-group comparison of control vs. VNS.
*P < 0.05 compared with PRE; †P < 0.05 VNS vs. Control.
Biomarker levels in plasma
Induction of heart failure caused a significant worsening for all plasma biomarkers (Figure 2). After 6 months, there were significant differences in all biomarkers when comparing the changes in VNS with the control.
Figure 2.

Protein levels in plasma for NT-proBNP, pro atrial natriuretic peptide (proANP), C-reactive protein (CRP), interleukin-6 (IL-6), and tumour necrosis factor-α (TNF-α) for the 6-month longitudinal study. Solid lines, vagus nerve stimulation (VNS) (n = 7); dashed lines, control (n = 7). *P < 0.05 vs. VNS.
Protein expression in left ventricular tissue
The results of the determination of protein levels in LV tissue in canines included in the 6 month study are shown in Table 3, along with results from healthy control canines (n =6) for comparison. Compared with healthy canines, TNF-α, IL-6, BCL-2, caspase-3, CNX-43, iNOS, and nNOS levels were significantly increased and eNOS was significantly decreased in HF control canines. Canines treated with VNS had TNF-α, IL-6, caspase-3, CNX-43, iNOS, and nNOS levels that were significantly lower and eNOS levels significantly higher than in the control canines. The results for the biomarkers did not change when normalized for GAPDH, a housekeeping protein.
Table 3.
Protein expression in left ventricular tissue in canines included in the 6-month longitudinal study
| Normal (n = 6) | Control (n = 7) | VNS (n = 7) | |
|---|---|---|---|
| GAPDH | 1.38 ± 0.10 | 1.36 ± 0.09 | 1.41 ± 0.09 |
| TNF-α | 0.94 ± 0.17 | 3.51 ± 0.46* | 1.44 ± 0.25† |
| IL-6 | 0.36 ± 0.03 | 1.33 ± 0.15* | 0.57 ± 0.06*† |
| BCL-2 | 0.14 ± 0.02 | 0.26 ± 0.04* | 0.52 ± 0.09*† |
| Caspase-3 | 0.20 ± 0.01 | 0.44 ± 0.04* | 0.34 ± 0.02*† |
| CNX-43 | 2.08 ± 0.12 | 0.37 ± 0.03* | 0.55 ± 0.05*† |
| eNOS | 0.53 ± 0.05 | 0.18 ± 0.03* | 0.33 ± 0.02*† |
| iNOS | 0.21 ± 0.02 | 1.12 ± 0.11* | 0.49 ± 0.06*† |
| nNOS | 0.25 ± 0.05 | 1.03 ± 0.19* | 0.46 ± 0.16† |
All measurements are in densitometric unit (dU). BCL-2, B-cell lymphoma 2; CNX-43, connexin-43; eNOS, endothelial nitric oxide synthase; iNOS, inducible nitric oxide synthase; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; IL-6, interleukin-6; nNOS; neuronal nitric oxide synthase; TNF-α, tumour necrosis factor-α; VNS, vagus nerve stimulation.
*P < 0.05 vs. normal; †P < 0.05 vs. control.
Crossover study results
Ventriculographic measurements
There were no significant differences between the two study groups for any of the measurements obtained at Baseline or PRE (Figure 3). At the 3-month time point (prior to crossover), treatment with VNS significantly improved LV ΔESV when compared with control. ΔEF values were also significantly improved with treatment. LVEDV was unchanged. Initiation of therapy in the C ×VNS group at 3 months resulted in a significant difference in ΔEF and ΔESV. Compared with the pre-randomization time point, there were no differences between the groups for LV ΔESV and ΔEF after 6 months.
Figure 3.
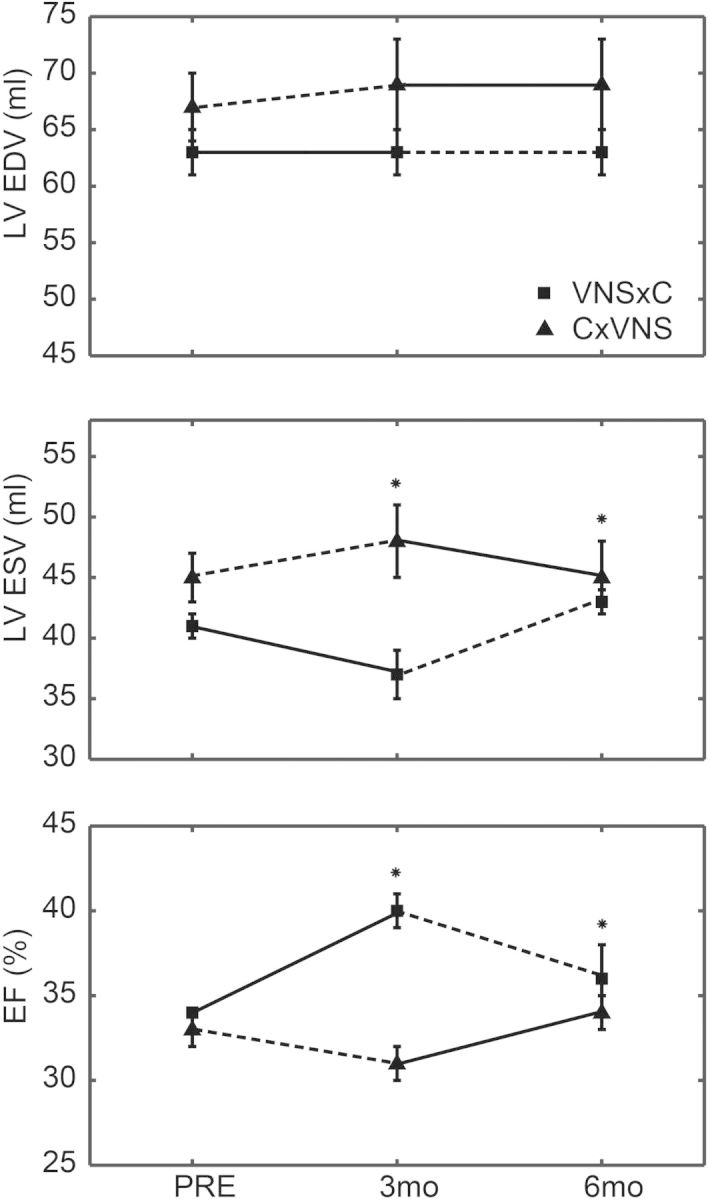
Crossover LV end-diastolic volume (LVEDV), LV end-systolic volume (LVESV), and LVEF data at pre-treatment (PRE), 3 months, and 6 months. The no therapy period is represented by the dashed line. *P < 0.05 vs. C × VNS. C × VNS, no therapy crossed-over to vagus nerve stimulation (VNS) therapy.
Biomarker levels in plasma
As shown in Table 4, by 3 months there was a significant difference between the VNS ×C vs. C ×VNS for CRP, proANP, IL-6, NT-proBNP, and TNF-α. After the crossover period at 3 months, canines in the VNS ×C group had a significant worsening in plasma levels for all biomarkers except CRP. The C ×VNS group showed significant improvement in IL-6, CRP, and NT-proBNP, but not for TNF-α or proANP.
Table 4.
Protein levels in plasma of canines included in the crossover study
| VNS × C | 3 months VNS ON | 6 months VNS OFF | Δ3 months to 6 months |
|---|---|---|---|
| TNF-α (pg/mL) | 4.0 ± 0.2* | 8.9 ± 0.9 | 4.9 ± 0.46† |
| IL-6 (pg/mL) | 44.9 ± 4.0* | 113.6 ± 8.7 | 68.7 ± 5.5† |
| CRP (ng/mL) | 11139 ± 692* | 13083 ± 1107 | 1944 ± 1488.7 |
| ProANP (pg/mL) | 2336 ± 285* | 4078 ± 218 | 1742 ± 372.2† |
| NT-proBNP (pg/mL) | 694 ± 31* | 1282 ± 21 | 587.5 ± 36.9† |
| C × VNS | 3 months VNS OFF | 6 months VNS ON | Δ3 months to 6 months |
| TNF-α (pg/mL) | 8.8 ± 0.7 | 9.4 ± 0.6 | 0.56 ± 0.3 |
| IL-6 (pg/mL) | 90.0 ± 5.7 | 77.3 ± 4.6 | –12.7 ± 4.2‡ |
| CRP (ng/mL) | 23 699 ± 1148 | 13 506 ± 1128 | –10 193 ± 2126.7‡ |
| ProANP (pg/mL) | 8349 ± 230 | 8786 ± 181 | 437 ± 286.8‡ |
| NT-proBNP (pg/mL) | 1378 ± 37 | 1130 ± 39 | –248 ± 61.6‡ |
Results are shown for 3 months (initial randomization) and 6 months (3 months post-crossover).
C × VNS, no therapy crossed-over to VNS therapy; CRP, C-reactive protein; IL-6, interleukin-6; proANP, pro atrial natriuretic peptide; TNF-α, tumour necrosis factor-α; VNS, vagus nerve stimulation; VNS × C, VNS therapy crossed-over to no therapy.
*P < 0.05 vs. 3 months VNS OFF; †P < 0.05 Δ 3 months VNS ON to 6 months VNS OFF; ‡P < 0.05 Δ 3 months VNS ON to 6 months VNS OFF.
Holter data
The 24 h Holter data shown in Table 5 indicate there were no significant differences within or between group means for the crossover study.
Table 5.
Ambulatory Holter data for the crossover study
| Arm | PRE | 3 months | 6 months | |
|---|---|---|---|---|
| HRmean | C × VNS | 80 ± 9 | 77 ± 9 | 79 ± 4 |
| VNS × C | 99 ± 6 | 89 ± 3 | 80 ± 5 |
Crossovers occurred at the 3-month follow-up.
C × VNS, no therapy crossed-over to VNS therapy; HR, heart rate; PRE, 24 h immediately before randomization; VNS, vagus nerve stimulation; VNS × C, VNS therapy crossed-over to no therapy.
Discussion
The results of this study indicate that long-term, open-loop VNS improves LV function, prevents LV dilation, and favourably alters cardiac biomarkers in canines with heart failure. This investigation had three significant findings: (i) VNS results in continued improvement in LV function with 6 months of continuous therapy; (ii) data obtained from the crossover investigation provide evidence that continuous delivery of VNS may be required for prolonged therapeutic efficacy; and (iii) open-looped, bi-directional (efferent and afferent) VNS is effective. The canine model of heart failure used in this study manifests many of the haemodynamic and neurohormonal sequelae of heart failure observed in humans (i.e. marked and progressive depression of LV systolic and diastolic function and reduced CO). This is an important distinction from other models such as rapid pacing which may result in spontaneous improvement when the pacing is stopped.17 The model used in this study produces a progressive condition of worsening heart failure, even after coronary microembolization is stopped. Thus, the findings from this study offer an important step forward in translating the results to clinical trial. All animals which received active VNS therapy during the investigation responded to the therapy as supported by the changes in LV remodelling and LV systolic function, and, although there was only a modest effect on EDV, the observed changes in LVEDP, PE/PA, DT, and EDWS suggest that VNS therapy improves LV diastolic function as well. Control canines not receiving therapy underwent progressive worsening of LV remodelling and LV systolic function, and a deterioration in diastolic function.
Animals first assigned to the control group whose LV function significantly decreased over the first 3 months of the investigation still responded to VNS therapy at crossover, suggesting that VNS may be effective along a continuum of the failing heart. Importantly, when VNS therapy was inactivated in animals first assigned to therapy, LV improvement ceased over the next 3 months, suggesting continued VNS therapy is required for sustained benefit.
The vagus nerve is known to have important effects over the control of HR. A prevailing theory has been that increasing vagal traffic to the heart at an intensity sufficient to reduce HR could positively impact sympathetic drive, pro-inflammatory cytokines, nitric oxide (NO) elaboration, and myocardial expression of gap junction proteins.18 Indeed, previous investigations using vagal stimulation to explore the impact on heart failure have used HR reduction as a part of a closed-loop system for titration and therapy delivery.9,10,19 However, the precise role of VNS-evoked bradycardia in the treatment of heart failure remains unclear,11,20 but the amplitude and frequency at which the HR is reduced has been used as a reference point for setting the stimulation parameters for VNS. Previous experiments have examined the activation (recruitment) of myelinated and unmyelinated fibres by electrical stimulation of the cervical vagus nerve in anaesthetized canines.21 Using the same bipolar electrode as in the current study, it was found that the stimulus amplitude required to evoke bradycardia (4.3 ±1.2 mA, n =4) was consistently equal to or greater than the threshold for activating B-fibres of the vagus nerve. This finding is consistent with other investigations which report that B-fibres have a cardiac slowing effect but the magnitude of the slowing is significantly lower than that achieved when C-fibres are also stimulated.21 In the present study, stimulation amplitudes were well below the amplitudes previously reported to evoke bradycardia. Furthermore, the lack of an observed HR response with ambulatory Holter monitoring in the present investigation provides further evidence that the present study used sufficiently low amplitudes so as to avoid any bradycardic effects from VNS. There was a significant reduction in the mean HR for the VNS group after 6 months of therapy, but this reduction is probably due to an improvement in heart failure, as there was no change in Holter values performed over two consecutive 24 h periods with therapy on vs. off.
This is also the first investigation to report VNS efficacy in heart failure where stimulation was delivered in an open-loop fashion not gated by sensing HR or a HR reduction, thereby eliminating the need for a cardiac lead and sensing. In the present study, the therapeutic effect of VNS was independent of HR. This provides an important step in our understanding of the underlying mechanism of vagal stimulation, in that HR reduction may not be essential to achieve a beneficial effect on the progression of heart failure. The magnitude of the VNS benefit, however, may be higher if HR reduction is also present.
Whether the effect is mediated by afferent or efferent mechanisms is not well understood. In previously published work, a specially designed tripolar electrode was used preferentially to deliver efferent stimulation and minimize neural traffic to the brain.11,19 The assumption appears to be that the effects mediating an improvement in heart failure are largely efferent in nature. In the current study, a bipolar electrode was used to deliver stimulation, and both efferent and afferent neural traffic is generated as a result.18,22,23 This study submits evidence that unidirectional nerve stimulation is not a requirement for a therapeutic effect.
Whether or not VNS must be delivered at precise times in certain physiological conditions is also an important consideration. In the previous pre-clinical work,18,19 the system required an implanted cardiac lead which would sense each beat and deliver the stimulus at a fixed interval within the cardiac cycle. This is presumably because spontaneous vagal reflexes have been shown to occur within certain phases of the cardiac and respiratory cycles.23 Thus, by delivering the stimulus at just the right time in the cardiac cycle, it could be argued that this restores the intrinsic discharge pattern of the vagus nerve. However, our current study suggests that such sophisticated timing within the cardiac cycle is not necessary to provide a therapeutic benefit.
The present investigation cannot provide a definitive mechanism for the observed improvements in LV systolic function from VNS. Antiapoptotic cell signalling, increased production of NO, and an anti-inflammatory effect may have contributed to the reverse remodelling observed in this investigation, but it is unclear if these markers are at the beginning or the end of a cascade of effects. However, a key component in the cascade may be NO. Vagal stimulation is known to have profound and immediate effects on the release of NO.24 Nitric oxide is derived from three different isoforms of NOS: iNOS, eNOS, and nNOS. In heart failure, it is known that NOS is adversely impacted.25 In the present investigation, all three isoforms of NOS were significantly improved. Interestingly, these results are consistent with previous findings in which a much higher dose of vagal stimulation was provided.26 Finally, it has been shown than VNS in mice with experimental heart failure can reduce the production of reactive oxygen species (ROS).27 When NO is blocked, the ROS suppression is blunted, suggesting a role for VNS- and NO-mediated effects on myocardial metabolism.
The most effective mean of vagal stimulation is still a question that is largely unanswered, and the variables one must consider are quite complicated and numerous. When considering strategies for effective therapy delivery, one must study the optimal settings for stimulation frequency (Hz), amplitude (mA), and pulse width (PW), the amount of on vs. off time per minute, per hour, per day, time of day, selective stimulation of afferent vs. efferent fibres, and even the specific fibres (e.g. A-, B-, and C-fibres) within the vagus nerve that may be responsible for the effect. In addition, is the therapy most effective when applied to the right cervical vagus nerve as done in this investigation? Would it be more efficacious if the left vagus nerve were stimulated, or if bilateral stimulation were delivered? In the current study, a low amplitude (mA) of 20 Hz VNS was applied continuously for 10 s of every minute, 24 h per day, and was not timed to the cardiac cycle or gated off any physiological response to the stimulation. Although this was sufficient to produce a robust response, it is not known if adjusting the aforementioned parameters would have produced a greater effect.
A significant limitation to translating the present investigations to the clinical setting is the lack of background pharmacological therapies (i.e. beta-blockade or ACE inhibitors). Only VNS + beta-blockade has been explored previously.19 In addition, although a low level of VNS was delivered which did not reduce the HR, it is not known if HR reduction would have provided additional benefit. Finally, although it would have been useful to explore the effect of vagal stimulation on overall arrhythmia burden, there were very few arrhythmias in this study in either the control or VNS groups, and thus no analysis could be performed.
There are two reported, ongoing clinical trials exploring the efficacy of VNS in patients with HF; INOVATE-HF (NCT01303718) and NECTAR-HF (NCT01385176). An important distinction in these two trials is the difference in the implantable system. The INOVATE-HF system requires the use of an intracardiac lead for timing the vagal stimulation with the cardiac cycle, and uses a tripolar cuff that preferentially stimulates efferent vagal fibres. The NECTAR-HF system does not require an implanted cardiac lead, making the system easier to implant, and may reduce long-term complications. The NECTAR-HF system also uses a bipolar electrode which stimulates afferent and efferent vagal fibres.23 Whether the cardiac sensing and/or unidirectional stimulation are necessary for therapy efficacy will be important findings from these two trials.
In summary, long-term VNS therapy results in an improved LV function and LV remodelling in canines with HF. It also appears from the crossover design component of this investigation that VNS must be chronically delivered for continued therapeutic benefits. Structural and functional changes are supported by an improved profile of inflammatory, apoptotic, and NO biomarkers. These results provide additional support for VNS therapy as promising therapy for chronic HF.
Funding
A research grant from Boston Scientific Corporation; the National Heart, Lung, and Blood Institute PO1 HL074237-09.
Conflict of interest: H.N.S. is a consultant to, and has received research grant funding from, Boston Scientific Corporation. J.J.H., S.B.R., and C.S are full-time employees of Boston Scientific Corporation. All other authors have no conflicts to declare.
Acknowledgements
The authors wish to thank Mr Nick Wold for his help in performing the statistical analysis for this manuscript, and Ms Viktoria Averina and Dr Shibaji Shome for their help in preparing the figures.
References
- 1.Schwartz PJ, De Ferrari GM. Sympathetic–parasympathetic interaction in health and disease: abnormalities and relevance in heart failure. Heart Fail Rev. 2011;16:101–107. doi: 10.1007/s10741-010-9179-1. [DOI] [PubMed] [Google Scholar]
- 2.Mortara A, La Rovere MT, Pinna GD, Prpa A, Maestri R, Febo O, Pozzoli M, Opasich C, Tavazzi L. Arterial baroreflex modulation of heart rate in chronic heart failure: clinical and hemodynamic correlates and prognostic implications. Circulation. 1997;96:3450–3458. doi: 10.1161/01.cir.96.10.3450. [DOI] [PubMed] [Google Scholar]
- 3.Zucker IH, Wang W, Brandle M, Schultz HD, Patel KP. Neural regulation of sympathetic nerve activity in heart failure. Prog Cardiovasc Dis. 1995;37:397–414. doi: 10.1016/s0033-0620(05)80020-9. [DOI] [PubMed] [Google Scholar]
- 4.Kishi T, Hirooka Y. Central mechanisms of abnormal sympathoexcitation in chronic heart failure. Cardiol Res Pract. 2012;2012:7. doi: 10.1155/2012/847172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang BS, Leenen FH. The brain renin–angiotensin–aldosterone system: a major mechanism for sympathetic hyperactivity and left ventricular remodeling and dysfunction after myocardial infarction. Curr Heart Fail Rep. 2009;6:81–88. doi: 10.1007/s11897-009-0013-9. [DOI] [PubMed] [Google Scholar]
- 6.Cleland JG, McGowan J, Clark A, Freemantle N. The evidence for β blockers in heart failure. BMJ. 1999;318:824–825. doi: 10.1136/bmj.318.7187.824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.La Rovere MT, Bigger JT, Jr., Marcus FI, Mortara A, Schwartz PJ. Baroreflex sensitivity and heart-rate variability in prediction of total cardiac mortality after myocardial infarction. ATRAMI (autonomic tone and reflexes after myocardial infarction) investigators. Lancet. 1998;351:478–484. doi: 10.1016/s0140-6736(97)11144-8. [DOI] [PubMed] [Google Scholar]
- 8.Lechat P, Hulot JS, Escolano S, Mallet A, Leizorovicz A, Werhlen-Grandjean M, Pochmalicki G, Dargie H. Heart rate and cardiac rhythm relationships with bisoprolol benefit in chronic heart failure in CIBIS II trial. Circulation. 2001;103:1428–1433. doi: 10.1161/01.cir.103.10.1428. [DOI] [PubMed] [Google Scholar]
- 9.Li M, Zheng C, Sato T, Kawada T, Sugimachi M, Sunagawa K. Vagal nerve stimulation markedly improves long-term survival after chronic heart failure in rats. Circulation. 2004;109:120–124. doi: 10.1161/01.CIR.0000105721.71640.DA. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Y, Popovic ZB, Bibevski S, Fakhry I, Sica DA, Van Wagoner DR, Mazgalev TN. Chronic vagus nerve stimulation improves autonomic control and attenuates systemic inflammation and heart failure progression in a canine high-rate pacing model. Circ Heart Fail. 2009;2:692–699. doi: 10.1161/CIRCHEARTFAILURE.109.873968. [DOI] [PubMed] [Google Scholar]
- 11.De Ferrari GM, Crijns HJ, Borggrefe M, Milasinovic G, Smid J, Zabel M, Gavazzi A, Sanzo A, Dennert R, Kuschyk J, Raspopovic S, Klein H, Swedberg K, Schwartz PJ. Chronic vagus nerve stimulation: a new and promising therapeutic approach for chronic heart failure. Eur Heart J. 2011;32:847–855. doi: 10.1093/eurheartj/ehq391. [DOI] [PubMed] [Google Scholar]
- 12.Position of the American Heart Association on research animal use. Circulation. 1985;71:849A–850A. [PubMed] [Google Scholar]
- 13.Sabbah HN, Stein PD, Kono T, Gheorghiade M, Levine TB, Jafri S, Hawkins ET, Goldstein S. A canine model of chronic heart failure produced by multiple sequential coronary microembolizations. Am J Physiol. 1991;260 doi: 10.1152/ajpheart.1991.260.4.H1379. H1379–H1384. [DOI] [PubMed] [Google Scholar]
- 14.Dodge HT, Sandler H, Baxley WA, Hawley RR. Usefulness and limitations of radiographic methods for determining left ventricular volume. Am J Cardiol. 1966;18:10–24. doi: 10.1016/0002-9149(66)90191-3. [DOI] [PubMed] [Google Scholar]
- 15.Rastogi S, Mishra S, Zaca V, Alesh I, Gupta RC, Goldstein S, Sabbah HN. Effect of long-term monotherapy with the aldosterone receptor blocker eplerenone on cytoskeletal proteins and matrix metalloproteinases in dogs with heart failure. Cardiovasc Drugs Ther. 2007;21:415–422. doi: 10.1007/s10557-007-6057-8. [DOI] [PubMed] [Google Scholar]
- 16.Rastogi S, Sharov VG, Mishra S, Gupta RC, Blackburn B, Belardinelli L, Stanley WC, Sabbah HN. Ranolazine combined with enalapril or metoprolol prevents progressive LV dysfunction and remodeling in dogs with moderate heart failure. Am J Physiol Heart Circ Physiol. 2008;295 doi: 10.1152/ajpheart.00728.2008. H2149–H2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moe GW, Armstrong P. Pacing-induced heart failure: a model to study the mechanism of disease progression and novel therapy in heart failure. Cardiovasc Res. 1999;42:591–599. doi: 10.1016/s0008-6363(99)00032-2. [DOI] [PubMed] [Google Scholar]
- 18.Sabbah HN. Electrical vagus nerve stimulation for the treatment of chronic heart failure. Cleve Clin J Med. 2011;78(Suppl 1) doi: 10.3949/ccjm.78.s1.04. S24–S29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sabbah HN, Imai M, Zaretsky A, Rastogi S, Wang M, Jiang A, Zaca V. Therapy with vagus nerve electrical stimulation combined with beta-blockade improves left ventricular systolic function in dogs with heart failure beyond that seen with beta-blockade alone. Eur J Heart Fail Suppl. 2007;6:114. [Google Scholar]
- 20.Schwartz PJ, De Ferrari GM, Sanzo A, Landolina M, Rordorf R, Raineri C, Campana C, Revera M, Ajmone-Marsan N, Tavazzi L, Odero A. Long term vagal stimulation in patients with advanced heart failure: first experience in man. Eur J Heart Fail. 2008;10:884–891. doi: 10.1016/j.ejheart.2008.07.016. [DOI] [PubMed] [Google Scholar]
- 21.Tosato M, Yoshida K, Toft E, Nekrasas V, Struijk JJ. Closed-loop control of the heart rate by electrical stimulation of the vagus nerve. Med Biol Eng Comput. 2006;44:161–169. doi: 10.1007/s11517-006-0037-1. [DOI] [PubMed] [Google Scholar]
- 22.Yoo PB, Lubock NB, Hincapie JG, Ruble SB, Hamann JJ, Grill WM. High-resolution measurement of electrically-evoked vagus nerve activity in the anesthetized dog. J Neural Eng. 2013;10:026003. doi: 10.1088/1741-2560/10/2/026003. [DOI] [PubMed] [Google Scholar]
- 23.Jewett DL. Activity of single efferent fibres in the cervical vagus nerve of the dog, with special reference to possible cardio-inhibitory fibres. J Physiol. 1964;175:321–357. doi: 10.1113/jphysiol.1964.sp007520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brack KE, Patel VH, Mantravardi R, Coote JH, Ng GA. Direct evidence of nitric oxide release from neuronal nitric oxide synthase activation in the left ventricle as a result of cervical vagus nerve stimulation. J Physiol. 2009;587:3045–3054. doi: 10.1113/jphysiol.2009.169417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haywood GA, Tsao PS, von der Leyen HE, Mann MJ, Keeling PJ, Trindade PT, Lewis NP, Byrne CD, Rickenbacher PR, Bishopric NH, Cooke JP, McKenna WJ, Fowler MB. Expression of inducible nitric oxide synthase in human heart failure. Circulation. 1996;93:1087–1094. doi: 10.1161/01.cir.93.6.1087. [DOI] [PubMed] [Google Scholar]
- 26.Gupta R, Mishra S, Rastogi S, Imai M, Zaca V, Sabbah H. Chronic therapy with electric vagus nerve stimulation normalizes mrna and protein expression of nitric oxide synthase in myocardium of dogs with heart failure. Eur Heart J. 2006;27:477. [Google Scholar]
- 27.Tsutsumi T, Ide T, Yamato M, Kudou W, Andou M, Hirooka Y, Utsumi H, Tsutsui H, Sunagawa K. Modulation of the myocardial redox state by vagal nerve stimulation after experimental myocardial infarction. Cardiovasc Res. 2008;77:713–721. doi: 10.1093/cvr/cvm092. [DOI] [PubMed] [Google Scholar]