

Abstract
Pharmacologic strategies have provided modest improvement in the devastating muscle-wasting disease, Duchenne muscular dystrophy (DMD). Pre-clinical gene therapy studies have shown promise in the mdx mouse model; however, studies conducted after disease onset fall short of fully correcting muscle strength or protecting against contraction-induced injury. Here we examine the treatment effect on muscle physiology in aged dystrophic mice with significant disease pathology by combining two promising therapies: micro-dystrophin gene replacement and muscle enhancement with follistatin, a potent myostatin inhibitor. Individual treatments with micro-dystrophin and follistatin demonstrated marked improvement in mdx mice but were insufficient to fully restore muscle strength and response to injury to wild-type levels. Strikingly, when combined, micro-dystrophin/follistatin treatment restored force generation and conferred resistance to contraction-induced injury in aged mdx mice. Pre-clinical studies with miniature dystrophins have failed to demonstrate full correction of the physiological defects seen in mdx mice. Importantly, the addition of a muscle enhancement strategy with delivery of follistatin in combination with micro-dystrophin gene therapy completely restored resistance to eccentric contraction-induced injury and improved force. Eccentric contraction-induced injury is a pre-clinical parameter relevant to the exercise induced injury that occurs in DMD patients, and herein, we demonstrate compelling evidence for the therapeutic potential of micro-dystrophin/follistatin combinatorial therapy.
INTRODUCTION
Duchenne muscular dystrophy (DMD) is the most severe form of childhood muscular dystrophy. As a monogenic disease with the absence of dystrophin, DMD is potentially amenable to gene replacement strategies. Proof-of-principle studies with mini- and micro-dystrophins have shown promise with marked improvements in tetanic force and protection from eccentric contraction-induced injury in the mdx mouse model for DMD. However, these studies have been conducted in transgenic mice (1) or in young mice prior to the evolution to the more severe dystrophic process which includes fibrosis, inflammation and fat replacement (2–5). Moreover, regardless of the delivery methods and levels of transduction, adeno-associated virus (AAV)-mediated delivery of micro-dystrophin alone has been insufficient to fully correct the force deficits seen in mdx mice (5–7). Lastly, recently modified miniature dystrophin cassettes used for AAV-mediated gene transfer that include either the C-terminus (8) or nNOS binding site (9,10) have also failed to show complete restoration to wild-type function. Other treatment strategies, including pharmacologic approaches to increase dystrophin levels that demonstrated promise in pre-clinical rodent studies, have failed to produce clinically meaningful outcomes in patients (11–13). Corticosteroids have been the only class of drugs to modestly alter the natural history of the disease (14) but do not provide sustained improvement in force generation, as illustrated in prednisone or prednisolone-treated mdx mice (15–17). These studies highlight the challenges of therapeutic strategies to ameliorate DMD.
Recently, myostatin inhibition has arisen as a promising approach for treatment of muscle disease (18). Myostatin is a member of the transforming growth factor-β family and plays an important role in regulating skeletal muscle growth (19,20). We have previously demonstrated the therapeutic potential of follistatin-344 (FS344), a potent myostatin inhibitor using a gene therapy strategy (21,22). These studies conducted in both mdx mice (21) and non-human primates (22) showed both histological improvement and increased muscle mass and strength for >2 years. Previous studies that have assessed the potential for functional improvement in DMD mice with myostatin inhibition by AAV delivery of the myostatin propeptide (23) or in the myostatin knock-out mouse (24) have found modest improvements in absolute tetanic force and no improvements in specific tetanic force (force per cross-sectional area). However, transgenic studies that expressed a myostatin inhibitor derived from follistatin (25) or by AAV-follistatin-344 delivery demonstrated promise by grip strength analysis (21) and increased twitch force (22). Another study utilized FS288, a muscle-bound isoform and showed an increase in muscle size and isometric force in the tibialis anterior (TA) muscle (26). Together these studies suggest that follistatin-based therapies may be a better therapeutic target than direct removal of myostatin.
Some attempts have been made to use combinatorial approaches to restore dystrophin and enhance muscle size (27–30). All demonstrated improvements in specific force in mdx mice but failed to restore the essential pre-clinical parameter of resistance to eccentric contraction-induced injury (27–30). We hypothesized that by combining the most promising myostatin inhibitor, FS344 (21,22) and most efficient micro-dystrophin cassette for long-term expression, MCK.µDys (5), we would achieve an additive effect on force improvement in mdx mice. We chose to test this in a rigorous paradigm by treating aged mdx mice with overt pathology that would most closely resemble the severe pathology seen in DMD patients. Herein, we demonstrate that FS344/µDys combinatorial therapy leads to restoration of tetanic force and protection from eccentric contraction-induced injury in 2-year-old mdx mice, thus providing rationale for considering a combinatorial therapy for clinical development.
RESULTS
Micro-dystrophin (µ-Dys) or follistatin-344 (FS) individual treatment improves force deficits in mdx mice
To more closely mimic a clinical paradigm where the dystrophic process is clearly underway, we chose to treat mdx animals at 6 months of age and test efficacy six months later when histopathology was more overt. To test the effectiveness of micro-dystrophin or follistatin treatment alone, we administered 1 × 1011 vg of rAAVrh.74.µ-Dys or rAAV1.FS by intramuscular (IM) injection into the TA/EDL unilaterally. When the mice reached 1 year of age, we isolated the EDL from each subject and performed in vitro force measurements. We chose to utilize muscle physiology measurements as our primary outcome given its highly sensitive non-biased readouts. Comparing absolute tetanic force between groups, we found µ-Dys or FS treatment resulted in significant improvements compared with PBS-treated mdx mice (P < 0.001, t-test) and they were not different than wild-type (Fig. 1A). To assess the specific force, we then normalized per cross-sectional area (CSA) and found both µ-Dys and FS therapies improved function compared with untreated mdx but only µ-Dys reached statistical significance (t-test, P < 0.01) (Fig. 1B) and both were below wild-type animals.
Figure 1.

Single treatment with µ-Dys or FS improves force generation and resistance to damage by eccentric contractions. (A) Tetanic force (N) was improved with µ-Dys or FS versus PBS-treated mdx animals but was not significantly different than wild-type force. Values are presented as the means ± SD (P < 0.01). (B) Specific force (mN/mm2) (normalized for cross-sectional muscle area) was improved with µ-Dys or FS over mdx, where µ-Dys was significant (P < 0.05) but was significantly lower than age-matched controls. (C) EDL muscles from mdx mice treated with FS or µ-Dys were subjected to a protocol of ten eccentric contractions to assess resistance to damage. Single treatment conferred resistance to eccentric contractions versus mdx muscles (ANOVA repeated measures P < 0.001), but they were still significantly different from age-matched controls. (D) A comparison of the fourth versus the first contraction demonstrates response to injury following µ-Dys or FS treatment was improved over mdx with either treatment but still different than wild-type. n = 8 per group.
We next subjected each EDL to a series of repeated eccentric contractions. Cohorts receiving either µ-Dys or FS were protected from damage (Fig. 1C). Comparing decay curves by ANOVA with repeated measures, both treatment groups were significantly improved (P < 0.01). A comparison of the fourth versus the first contraction demonstrates that response to injury following µ-Dys or FS treatment was increased over untreated mdx but still did not reach wild-type levels on post-hoc analysis (Fig. 1D). Thus, although µ-Dys and FS demonstrate an overall improvement in resistance to eccentric contraction-induced injury, neither restored function to wild-type levels as single therapies, suggesting that each therapy by itself was insufficient for maximal therapeutic benefit.
Combinatorial treatment with µ-Dys and FS restores skeletal muscle physiology
We next asked whether combining µ-Dys and FS treatment would demonstrate a synergistic effect that would in turn fully restore the physiologic deficits in mdx mice. We again treated mdx mice at 6 months of age with 1 × 1011 vg of rAAVrh.74.µ-Dys and rAAV1.FS by IM injection to the TA/EDL compartment unilaterally. To mirror the first study, the in vitro force measurements were performed at 365 days. Co-gene delivery significantly improved both absolute tetanic and specific force (P < 0.05, t-test) (Fig. 2A and B). The findings for absolute tetanic force reached wild-type levels (Fig. 2A). Importantly, the response to eccentric contraction-induced injury with co-delivery of µ-Dys and FS restored physiologic responses to wild-type levels (mdx vs. co-delivery, P < 0.001, ANOVA repeated measures, P < 0.05, t-test at each contraction) (Fig. 2C), demonstrating that this combination resulted in full correction and optimized therapeutic effect.
Figure 2.
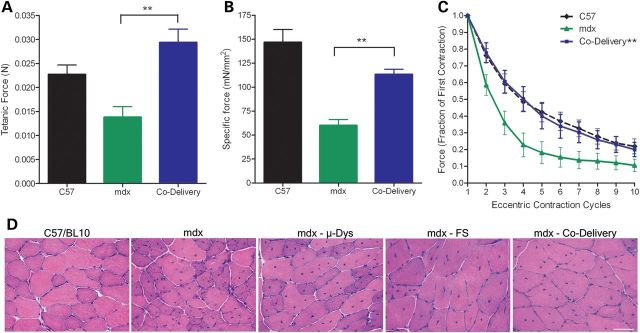
Co-delivery of µ-Dys and FS restores force and resistance to injury. (A and B) Tetanic force and specific force were significantly improved in 1-year-old mdx mice treated with µ-Dys/FS co-delivery (P < 0.01). (C) Co-delivery of µ-Dys/FS restored protection from eccentric contraction-induced injury to normal (mdx vs. co-delivery, ANOVA repeated measures P < 0.001). (D) Hematoxylin and Eosin (H&E) analysis reveals an improvement of dystrophic histopathology. H&E staining of 12 µm sections from each group revealed an overall improvement in fiber size (further analyzed in Fig. 3 with Fiber type analysis) in all treated mdx mice (n = 8 per group). Scale bar = 50 µm.
Gene expression and histopathological improvement
Although functional assessment was the primary outcome of the study, we examined the histopathological consequences of both single and combination treatments at the termination of the study when the animals were 365 days of age. First, the number of muscle fibers expressing µ-Dys and serum levels for FS measured by ELISA was used to assess efficacy of transgene delivery. We found robust μ-dys expression (Fig. 3) that was not different between cohorts treated with µ-Dys alone (81.3 ± 7.0%) compared with µ-Dys/FS combination therapy (85.7 ± 6.2%, Supplementary Material, Fig. S1A–C). FS serum levels were also the same whether delivered alone or in combination with µ-Dys (Supplementary Material, Fig. S1D). Histological evaluation of all treatment groups revealed a decrease in inflammatory infiltrates (Fig. 2D) and a significant increase in fiber size (Fig. 3, P < 0.001, ANOVA). µ-Dys/FS combination treatment demonstrated an increase in average fiber size across all fiber types. Comparing mdx controls with µ-Dys/FS-treated mdx, the average diameter of type I fibers increased from 31.0 to 43.6 µm. Type IIa fiber diameters increased from 40.5 to 51.6 µm and type IIb fiber diameters increased from 37.5 to 53.7 µm (Fig. 3). The co-delivery paradigm produced a significant shift toward wild-type fiber size distribution in all groups treated. There was no decrease in the number of centralized nuclei consistent with repeated cycles of regeneration prior to gene transfer (data not shown).
Figure 3.
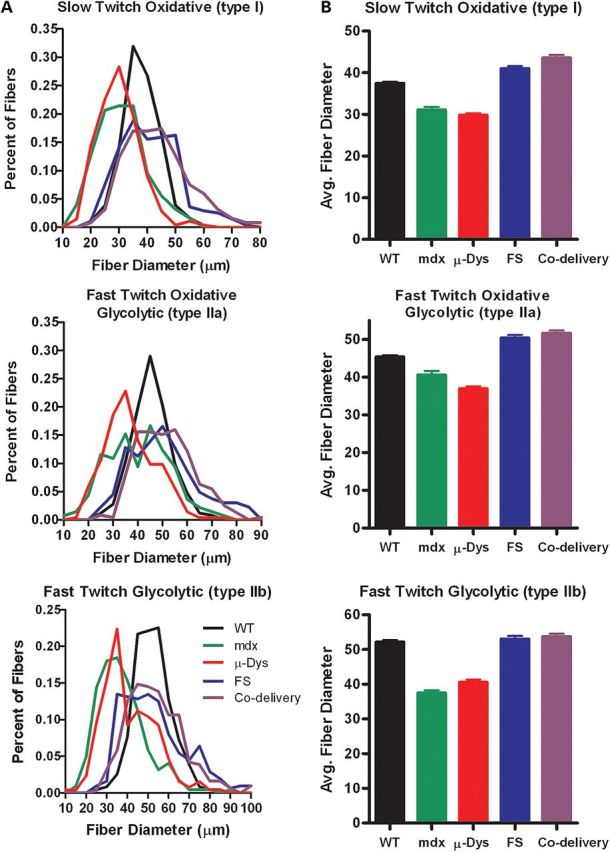
Co-delivery of µ-Dys and FS treatment restores average fiber size. TA sections from all groups were stained with succinic dehydrogenase to delineate mitochondria-enriched fibers representing slow twitch oxidative (type I), fast twitch oxidative glycolytic (IIa) and fast twitch glycolytic (IIb) fiber types. Four random 20× fields for each muscle were captured, and the smallest diameter was measured. (A) A frequency distribution was performed to represent the percent number of fibers within 10 μm intervals for each fiber type. (B) Average fiber size in µ-Dys/FS co-delivery treated groups was significantly larger than mdx (n = 6 per group, P < 0.001).
Co-delivery of µ-Dys and FS demonstrates efficacy in dystrophic mice at 600 days of age
To address whether the co-delivery paradigm could still demonstrate a robust effect in even older mdx mice manifesting more dystrophic pathology, we treated animals at 14 months of age and performed in vitro force measurements six months post-treatment (600 days of age). Remarkably, both absolute tetanic and specific force were significantly improved by co-delivery of μ-Dys/FS, P < 0.01, t-test (Fig. 4A and B). The most clinically significant measure, protection against eccentric contraction-induced injury, demonstrated full restoration with co-µ-Dys/FS treatment (P < 0.001 ANOVA repeated measures) (Fig. 4C). Importantly, there was a correlation between function and muscle histopathology demonstrated by a reduction in inflammation (Fig. 4D) and a significant increase in muscle fiber diameter (P < 0.05, unpaired T-test, Fig. 4E). These data provide evidence that co-delivery of µ-Dys and FS may be a viable treatment modality for DMD patients even in the face of overt pathology.
Figure 4.
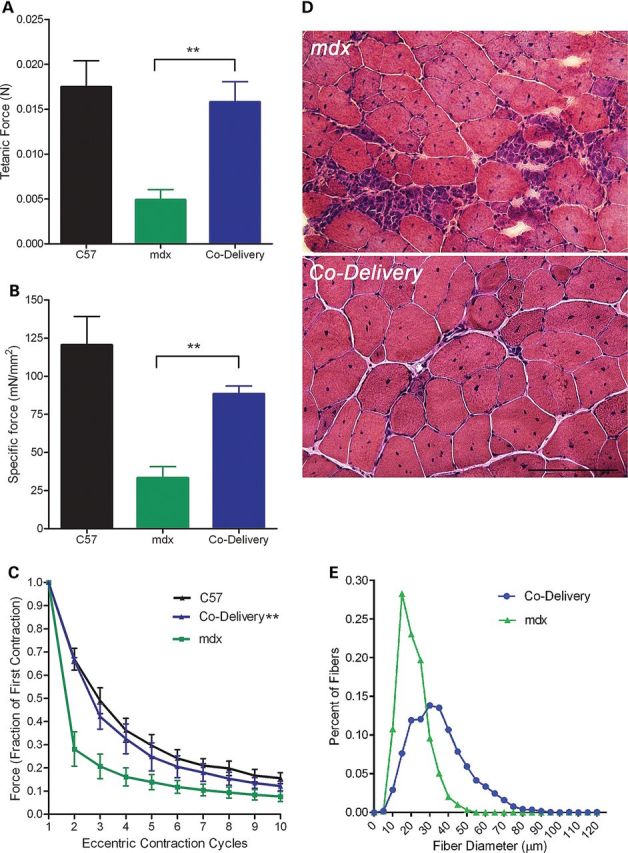
Co-delivery of µ-Dys and FS restores force and resistance to injury in old mdx with overt pathology. (A and B) Tetanic force and specific force were significantly improved in 600-day-old mdx mice treated with µ-Dys/FS co-delivery (P < 0.01). (C) Co-delivery of µ-Dys/FS restored protection from eccentric contraction-induced injury (mdx vs. co-delivery, ANOVA repeated measures P < 0.001). (D) Hematoxylin and Eosin (H&E) analysis reveals an improvement of dystrophic histopathology. H&E staining of 12 µm sections from each group revealed an overall improvement in inflammatory infiltrates. (E) Muscle fiber diameter was significantly increased (P < 0.05, unpaired T-test). Scale bar = 100 µm.
DISCUSSION
Gene delivery strategies to treat DMD have been under investigation for over a decade without resolution. The biggest hurdles for an effective treatment are the inability to transfer full-length dystrophin and identifying treatments that will provide full functional restoration in the presence of a well-established dystrophic process. Our work investigated whether we could enhance functional recovery by using a co-delivery paradigm with AAV.µDys and AAV.FS344. The study was conducted in dystrophic mdx mice with completion at 12 and 20 months of age to more closely simulate clinical challenges. The results presented demonstrate that co-delivery of µ-Dys/FS344 leads to restoration of force generation and protection against contraction-induced injury even in the face of a dystrophic environment. Combinatorial treatment with AAV.µ-Dys/FS344 was significantly better than either treatment alone and provides proof-of-principle as a potential translational treatment for DMD. We are currently conducting a clinical trial with AAV1.FS in Becker muscular dystrophy patients and preparing an IND application for AAVrh.74.micro-dystrophin. The AAVrh.74 serotype works well by both IM and vascular routes thereby providing a path forward to deliver by multiple routes. Thus, both AAV1.FS and AAVrh.74.micro-dystrophin will have been tested in the clinic individually prior to bringing co-delivery treatment to a clinical trial, providing valuable safety and efficacy data.
Prior studies show improvement in force deficits in the mdx mouse using micro-dystrophin constructs when experiments are conducted in young mice prior to the onset of the dystrophic process. However, even in these mice, full muscle strength could not be restored (1,2,4,5,31). The failure may be multi-factorial: due in part to missing structural, or protein binding domains, or the inability to prevent or reverse the dystrophic process, once it has occurred.
Myostatin inhibition has shown promise as a therapy to increase muscle mass for a variety of muscle-wasting disorders (21,22,32–34). The study presented here is the first to directly test the ability of AAV.FS344 to improve skeletal muscle strength in aged dystrophic mice by performing in vitro force measurements in the EDL. A previous report assessing EDL force measurements in the myostatin knock-out mouse casts doubt as to whether increased fiber size correlated with increased strength (35). A more recent study that tested the potential therapeutic effect of a muscle-bound isoform of follistatin, FS288 delivered by AAV, showed an improvement in isometric force (26) and further showed the results to be dependent on Smad3 and mTOR signaling independent of myostatin. In this study, we showed an improvement in force with FS alone even after normalizing for cross-sectional area, indicating that the mere increase in fiber size does not account for increased strength. In addition, FS conferred resistance to eccentric contraction-induced injury. This data speak to the multi-dimensional role FS plays in muscle. In addition to myostatin, FS has also been shown to inhibit activin to induce muscle hypertrophy through satellite cell proliferation when overexpressed (36). This is demonstrated by the quadrupling of muscle mass that occurs by overexpressing FS in myostatin knock-out mice (37). FS has also been implicated in suppression of muscle fibrosis, evidenced by the regulatory role of myostatin in muscle fibroblast proliferation (38).
Three previous studies have tested various forms of combination therapy with gene replacement and muscle enhancement in DMD mice. Importantly, none of these studies were able to restore force and resistance to injury and were not conducted in aged mice with long-term treatment (27,29,30). In a sentinel study utilizing co-delivery of micro-dystrophin and Igf1, Abmayr and colleagues showed improvements in tetanic force and force retention over mdx but no improvement in specific force (27). A second strategy combined exon skipping using an AAV-U7-Dys construct and RNA interference directed against the activin receptor IIb to inhibit the myostatin absolute tetanic and the specific force but did not assess response to injury (29). A third study investigated a dual exon-skipping approach with myostatin and dystrophin in a DMD cell culture model that focused only on RT-PCR as an outcome (30). Although each of these studies demonstrated varying degrees of efficacy, our study showed complete restoration of absolute force and most importantly resistance to eccentric contraction-induced damage in older mice with dystrophic pathology. This potentially sets the stage for a combinational clinical gene therapy approach based on the principles observed in this experimental paradigm of co-delivery of μ-Dys/FS344.
MATERIALS AND METHODS
All procedures were approved by the Institutional Animal Care and Use Committee at the Research Institute at Nationwide Children's Hospital. Mdx mice and normal age-matched C57/BL10 were used for IM delivery and force generation studies of the extensor digitorum longus (EDL) muscle (n = 8 per group). For IM injections, mice were anesthetized and maintained on 1–4% isoflurane (in Oxygen). Both hindlimbs were shaved, and the anterior compartment of the lower limb [EDL/TA compartment] was injected with 1 × 1011 vg of rAAVrh74.MCK.micro-dystrophin (5), rAAV1.CMV.follistatin (21,22) or both (50 μl total volume) using a 30-gauge ultra-fine insulin needle and syringe. PBS was used to inject sham control animals because we had previously shown that injection of empty capsid particles demonstrated no effect on functional outcomes (Table 1; Supplementary Material, Fig. S2).
Table 1.
CSA of the EDL muscle following treatment
| Group | n | EDL weight (mg) | EDL CSA (mm2) |
|---|---|---|---|
| C57/BL10 | 8 | 13.6 ± 0.88 | 1.5 ± 0.08 |
| Mdx | 8 | 18.6 ± 3.1a | 2.2 ± 0.45a |
| mdx-µDys | 8 | 19.0 ± 2.87 | 2.4 ± 0.46 |
| mdx-FS | 8 | 26.6 ± 3.52b | 3.1 ± 0.50b |
| mdx-co-delivery | 8 | 27.1 ± 4.5b | 3.5 ± 0.64b |
amdx are significantly different than C57/BL10 mice.
bmdx mice treated with FS alone or by co-delivery of FS and µDys were significantly larger than saline treated mdx. Mdx mice treated with µDys alone were not different than saline treated mdx. ANOVA, P ≤ 0.05.
Micro-dystrophin and follistatin vector construction
The murine micro-dystrophin construct possessed the (R4-R23/Δ71–78) domains as previously described (1,5). The cDNA was codon optimized for rodents and synthesized by GenScript Inc. It includes a consensus Kozak sequence, an SV40 intron and synthetic polyadenylation site (53 bp). An MCK promoter/enhancer (GenBank Accession No. M21390)-derived sequence was used to drive muscle-specific gene expression (39). The MCK micro-dystrophin expression cassette was cloned between AAV2 ITRs using flanking Xba I restriction enzyme sites in plasmid derived from pCMVβ (Clontech). Msc I/Sma I restriction enzyme digestions were used to confirm ITR integrity. The cDNA for the human FS344 gene was obtained from Origene and cloned by Xho l/Bam HI restriction into a second AAV vector plasmid containing the CMV promoter.
rAAV vector production
rAAV vectors were produced by a modified cross-packaging approach whereby the AAV type 2 vector genome can be packaged into multiple AAV capsid serotypes (40). Production was accomplished using a standard 3 plasmid DNA/CaPO4 precipitation method using HEK293 cells. The HEK293 cells were maintained in DMEM supplemented with 10% fetal bovine serum and penicillin and streptomycin. The production plasmids were as follows: (i) pAAV.MCK.microdys of pAAV.CMV.FS, (ii) rep2-capX modified AAV helper plasmids encoding cap serotypes 1- or an 8-like isolate rh.74 and (iii) an adenovirus type 5 helper plasmid (pAdhelper) expressing adenovirus E2A, E4 ORF6 and VA I/II RNA genes. To allow comparisons between serotypes, a quantitative PCR-based titration method was used to determine an encapsidated vector genome (vg) titer utilizing a Prism 7500 Taqman detector system (PE Applied Biosystems) (41). The primer and fluorescent probe targeted the MCK promoter and were as follows: MCK forward primer, 5'-CCCGAGATGCCTGGTTATAATT-3'; MCK reverse primer, 5'-GCTCAGGCAGCAGGTGTTG-3'; and MCK probe, 5'-FAM-CCAGACATGTGGCTGCTCCCCC -TAMRA-3' and CMV promoter and were as follows: CMV forward primer, 5'-TGGAAATCCCCGTGAGTCAA-3'; CMV reverse primer, 5'-CATGGTGATGCGGTTTTGG-3'; and CMV probe, 5'-FAM-CCGCTATCCACGCCCATTGATG – TAMRA-3'.
Gene expression analysis
EDL, TA and gastrocnemius skeletal muscles were collected from mdx treated and contralateral control limbs 180 days post-treatment. Muscles were embedded in 7% gum tragacanth and flash frozen in isopentane cooled in liquid nitrogen. Cryostat sections (12 μm) for immunostains were incubated with the N-terminal Manex1a primary antibody (Developmental Studies Hybridoma Bank), at a dilution of 1:50 in blocking buffer (PBS, 10% goat serum, 0.1% Triton X-100) for 1 h at room temperature in a wet chamber. Sections were then washed with PBS three times, each for 20 min and re-blocked. Visualization was achieved by incubation for 30 min at room temperature with an Alexa 488 at a 1:300 dilution (Molecular Probes). Sections were washed in PBS three times for 20 min and mounted with Vectashield mounting medium (Vector Laboratories). Fluorescence staining for micro-dystrophin was visualized at an excitation wavelength of 488 nm using a Zeiss Axioskop2 Plus Microscope, and images were captured with a Zeiss AxioCam MRC5 camera. The number of fibers with sarcolemmal dystrophin staining was expressed as percent of all dystrophin positive fibers. Serum follistatin was evaluated with a human follistatin immunoassay kit (Quantikine; R&D Systems) according to the manufacturer's instructions. Briefly, 100 µl of serum was loaded per well, and serum follistatin concentrations were determined against a standard curve made with recombinant human follistatin provided by the manufacturer.
Morphometrics
Centralized nuclei counts were performed on sections of TA muscles stained with hematoxylin and Eosin (H&E) from treated mdx animals. Five random 20× fields of 12 μm sections for each muscle were captured and the number of fibers with central nuclei counted. Fiber diameter measurements were also performed on TA muscles from treated mdx animals stained with succinic dehydrogenase to delineate mitochondria-enriched fibers representing slow twitch oxidative (type 1), fast twitch oxidative glycolytic (type 2A) and fast twitch glycolytic (type 2B) fiber types. Controls were PBS-treated TA muscles. Five random 20× fields of 12 μm sections for each muscle were captured with a Zeiss AxioCam MRC5 camera. For each fiber type, the smallest diameter was measured using Zeiss Axiovision LE4 software (400–500 fibers were measured per animal). A frequency distribution was performed to represent the percent of fibers within 10 μm intervals.
Force generation and protection from eccentric contractions
Measurements of force generation and protection from eccentric contraction-induced injury were performed as previously described (5,7). Six months post-gene transfer, mice were euthanized and the EDL muscle was removed, weighed and bathed in oxygenated Krebs-Henseleit solution (95% O2/5% CO2 (pH 7.4), 118 mm NaCl, 25 mm NaHCO3, 5 mm KCl, 1 mm KH2PO4, 2.5 mm CaCl2, 1 mm MgCl2, 5 mm Glucose) at 30°C in a circulating bath. 6.0 silk suture was tied to the tendon at each end of the muscle. One end of the muscle was tied to a force transducer and the other to a high-speed linear servo-controlled motor. Muscle length was changed at predetermined values and speeds using customized software. Stimulation was delivered via two parallel platinum–iridium electrodes on either side of the muscle. Muscles were adjusted to optimum length (L0), defined as the length for maximal twitch. Following a ten-minute rest period, muscles were subjected to an isometric tetani of 150 Hz for 500 ms. Following a 5-min rest period, the muscles were subjected to an eccentric contraction protocol consisting of a series of 10 isometric 700 ms tetani, at 2 min intervals, with a 5% lengthening of the muscles (0.5 fiber length per second for duration of 200 ms) when maximal force has developed at 500 ms. After the tetanus ended (at t = 700 ms), the muscle was brought back to initial length (at the same speed as the stretch), allowing for full relaxation to the initial length. When measurements were complete, the muscle was removed from the set-up, and the sutures were removed. The muscle was blotted dry carefully to retain integrity for histology and weighed. For comparative purposes, all force measurements were expressed per unit CSA (normalized isometric force or tension, mN/mm2). CSA was calculated using the equation,
where muscle density is 1.06 g/cm3.
Statistical analyses
Data were analyzed with standard statistical methods performed in GraphPad Prism 5 (GraphPad Software, La Jolla, CA) using indicated statistical tests.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
Conflict of Interest statement. B.K. is a founder, shareholder and consultant for Milo Biotechnology, which is a muscle gene delivery company. The work presented here was not funded by Milo Biotechnology.
FUNDING
This work has been supported by Jesse's Journey Foundation for Gene and Cell Therapy, Ruth L. Kirschstein National Research Service Award (1F32AR055008) to L.R.R.-K.
Supplementary Material
Acknowledgements
We thank Anil Birdi, Amy Eagle and Nancy Davis for technical assistance and RINCH Viral Vector Core for assistance with AAV production.
REFERENCES
- 1.Harper S.Q., Hauser M.A., DelloRusso C., Duan D., Crawford R.W., Phelps S.F., Harper H.A., Robinson A.S., Engelhardt J.F., Brooks S.V., et al. Modular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophy. Nat. Med. 2002;8:253–261. doi: 10.1038/nm0302-253. [DOI] [PubMed] [Google Scholar]
- 2.Gregorevic P., Blankinship M.J., Allen J.M., Crawford R.W., Meuse L., Miller D.G., Russell D.W., Chamberlain J.S. Systemic delivery of genes to striated muscles using adeno-associated viral vectors. Nat. Med. 2004;10:828–834. doi: 10.1038/nm1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu M., Yue Y., Harper S.Q., Grange R.W., Chamberlain J.S., Duan D. Adeno-associated virus-mediated microdystrophin expression protects young mdx muscle from contraction-induced injury. Mol. Ther. 2005;11:245–256. doi: 10.1016/j.ymthe.2004.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yoshimura M., Sakamoto M., Ikemoto M., Mochizuki Y., Yuasa K., Miyagoe-Suzuki Y., Takeda S. AAV vector-mediated microdystrophin expression in a relatively small percentage of mdx myofibers improved the mdx phenotype. Mol. Ther. 2004;10:821–828. doi: 10.1016/j.ymthe.2004.07.025. [DOI] [PubMed] [Google Scholar]
- 5.Rodino-Klapac L.R., Janssen P.M., Montgomery C.L., Coley B.D., Chicoine L.G., Clark K.R., Mendell J.R. A translational approach for limb vascular delivery of the micro-dystrophin gene without high volume or high pressure for treatment of Duchenne muscular dystrophy. J. Transl. Med. 2007;5:45. doi: 10.1186/1479-5876-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gregorevic P., Allen J.M., Minami E., Blankinship M.J., Haraguchi M., Meuse L., Finn E., Adams M.E., Froehner S.C., Murry C.E., et al. rAAV6-microdystrophin preserves muscle function and extends lifespan in severely dystrophic mice. Nat. Med. 2006;12:787–789. doi: 10.1038/nm1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martin P.T., Xu R., Rodino-Klapac L.R., Oglesbay E., Camboni M., Montgomery C.L., Shontz K., Chicoine L.G., Clark K.R., Sahenk Z., et al. Overexpression of Galgt2 in skeletal muscle prevents injury resulting from eccentric contractions in both mdx and wild-type mice. Am. J. Physiol. Cell. Physiol. 2009;296:C476–C488. doi: 10.1152/ajpcell.00456.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Odom G.L., Gregorevic P., Allen J.M., Chamberlain J.S. Gene therapy of mdx mice with large truncated dystrophins generated by recombination using rAAV6. Mol. Ther. 2011;19:36–45. doi: 10.1038/mt.2010.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shin J.H., Pan X., Hakim C.H., Yang H.T., Yue Y., Zhang K., Terjung R.L., Duan D. Microdystrophin Ameliorates Muscular Dystrophy in the Canine Model of Duchenne Muscular Dystrophy. Mol. Ther. 2013;4:750–757. doi: 10.1038/mt.2012.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lai Y., Thomas G.D., Yue Y., Yang H.T., Li D., Long C., Judge L., Bostick B., Chamberlain J.S., Terjung R.L., et al. Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J. Clin. Invest. 2009;119:624–635. doi: 10.1172/JCI36612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wagner K.R., Hamed S., Hadley D.W., Gropman A.L., Burstein A.H., Escolar D.M., Hoffman E.P., Fischbeck K.H. Gentamicin treatment of Duchenne and Becker muscular dystrophy due to nonsense mutations. Ann. Neurol. 2001;49:706–711. [PubMed] [Google Scholar]
- 12.Malik V., Rodino-Klapac L.R., Viollet L., Mendell J.R. Aminoglycoside-induced mutation suppression (stop codon readthrough) as a therapeutic strategy for Duchenne muscular dystrophy. Ther. Adv. Neurol. Disord. 2010;3:379–389. doi: 10.1177/1756285610388693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Malik V., Rodino-Klapac L.R., Viollet L., Wall C., King W., Al-Dahhak R., Lewis S., Shilling C.J., Kota J., Serrano-Munuera C., et al. Gentamicin-induced readthrough of stop codons in Duchenne muscular dystrophy. Ann. Neurol. 2010;67:771–780. doi: 10.1002/ana.22024. [DOI] [PubMed] [Google Scholar]
- 14.Mendell J.R., Moxley R.T., Griggs R.C., Brooke M.H., Fenichel G.M., Miller J.P., King W., Signore L., Pandya S., Florence J., et al. Randomized, double-blind six-month trial of prednisone in Duchenne's muscular dystrophy. N. Engl. J. Med. 1989;320:1592–1597. doi: 10.1056/NEJM198906153202405. [DOI] [PubMed] [Google Scholar]
- 15.Carre-Pierrat M., Lafoux A., Tanniou G., Chambonnier L., Divet A., Fougerousse F., Huchet-Cadiou C., Segalat L. Pre-clinical study of 21 approved drugs in the mdx mouse. Neuromuscul. Disord. 2011;21:313–327. doi: 10.1016/j.nmd.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 16.Baltgalvis K.A., Call J.A., Nikas J.B., Lowe D.A. Effects of prednisolone on skeletal muscle contractility in mdx mice. Muscle Nerve. 2009;40:443–454. doi: 10.1002/mus.21327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sali A., Guerron A.D., Gordish-Dressman H., Spurney C.F., Iantorno M., Hoffman E.P., Nagaraju K. Glucocorticoid-treated mice are an inappropriate positive control for long-term preclinical studies in the mdx mouse. PLoS One. 2012;7:e34204. doi: 10.1371/journal.pone.0034204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rodino-Klapac L.R., Haidet A.M., Kota J., Handy C., Kaspar B.K., Mendell J.R. Inhibition of myostatin with emphasis on follistatin as a therapy for muscle disease. Muscle Nerve. 2009;39:283–296. doi: 10.1002/mus.21244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McPherron A.C., Lawler A.M., Lee S.J. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature. 1997;387:83–90. doi: 10.1038/387083a0. [DOI] [PubMed] [Google Scholar]
- 20.Lee S.J. Regulation of muscle mass by myostatin. Annu. Rev. Cell Dev. Biol. 2004;20:61–86. doi: 10.1146/annurev.cellbio.20.012103.135836. [DOI] [PubMed] [Google Scholar]
- 21.Haidet A.M., Rizo L., Handy C., Umapathi P., Eagle A., Shilling C., Boue D., Martin P.T., Sahenk Z., Mendell J.R., et al. Long-term enhancement of skeletal muscle mass and strength by single gene administration of myostatin inhibitors. Proc. Natl. Acad. Sci. U. S. A. 2008;105:4318–4322. doi: 10.1073/pnas.0709144105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kota J., Handy C.R., Haidet A.M., Montgomery C.L., Eagle A., Rodino-Klapac L.R., Tucker D., Shilling C.J., Therlfall W.R., Walker C.M., et al. Follistatin gene delivery enhances muscle growth and strength in nonhuman primates. Sci. Transl. Med. 2009;1:6ra15. doi: 10.1126/scitranslmed.3000112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qiao C., Li J., Jiang J., Zhu X., Wang B., Li J., Xiao X. Myostatin propeptide gene delivery by adeno-associated virus serotype 8 vectors enhances muscle growth and ameliorates dystrophic phenotypes in mdx mice. Hum. Gene Ther. 2008;19:241–254. doi: 10.1089/hum.2007.159. [DOI] [PubMed] [Google Scholar]
- 24.Mendias C.L., Kayupov E., Bradley J.R., Brooks S.V., Claflin D.R. Decreased specific force and power production of muscle fibers from myostatin-deficient mice are associated with a suppression of protein degradation. J. Appl. Physiol. 2011;111:185–191. doi: 10.1152/japplphysiol.00126.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakatani M., Takehara Y., Sugino H., Matsumoto M., Hashimoto O., Hasegawa Y., Murakami T., Uezumi A., Takeda S., Noji S., et al. Transgenic expression of a myostatin inhibitor derived from follistatin increases skeletal muscle mass and ameliorates dystrophic pathology in mdx mice. FASEB. J. 2008;22:477–487. doi: 10.1096/fj.07-8673com. [DOI] [PubMed] [Google Scholar]
- 26.Winbanks C.E., Weeks K.L., Thomson R.E., Sepulveda P.V., Beyer C., Qian H., Chen J.L., Allen J.M., Lancaster G.I., Febbraio M.A., et al. Follistatin-mediated skeletal muscle hypertrophy is regulated by Smad3 and mTOR independently of myostatin. J. Cell Biol. 2012;197:997–1008. doi: 10.1083/jcb.201109091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abmayr S., Gregorevic P., Allen J.M., Chamberlain J.S. Phenotypic improvement of dystrophic muscles by rAAV/microdystrophin vectors is augmented by Igf1 codelivery. Mol. Ther. 2005;12:441–450. doi: 10.1016/j.ymthe.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 28.Brunelli S., Sciorati C., D'Antona G., Innocenzi A., Covarello D., Galvez B.G., Perrotta C., Monopoli A., Sanvito F., Bottinelli R., et al. Nitric oxide release combined with nonsteroidal antiinflammatory activity prevents muscular dystrophy pathology and enhances stem cell therapy. Proc. Nat.l Acad. Sci. U. S. A. 2007;104:264–269. doi: 10.1073/pnas.0608277104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dumonceaux J., Marie S., Beley C., Trollet C., Vignaud A., Ferry A., Butler-Browne G., Garcia L. Combination of myostatin pathway interference and dystrophin rescue enhances tetanic and specific force in dystrophic mdx mice. Mol. Ther. 2010;18:881–887. doi: 10.1038/mt.2009.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kemaladewi D.U., Hoogaars W.M., van Heiningen S.H., Terlouw S., de Gorter D.J., den Dunnen J.T., van Ommen G.J., Aartsma-Rus A., ten Dijke P., t Hoen P.A. Dual exon skipping in myostatin and dystrophin for Duchenne muscular dystrophy. BMC Med. Genomics. 2011;4:36. doi: 10.1186/1755-8794-4-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Watchko J., O'Day T., Wang B., Zhou L., Tang Y., Li J., Xiao X. Adeno-associated virus vector-mediated minidystrophin gene therapy improves dystrophic muscle contractile function in mdx mice. Hum. Gene Ther. 2002;13:1451–1460. doi: 10.1089/10430340260185085. [DOI] [PubMed] [Google Scholar]
- 32.Bogdanovich S., Krag T.O., Barton E.R., Morris L.D., Whittemore L.A., Ahima R.S., Khurana T.S. Functional improvement of dystrophic muscle by myostatin blockade. Nature. 2002;420:418–421. doi: 10.1038/nature01154. [DOI] [PubMed] [Google Scholar]
- 33.Parsons S.A., Millay D.P., Sargent M.A., McNally E.M., Molkentin J.D. Age-dependent effect of myostatin blockade on disease severity in a murine model of limb-girdle muscular dystrophy. Am. J. Pathol. 2006;168:1975–1985. doi: 10.2353/ajpath.2006.051316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bartoli M., Roudaut C., Martin S., Fougerousse F., Suel L., Poupiot J., Gicquel E., Noulet F., Danos O., Richard I. Safety and efficacy of AAV-mediated calpain 3 gene transfer in a mouse model of limb-girdle muscular dystrophy type 2A. Mol. Ther. 2006;13:250–259. doi: 10.1016/j.ymthe.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 35.Amthor H., Macharia R., Navarrete R., Schuelke M., Brown S.C., Otto A., Voit T., Muntoni F., Vrbova G., Partridge T., et al. Lack of myostatin results in excessive muscle growth but impaired force generation. Proc. Nat.l Acad. Sci. U. S. A. 2007;104:1835–1840. doi: 10.1073/pnas.0604893104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gilson H., Schakman O., Kalista S., Lause P., Tsuchida K., Thissen J.P. Follistatin induces muscle hypertrophy through satellite cell proliferation and inhibition of both myostatin and activin. Am. J. Physiol. Endocrinol. Metab. 2009;297:E157–E164. doi: 10.1152/ajpendo.00193.2009. [DOI] [PubMed] [Google Scholar]
- 37.Lee S.J. Quadrupling muscle mass in mice by targeting TGF-beta signaling pathways. PLoS One. 2007;2:e789. doi: 10.1371/journal.pone.0000789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li Z.B., Kollias H.D., Wagner K.R. Myostatin directly regulates skeletal muscle fibrosis. J. Biol. Chem. 2008;283:19371–19378. doi: 10.1074/jbc.M802585200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heller K.N., Montgomery C.L., Janssen P.M., Clark K.R., Mendell J.R., Rodino-Klapac L.R. AAV-mediated Overexpression of Human α7 Integrin Leads to Histological and Functional Improvement in Dystrophic Mice. Mol. Ther. 2013;21:520–525. doi: 10.1038/mt.2012.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rabinowitz J.E., Rolling F., Li C., Conrath H., Xiao W., Xiao X., Samulski R.J. Cross-packaging of a single adeno-associated virus (AAV) type 2 vector genome into multiple AAV serotypes enables transduction with broad specificity. J. Virol. 2002;76:791–801. doi: 10.1128/JVI.76.2.791-801.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clark K.R., Liu X., McGrath J.P., Johnson P.R. Highly purified recombinant adeno-associated virus vectors are biologically active and free of detectable helper and wild-type viruses. Hum. Gene Ther. 1999;10:1031–1039. doi: 10.1089/10430349950018427. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.