

Abstract
Mice with cytotoxic lesions of the dorsal hippocampus (DH) underestimated 15 s and 45 s target durations in a bi-peak procedure as evidenced by proportional leftward shifts of the peak functions that emerged during training as a result of decreases in both ‘start’ and ‘stop’ times. In contrast, mice with lesions of the ventral hippocampus (VH) displayed rightward shifts that were immediately present and were largely limited to increases in the ‘stop’ time for the 45 s target duration. Moreover, the effects of the DH lesions were congruent with the scalar property of interval timing in that the 15 s and 45 s functions superimposed when plotted on a relative timescale, whereas the effects of the VH lesions violated the scalar property. Mice with DH lesions also showed enhanced reversal learning in comparison to control and VH lesioned mice. These results are compared with the timing distortions observed in mice lacking δ-opioid receptors (Oprd1−/−) which were similar to mice with DH lesions. Taken together, these results suggest a balance between hippocampal–striatal interactions for interval timing and demonstrate possible functional dissociations along the septotemporal axis of the hippocampus in terms of motivation, timed response thresholds and encoding in temporal memory.
Keywords: time perception, temporal memory, scalar property, motivation, basal ganglia, hippocampal–striatal interactions
1. Introduction
Timing and time perception are fundamental properties of cognition [1–4], with numerous studies conducted to investigate their neural basis [5–11]. In particular, considerable attention has been directed to cortical–striatal pathways that are essential in maintaining timing abilities in the range of hundreds of milliseconds to multi-seconds [12–16]. Interestingly, although neglected by many of the current models of interval timing [17], the hippocampus has long been considered a site of spatial–temporal interaction, which provides a basis for generation, maintenance and retrieval of episodic memories [18,19]. Since the initial evaluation of the effects of post-training fimbria–fornix lesions on timing and temporal memory [20], studies investigating the role of the hippocampus in the temporal control of behaviour have generated consistent, though not always conclusive, results [21–24]. Specifically, both humans and rodents with a variety of different types of hippocampal lesions have been shown to underestimate target durations, and/or to exhibit increased sensitivity to signal duration [25,26]. However, to date, no studies have examined the concurrent timing of multiple target durations as a function of pre-training lesions to the dorsal hippocampus (DH) or ventral hippocampus (VH). As a consequence, adequate explanations for the source of the changes in timing behaviour observed after selective hippocampal lesions are still lacking due to the inability to evaluate the scalar property of interval timing which posits that the sensitivity in temporal processing as measured by the standard deviation of timed performance increases in proportion to the target durations. Distortions in timing and time perception are often revealed by changes both in accuracy and precision, thus requiring that multiple durations be examined in order to determine the nature of the distortion [27–29].
A major question is whether the dorsal and ventral regions of the hippocampus play functionally distinct roles in timing and other types of behaviour [30,31]. It is known that these two hippocampal regions project differentially to other brain areas. The VH connects, for example, to the prefrontal cortex, amygdala, as well as the ventral and rostral sections of the nucleus accumbens (NAc)—ventral striatum (VS) shell associated with the regulation of context-dependent learning and emotional behaviour [32]. In contrast, the DH connects to the rostral part of the dorsal striatum (DS), including both the core and shell, dorsal lateral septum, mammillary bodies, anterior thalamus, ventral medial hypothalamus and anterior cingulate cortex [30]. Highly processed information from the sensory cortices enters the hippocampus, primarily through its dorsal section [33]. As a consequence, the DH is thought to subserve memory and other cognitive functions [30]. Moreover, examination of the behavioural functions of the DH and DS have recently come to focus on the competition and cooperation between distinct memory systems represented by these brain structures [34,35].
The importance of hippocampal delta (δ)-opioid receptors to learning and memory (but not morphine reinforcement) has recently been demonstrated [36,37]. Mice lacking δ-opioid receptors (Oprd1−/−), for example, exhibit increased motor impulsivity, but no obvious alteration of pain perception [38]. In the hippocampus, these receptors are mainly present in gamma-aminobutyric acid (GABA)ergic neurons of the DH, e.g. Ammon's horn and dentate gyrus [39], suggesting their involvement in neural plasticity. Moreover, it has recently been reported that Oprd1−/− mice display decreased ability to solve hippocampal-dependent tasks, e.g. novel object/spatial location recognition, and facilitated striatum-dependent responses, e.g. egocentric response learning and rotarod procedural/motor skill learning [36]. The functional importance of δ-opioid receptors in the hippocampus probably involves the enkephalins, which are released in the lateral perforant path, where δ-receptor activation is required for high-frequency-induced long-term potentiation (LTP)—thought to be crucial for hippocampal-dependent learning [40,41]. Consequently, inactivation of δ-opioid receptors in the hippocampus would prevent their endogenous ligands from inhibiting GABAergic interneurons, and thereby reducing the probability for LTP and associated neural plasticity. In contrast, decreased hippocampal activity has been shown to facilitate striatal-based skills when the two systems compete with each other to control behaviour [35,42–45]. Consequently, impaired hippocampal function in Oprd1−/− mice may account for their facilitated performance in response and skill learning tasks, via alterations in hippocampal–striatal balance in favour of the striatum [46].
The current study sought to evaluate the behavioural effects of cytotoxic lesions of either the DH or VH on interval timing and to compare these performances to mice lacking the δ-opioid receptor in an effort to better characterize the nature of hippocampal–striatal interactions in timing and time perception in the multi-seconds range. A bi-peak procedure was used in order to evaluate changes in accuracy and precision as a function of multiple target durations [5,47,48].
2. Material and methods
(a). Subjects
The subjects used in the experiments were male C57BL (baseline)/6J mice (n = 66, Charles River Laboratories, Raleigh, NC) or male mice homozygous for the δ-opioid receptor (Oprd1−/−) backcrossed to C57BL/6J mice for at least 12 generations (n = 10), plus wild-type littermates (n = 10, Oprd1+/+; Jackson Laboratory, Bar Harbor, ME). All mice were between 5 and 7 weeks of age when first delivered to our climate-controlled animal colony with a 12 L : 12 D cycle (lights on at 07.00 h, off at 19.00 h). Mice were group housed (four to five mice per cage). Standard rodent chow (5001 – Purina LabDiet, Purina Mills, St Louis, MO) and water were available ad libitum in the home cages except during the food-restricted period of behavioural testing described in §2d(i–iv). Body weights were monitored on a daily basis throughout the course of the experiment.
Behavioural testing started at 8–9 weeks of age and occurred between 09.00 h and 17.00 h. The mice in each cohort were assigned to one of the 20 lever boxes with different lesion or genetic conditions randomly distributed—with mice trained in the same lever box at approximately the same time of day throughout the course of the experiment. Sessions were conducted 7 days/week unless otherwise stated. During behavioural training, mice were maintained at 85–90% of their ad libitum body weights by food restriction.
(b). Surgery
Mice were anaesthetized with an intraperitoneal injection of ketamine/xylazine cocktail (100/10 mg kg−1). The mouse's head was shaved and mounted into a standard stereotaxic instrument (David Kopf, Tujunga, CA). A sterile lubricant (ointment) was generously applied to the eyes. The scalp was incised, and the skin was retracted. The head was levelled by equating bregma and lambda in the dorsoventral plane. Four (DH) or eight (VH) small holes were drilled into the skull according to the coordinates measured from bregma as shown in table 1. A 1.0-µl Hamilton syringe (model 65458-01) was lowered into each of these holes, and N-Methyl-d-aspartic acid (NMDA; 20 ug µl−1; Sigma, St Louis, MO), dissolved in sterilized phosphate-buffered saline, was infused. An automatic syringe pump (Nanojet, Chemyx, Stafford, TX) attached to the Hamilton syringe mounted on the stereotaxic instrument was used to deliver 0.1 µl−1 NMDA over 3 min. The syringe remained in place for an additional 2 min to allow for diffusion of the drug. In sham mice, the syringe was lowered to the same sites as lesioned mice but no injection was placed in order to minimize the physical damage to the target brain areas. After the final infusion, the incision was closed with stainless steel wound clips. Mice were allowed to recover in a heating-pad-warmed cage with food and water easily accessible nearby. Upon awakening, DH mice (and their sham controls) were intraperitoneally injected with a single dose (approx. 0.03 ml−1) of diazepam (5 mg ml−1; Hospira, Lake Forest, IL) to help control potential seizures that would typically be observed after successful neurotoxic DH lesions (laboratory observation). No diazepam was needed for VH mice and their sham controls. After they regained full mobility and were actively running and consuming food and water for 12 h, mice were returned to their home cages and allowed to recover for 10 days before post-lesion behavioural training began.
Table 1.
Coordinates for dorsal/ventral hippocampal lesions. Reference: bregma and skull surface.
| location of lesions | anterior/posterior (mm) | medial/lateral (mm) | dorsal/ventral (mm) |
|---|---|---|---|
| DH | −1.3 | ±1.0 | −2.0 |
| −2.1 | ±1.5 | −2.2 | |
| VH | −2.9 | ±3.0 | −3.5 |
| −3.3 | ±3.6 | −4.0 | |
| −3.6 | ±2.85 | −4.0 | |
| −3.8 | ±2.75 | −4.8 |
(c). Behavioural procedures
(i). Apparatus
The experimental apparatus consisted of 10 matching lever boxes (Model ENV-307A, Med Associates, St. Albans, VT) housed in sound-attenuating chambers (Model ENV-021M; Med Associates). The dimensions of each lever box were 21.59 × 17.78 × 12.70 cm. The ceiling, side walls and door of each box were made from clear Plexiglas. The front and back walls were stainless-steel panels and the floor was made of parallel stainless-steel bars. The front wall of each box contained left and right retractable levers; a food cup was located between the levers; and a cue light was located directly above the food cup. A pellet dispenser delivered 20-mg grain-based food pellets (Research Diets, New Brunswick, NJ) into the food cup. The back wall of each box contained a house light (14 W, 100 mA) directed towards the ceiling. The operant chambers were controlled by the Med-pc iv software package. The fan was on throughout the session. An IBM-PC compatible computer attached to an electronic interface (MED Associates, models DIG-700 and SG-215) was used to control the experimental equipment and record the data. The time of each lever press was recorded to an accuracy of 10 ms and placed into 1 s time bins.
(d). Bi-peak timing procedure
(i). Lever-press training (sessions 1–10)
All mice were given ten daily sessions of lever-press training. During the session, one of the two side levers was continuously retracted and inserted in a 1 s cycle every 120 s to attract attention from mice. The delivery of a food pellet in the foodcup was primed every 90 s, which was signalled by the blinking of the cue light. In addition to the free food pellet delivered, a food pellet was delivered for every lever press (FR-1). Every ten lever presses resulted in the alternation of the two levers. Sessions ended after 3600 s and there was no limit for total pellets that could be earned within a single session (in reality, all mice earned between 40 and 150 pellets altogether per session). After seven sessions, the food pellets were withdrawn in order to further encourage mice to earn food by pressing the lever. All mice that participated in the experiment learned to press the lever for food pellets after this stage.
(ii). 15 s and 45 s fixed-interval training (sessions 11–20)
During these sessions, the onset of the house light was used as a signal for the duration to be timed, i.e. fixed-interval (FI) trials were signalled by the onset of the house light and the appropriate lever(s) was primed for reinforcement at the associated target duration(s). The target duration used on each trial (15 s or 45 s) was randomly selected with equal probability and no external cue was given to indicate which lever/duration resulted in the delivery of a food pellet, signal termination, and the onset of a variable inter-trial interval, range 30–150 s. The assignment of target durations to response levers was counterbalanced both within and across groups of mice. After seven sessions of FI training, two levers were set to be simultaneously available (inserted) throughout the session, and mice were trained to press the lever associated with the 15 s duration first and then to switch to the other lever associated with the 45 s duration.
(iii). 15 s and 45 s bi-peak training (sessions 21–40)
Bi-peak training was used to assess the start and stop times with which mice timed the target duration(s). Sessions consisted of two trial types: FI trials (as described in §2d(ii)) and unreinforced probe trials. The two levers were set to be simultaneously available throughout the session. During probe trials the house light was turned on for a minimum of 3× the longer target duration (45 s) plus an additional random amount of time with a mean of 20 s and a Gaussian distribution. No food was available for lever pressing on these unreinforced probe trials. FI and probe trials were ordered randomly with 50% probability each. Thus, one of the two target durations (15 s or 45 s) was presented in conjunction with non-reinforced probe trials in a random sequence. No external cue was provided to indicate which, if any, lever/target duration would be selected for reinforcement on any trial. Mice were free to respond on the lever(s) at any time during the session, though only responses made to the appropriate lever following the target duration during FI trials were reinforced.
(iv). Bi-peak reversal training (sessions 41–75)
The reversal experiments used the same bi-peak procedure as described in §2d(iii), with the exception that the association between the lever (left or right) and the target duration (15 s or 45 s) were switched for each individual mouse. For example, if a mouse was trained to associate the left lever with the 15 s target duration and to associate the right lever with the 45 s target duration, then during reversal learning, the left lever was switched to providing reinforcement for responding on a peak interval 45 s schedule of reinforcement and the right lever was switched to providing reinforcement for responding on a peak interval 15 s schedule of reinforcement. The adjustment in peak times of responding on the two levers was observed as a function of sessions. Previous studies have shown reversal learning for duration discrimination procedures to be sensitive to cortico-striatal-hippocampal damage [49].
(e). Dorsal hippocampus lesions: post-operative bi-peak training
(i). Pre-training dorsal hippocampus lesions
In order to examine the effects of DH lesions on interval timing, a bi-peak procedure (see §2d) was employed. Fifteen mice were randomly assigned to the sham control group (sham, n = 6) and the DH lesion group (pre-DH, n = 9). Surgeries were performed after mice were shaped to press the lever for food pellets in order to exclude any effect that DH lesions might have on instrumental learning per se [50,51]. After mice recovered from surgery, they were trained with the bi-peak procedure as described in §2d(iii).
(f). Ventral hippocampus lesions: post-operative bi-peak training
(i). Pre-training ventral hippocampus lesions
Similarly, we examined the effects of VH lesions on interval timing using the same bi-peak procedure. Fifteen mice were randomly assigned to the sham control group (sham, n = 7) and the VH lesion group (pre-VH, n = 8). Experiments were performed in an identical manner to the behavioural procedures used for the pre-DH condition described in §2d(i–iv).
(g). Dorsal hippocampus lesions: pre-operative bi-peak training
(i). Post-training dorsal hippocampus lesions
Ten mice were randomly assigned to the sham control (sham, n = 5) and the DH lesion (post-DH, n = 5) treatment groups. The experiment was performed in an identical manner to the previous two experiments, with the exception that surgeries were performed after the mice had already received FI training and 20 sessions of bi-peak training. Such post-training lesions are the most common procedure used to evaluate the effects of hippocampal damage on timing behaviour [20,52–55, but see 22,23]. Behavioural training was resumed after the mice recovered from the surgery and no reversal learning was conducted.
(h). δ-opioid receptor: bi-peak training
(i). Pre-training gene deletions
Twenty mice were evaluated and were assigned to either the control (Oprd1+/+, n = 10) or the experimental (Oprd1−/−, n = 10) treatment groups. Experiments were performed in an identical manner to the FI and bi-peak training described in §2d(i–iii) or the DH and VH lesion groups (§2e,f) except that no surgery was performed.
(i). Histology
After behavioural testing was complete, all mice in the lesion groups were deeply anaesthetized with ketamine and then intracardially perfused with saline and 4% paraformaldehyde. The brains were removed and stored in paraformaldehyde. Sections (100 µm) were cut coronally on a vibratome and stained with cresyl violet. Outlines of the relevant hippocampal tissue characteristics and lesions were examined under a Zeiss SteREO Lumar.V12 stereoscope and then traced onto line drawings of 16 coronal sections covering the entire hippocampus [56]. The outlines were then digitized to display the minimum and maximum lesions for each treatment group.
(j). Data analysis
Individual peak functions for each target duration (15 s and 45 s) were fit using a Gaussian curve with the addition of a linear ramp function to account for right-tailed skew. These fits accounted for over 90% of the variance for all groups of mice and did not reliably differ as a function of treatment condition. The Gaussian fits were used to obtain peak time (a measure of accuracy), peak spread (a measure of precision) and peak rate (a measure of motivation) as previously described [47,57,58]. Peak time divided by peak spread at the 50th percentile can be used as a measure of the relative standard deviation or sensitivity to time—also referred to as the Weber fraction (WF) or coefficient of variation [57,59]. The scalar property of interval timing predicts a constant WF across multiple target durations.
A rate index representative of the mean S1 response thresholds was also determined for FI response functions averaged over blocks of sessions. This rate index was calculated by taking the response rate in a specified interval (20% of the target duration) just prior to the observed peak time as a ratio of overall response rate within the first (S1) half of the trial as defined by the target duration. Higher S1 values indicate sharper FI timing functions and better duration discrimination [5,47].
(k). Single-trials analysis
Response states defined by ‘start’ (S1) and ‘stop’ (S2) response thresholds were identified on individual trials that contained at least five lever presses as previously described [57–60]. Briefly, in a single trial, the location of a ‘high, relatively continuous’ state of lever pressing during a trial is determined by fitting three contiguous, but non-overlapping horizontal lines to the response series over time during a single unreinforced probe trial. Therefore, the intercept of each line represents a response rate over an interval that is defined by the length of the line. The goal is to iteratively maximize the difference between the response rate defined by the middle horizontal line (‘high’ state) and the response rate defined during the flanking horizontal lines (‘low’ states). This calculation effectively fits a boxcar-like step function, referred to as a ‘low–high–low’ pattern of responding. The S1 and S2 times are defined as the time points in the fitted function at which the ‘high’ state begins and ends, respectively.
A single-trials analysis has certain limitations with this particular dataset due to the mice being able to switch back and forth between levers in the bi-peak procedure. In particular, switching between the 15 s and 45 s levers interferes with the determination of the ‘stop’ times for the 15 s target duration as well as the ‘start’ and ‘stop’ times for the 45 s target duration. As a consequence, the measures obtained from the single-trials analysis may not correspond with the measures obtained from the mean peak function as well as in other experiments using a single target duration [57]. Nevertheless, the application of a single-trials analysis is robust enough to look for group differences as well as session effects.
(l). Statistical analysis
Statistical analysis was performed using GraphPad Prism 5 (GraphPad Software, La Jolla, CA). Single-factor and repeated measures analyses of variance (ANOVA) were used as appropriate. The alpha level was set at p < 0.05 for all statistical analysis.
3. Results and discussion
(a). Effects of pre-training dorsal hippocampal lesions on the acquisition of temporal control in the bi-peak procedure
No significant differences in the S1 rate index were observed between the sham and pre-DH lesion groups during the ten sessions of FI training for either the 15 s (F1,13 = 0.51, p > 0.05) or the 45 s target duration (F1,13 = 1.09, p > 0.05).
The Gaussian + ramp functions fit to the mean response rate functions displayed in the upper portion of figure 1 revealed no significant group differences in peak time early in training (sessions 4–6, figure 1a) for either the 15 s or the 45 s target durations (F1,13 < 1.0, p > 0.05). In contrast, significantly lower peak times were observed for the pre-DH lesion group compared with the sham control group late in training (sessions 16–18, figure 1b) for both the 15 s and 45 s target durations (F1,13 = 4.68, p < 0.05 and F1,13 = 5.46, p < 0.05, respectively). Taken together, these data suggest that a leftward shift in peak times emerged over the course of post-operative bi-peak training in pre-DH lesioned mice. Peak time and peak rate measures for these conditions are reported in table 2.
Figure 1.

Bi-peak timing functions during post-operative sessions 4–6 and 16–18. The bi-peak functions for mice with pre-training cytotoxic lesions of the dorsal hippocampus (DH) are shown in (a,b) and the functions of mice with pre-training lesions of the ventral hippocampus (VH) are shown in (c,d). Black and grey lines represent the sham and the lesioned groups, respectively, whereas solid lines and dashed lines represent the 15 s and 45 s peak functions, respectively. Response rates as a function of time since signal onset were calculated from the pooled lever presses from all trials in a session and then normalized by the maximum response rate for each subject. Data were then averaged across three sessions for all the subjects in a group and then re-normalized to the maximum response rate.
Table 2.
Peak time (s) and peak rate (responses/minute) measures. Numbers are means ± s.e. s, seconds; r, response per minute.
| groups/sessions | sessions 4–6 |
sessions 16–18 |
||
|---|---|---|---|---|
| 15 s | 45 s | 15 s | 45 s | |
| sham (pre-DH) n = 6 |
13.62 s±1.11 71.26 r±14.30 |
45.43 s±2.79 72.70 r±15.39 |
16.06 s±1.12 84.59 r±15.85 |
46.78 s±2.22 63.64 r±13.44 |
| pre-DH lesion n = 9 |
13.30 s±0.98 73.04 r±12.83 |
44.52 s±1.28 77.34 r±7.01 |
11.89 s±0.90 53.63 r±8.87 |
36.00 s±2.84 72.02 r±10.62 |
| sham (pre-VH) n = 7 |
12.71 s±0.70 126.05 r±27.77 |
42.74 s±3.01 80.15 r±21.43 |
15.00 s±2.01 138.14 r±29.83 |
44.10 s±1.15 112.69 r±21.68 |
| pre-VH lesion n = 8 |
13.90 s±0.56 126.97 r±29.97 |
51.67 s±2.03 90.49 r±13.03 |
13.46 s±0.68 122.81 r±18.30 |
46.46 s±1.13 139.11 r±17.59 |
| sham (post-DH) n = 5 |
17.80 s±1.77 74.84 r±7.29 |
50.33 s±1.75 93.57 r±12.95 |
15.58 s±0.84 126.63 r±14.93 |
45.83 s±1.21 130.20 r±18.73 |
| post-DH lesion n = 5 |
14.40 s±1.81 116.27 r±27.05 |
51.00 s±2.18 112.62 r±16.15 |
14.13 s±0.90 130.26 r±42.33 |
41.13 s±1.83 147.18 r±27.78 |
| Oprd1+/+
n = 10 |
15.35 s±1.45 48.78 r±5.67 |
46.50 s±2.23 37.24 r±5.81 |
15.50 s±1.02 58.30 r±6.58 |
45.90 s±1.97 33.70 r±4.44 |
| Oprd1−/−
n = 10 |
15.15 s±1.86 44.23 r±5.67 |
44.30 s±2.56 39.76 r±3.94 |
12.50 s±0.95 54.30 r±5.25 |
37.30 s±2.08 38.60 r±3.75 |
Single-trials analyses for the 15 s and 45 s target durations as a function of blocks of training sessions are illustrated in the upper portion of figure 2. Significantly lower ‘start’ times were observed for the mice in the pre-DH lesion group compared to the sham control group for both the 15 s and 45 s target durations (F1,65 = 4.66, p < 0.05 and F1,65 = 5.91, p < 0.05, respectively). There was also a significant effect of session block (F5,65 ≥ 4.53, p < 0.05), but no reliable group×session block interaction (F5,65 < 1.0; figure 2a). In contrast, the apparent reductions in ‘stop’ times observed for the mice in the pre-DH lesion group were unreliable when compared with the sham control group for both the 15 s and 45 s target durations (F1,65 < 1.0, p > 0.05), with no reliable effects of session block or the group × session block interaction (figure 2b).
Figure 2.

Start (S1) and stop (S2) transition times obtained from the single-trials analysis plotted as a function of 15 s and 45 s peak-interval training sessions for mice with pre-training cytotoxic lesions of the dorsal (DH) or ventral (VH) hippocampus. The effects of pre-DH lesions are shown in (a,b) and the effects of pre-VH lesions are shown in (c,d). Empty circles and filled circles represent the sham and lesioned groups, respectively; whereas circles and squares represent the ‘start’ and ‘stop’ transition times for the 15 s and 45 s target duration, respectively. Data are means (±s.e.m.) averaged over three sessions. Note that when the errors bars are contained within the symbols used to plot the means they are effectively invisible.
(b). Effects of pre-training ventral hippocampal lesions on the acquisition of temporal control in the bi-peak procedure
No significant differences in the S1 rate index were observed between the sham and pre-VH lesion groups during the ten sessions of FI training for either the 15 s or the 45 s target duration (F1,13 < 1.0. p > 0.05).
The Gaussian + ramp functions fit to the mean response rate functions displayed in the lower portion of figure 1 revealed significant group differences in peak time between the pre-VH lesion and sham control groups during both early (sessions 4–6, figure 1c) and late (sessions 16–18, figure 1d) stages of training for the 45 s target duration (F1,13 > 4.74, p < 0.05), but not the 15 s target duration (F1,13 < 1.0, p > 0.05), although there was a trend of an effect for the 15 s target duration early in training (p < 0.06). Peak time and peak rate measures for these conditions are reported in table 1.
Single-trials analyses for the 15 s and 45 s target durations as a function of blocks of training sessions are illustrated in the lower portion of figure 2. No reliable differences in ‘start’ times were observed for the mice in the pre-VH lesion group compared to the sham control group for either the 15 s or the 45 s target durations (F1,65 < 1.0, p > 0.05; figure 2c). In contrast, significant differences in ‘stop’ times were observed for the mice in the pre-VH lesion group when compared with the sham control group for both the 15 s and 45 s target durations (F1,65 = 4.63, p < 0.05 and F1,65 = 12.17, p < 0.01, respectively; figure 2d). The effect of session block was significant for both ‘start’ and ‘stop’ times for the 15 s and 45 s target durations (F5,65 ≥ 4.78, p < 0.05), whereas the group × session block interactions were unreliable (F5,65 < 1.0). These data suggest that pre-VH lesions mainly affect ‘stop’ times as evident by the ‘elevated tails’ observed in the mean peak functions (figure 1c,d). The increase in ‘stop’ times for mice in the pre-VH lesion group diminished with continued training as demonstrated in figure 2d.
(c). Effects of pre-training dorsal/ventral hippocampal lesions on discrimination reversal learning
Discrimination reversal learning has been shown to be sensitive to impairments in timing associated with neurotoxic regimens of methamphetamine intoxication that contribute to neuronal death in the striatum and hippocampus [49,61]. The current experiment was conducted in order to evaluate the performance of pre-DH and pre-VH lesioned mice as a function of blocks of five sessions following the switch in reinforcement contingencies, e.g. 15 s to 45 s target durations and 45 s to 15 s target durations for the left and right response levers, respectively. The pre-DH lesioned mice displayed faster reversal learning (as indexed by changes in peak time) than their sham controls in both the 15 s to 45 s condition (F1,78 = 15.11, p < 0.01) and the 45 s to 15 s condition (F1,78 = 6.81, p < 0.05), as shown by the transitions in peak time displayed in figure 3a,b. In contrast, the pre-VH lesioned mice were not significantly different in reversal learning compared with their sham controls in either the 15 s to 45 s condition (F1,78 < 1.0, p > 0.05), as shown by the transitions in peak time displayed in figure 3c,d.
Figure 3.

Fitted peak times plotted as a function of the 15 s to 45 s and 45 s to 15 s reversal sessions for mice with pre-training cytotoxic lesions of the dorsal (DH) or ventral (VH) hippocampus. The effects of pre-DH lesions are shown in (a,b) and the effects of pre-VH lesions are shown in (c,d). Empty circles and filled squares represent the sham and lesioned groups, respectively. Data are means (±s.e.m.) averaged over five sessions. Before the reversal conditions, five sessions of bi-peak training with 15 s and 45 s target durations were used as a baseline (BL) for comparison.
A different pattern of results was observed between the pre-DH and pre-VH lesion groups, however, when response rates during reversal learning were compared. Peak rates calculated from the ‘high’ state of the single-trials analysis for pre-DH lesioned mice (101.3 ± 10.12 responses min−1) did not reliably differ from their sham controls (116.3 ± 22.63 responses min−1; F1,78 < 1.0, p > 0.05). In contrast, the pre-VH lesioned mice displayed significantly enhanced peak rates (187.7 ± 24.04 responses min−1) during all phases of reversal learning compared with their sham controls (111.8 ± 13.07 responses min−1; F1,78 ≥ 5.47, p < 0.05).
Taken together, these data demonstrate that pre-DH, but not pre-VH lesions lead to an enhancement of reversal learning in timing tasks, which suggest that DH may be more relevant to core timing mechanisms than the VH. Nevertheless, VH lesions had reliable effects on peak rate when peak times were in transition, suggesting a possible interaction between the motivational and memory components of timing behavior [61].
(d). Superimposition of 15 s and 45 s bi-peak functions as a result of dorsal and ventral hippocampal lesions
Figure 4 displays mean bi-peak functions for the 15 s and 45 s target durations for each of the treatment groups (sham DH and VH) plotted on a timescale normalized by the observed peak times for each mouse prior to averaging. If the scalar property of interval timing holds, then these bi-peak functions should superimpose when plotted on a relative timescale [47,62,63]. As one can see from visual inspection, the degree of superimposition is relatively good for the sham and DH-lesion groups, but characteristically fails for the VH-lesion group with the 45 s peak function being consistently sharper (i.e. narrower) than the 15 s peak function. This type of failure of the scalar property has been observed in Parkinson's disease patients tested off their dopaminergic medication [27,64] as well as in some types of timing procedures involving systematic error and impulsive responding in mice [65].
Figure 4.
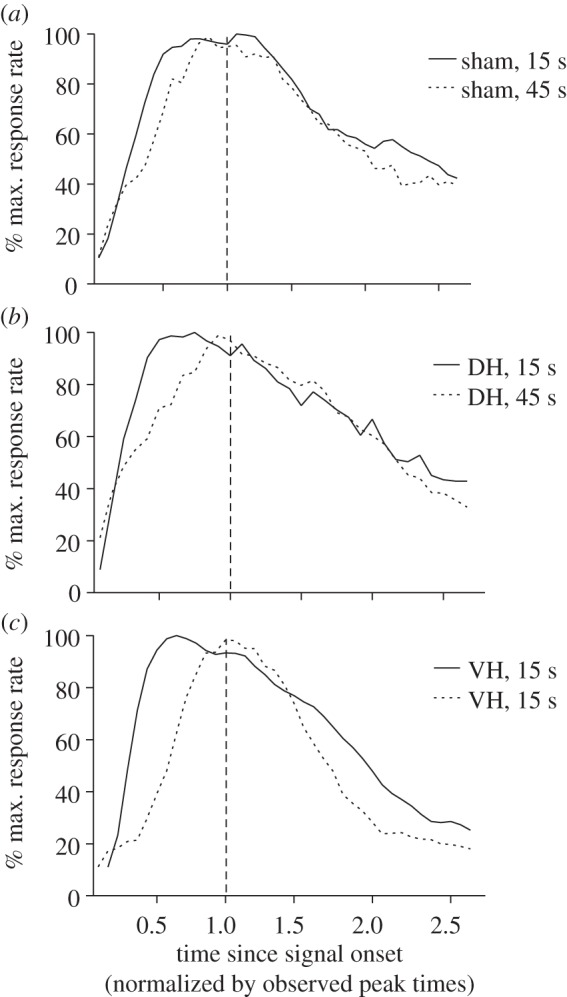
Superimposition plots of 15 s and 45 s bi-peak functions obtained from post-operative bi-peak sessions 16–18 for sham, dorsal (DH) and ventral (VH) hippocampal pre-training cytotoxic lesion groups. The 45 s peak functions were first normalized to the same relative percentage scale of the 15 s functions, and then re-normalized by their observed peak times.
(e). Effects of post-training dorsal hippocampal lesions on the maintenance of temporal control in the bi-peak procedure
Because training itself may recruit certain brain areas that may not be required after learning, we also performed post-training DH lesions to examine whether this hippocampal region is still essential after mice had learned the timing tasks. The Gaussian + ramp functions fit to the mean response rate functions displayed in the upper portion of figure S1 in the electronic supplementary material exhibited non-significant group differences in peak time between the post-DH lesion and sham control groups during early stages of training (sessions 4–6, figure S1a) for both the 15 s and 45 s target durations (F1,8 < 1.0, p > 0.05). In contrast, significant group differences were observed in peak time for the later stages of training (sessions 16–18, figure S1b) for both the 15 s and 45 s target durations (F1,8 ≥ 7.74, p < 0.05). Peak time and peak rate measures for these conditions are reported in table 1.
Single-trials analyses for the 15 s and 45 s target durations as a function of blocks of training sessions are illustrated in the lower portion of figure S1c,d for ‘start’ and ‘stop’ times, respectively. Interestingly, similar to the pre-training DH lesion experiment, significant differences were found between the sham and post-DH lesion groups for the ‘start’ times for both the 15 s and 45 s target durations (F1,8 ≥ 8.27, p = 0.05), but no significant differences were found for the ‘stop’ times (F1,8 < 1.0, p > 0.05). These data suggest that even after acquisition of the bi-peak procedure, post-DH lesions are still capable of producing gradual leftward shifts in the peak times of a magnitude similar to pre-DH lesions.
(f). Histology
Histology confirmed that both DH and VH lesions were complete and successful as illustrated in figure 5. This illustration displays the maximal extent of the lesions for each group. The lesions were consistent in their placement in either the DH or VH, and variability in the extent of the lesions did not correlate in any obvious way with the observed behavioural measures.
Figure 5.
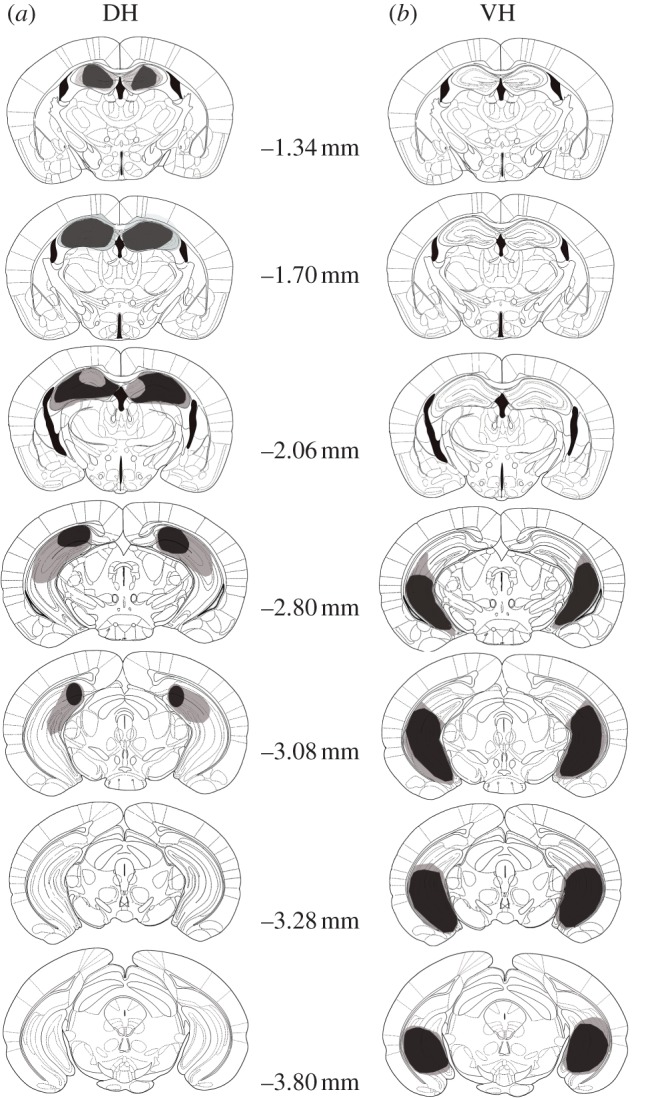
Schematic representations of the extent of (a) dorsal (DH) and (b) ventral (VH) hippocampal lesions on the coronal sections of the mouse brain [56]. The light grey areas represent the maximum scope of lesions within a lesion group and the dark grey areas represent the minimum scope of lesions within a lesion group.
(g). Effects of δ-opioid receptor (Oprd1−/−) gene deletion on the acquisition of temporal control in the bi-peak procedure
A repeated-measures ANOVA (one within and one between factor) was used to analyse the timing performance averaged over the first ten sessions of FI training. This analysis indicated that there were no significant differences in the S1 rate index between the wild type (0.63 ± 0.02) and Oprd1−/− (0.66 ± 0.02) groups (F1,18 = 0.86, p > 0.05) for either the 15 s (0.64 ± 0.02) or the 45 s (0.66 ± 0.01) target durations (F1,18 = 0.58, p > 0.05), nor was the target duration×genotype interaction significant (F1,18 = 0.04, p > 0.05).
The Gaussian + ramp functions fit to the mean response rate functions revealed non-significant differences in peak time and peak rate as a function of genotype, target duration and the target duration × genotype interaction early in training (sessions 4–6; F1,18 < 1.0, p > 0.05). In contrast, significant effects of genotype (F1,18 = 7.73, p < 0.05), target duration (F1,18 = 1034.84, p < 0.0001), and the target duration×genotype interaction (F1,18 = 10.65, p < 0.01) were observed later in training (sessions 16–18) as illustrated in the left-hand panel of figure 6. Peak time and peak rate measures as a function of genotype and target duration are reported in table 1 for both blocks of sessions.
Figure 6.
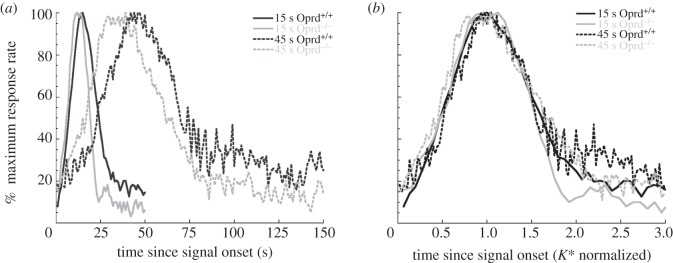
Bi-peak functions plotted on absolute (a) and relative (b) timescales for the Oprd mice. The bi-peak functions for the wild-type mice (Oprd+/+) are depicted in black and the bi-peak functions for the knock-out mice (Oprd−/−) are depicted in grey. The 15 s peak functions are displayed using solid lines and the 45 s peak functions are displayed using dashed lines. In (a), response rates are plotted as a function of time since signal onset. Lever presses were pooled across all peak trials in a session and then normalized by the maximum response rate for each subject. Data were then averaged across three sessions for all the subjects in a treatment group and then re-normalized to the maximum response rate. In (b), the 45 s peak functions were first normalized to the same relative percentage scale of the 15 s peak functions, and then the placement of both the 15 s and 45 s functions were re-normalized by their obtained peak times to determine the K* values.
The 15 s and 45 s PI response functions for Oprd1+/+ and Oprd1−/− groups are plotted on a timescale normalized by the observed peak times in the right-hand panel of figure 6. The high-degree of overlap among all four functions indicates excellent superimposition, a feature of the scalar property of interval timing indicating that timing variability (e.g. standard deviation or spread of response functions) increases in proportion to the duration of the interval being timed [47,57,63]. Interestingly, the only systematic deviation from superimposition is observed on the right-hand tail of the response functions which is thought to reflect a level of impulsive responding uncontrolled by the timing of the target duration [66,67].
4. Discussion
(a). Dorsal and ventral hippocampal lesions differentially affect bi-peak performance
In previous work, rats with hippocampal damage (e.g. fimbria–fornix lesions or complete hippocampal cytotoxic lesions) responded earlier than the scheduled time of reinforcement in a variety of peak-interval procedures [20,68], suggesting that the hippocampus plays a role in temporal memory [25]. Hippocampal lesions also disrupt responding in differential reinforcement of low rates (DRL) schedules. In DRL, rats are trained to withhold responding for food until after a set time has elapsed (e.g. more than 15 s). Rats with dorsal, ventral or complete hippocampal lesions are highly inefficient in this task because they are less able to wait for the defined temporal interval to elapse [69]. Consequently, the hippocampus has been proposed to play a role in temporal memory and/or inhibitory processes [26].
Hippocampal lesions have been suggested to affect peak times in the peak-interval procedure and the subjective equivalence points in the temporal bisection procedure [20,21,53,70]. Therefore, our first priority was to determine whether DH and VH lesions affect the peak times in the bi-peak procedure, and whether the ‘start’ and ‘stop’ times during individual trials were equally affected by the lesions.
Importantly, both pre-training and post-training DH lesions produced leftward shifts in peak times, confirming previous investigations and suggesting a possible role for the DH in the cortical–striatal-based timing mechanisms [16,20,22,23,25,70]. Importantly, examination of the individual-trial performance revealed that earlier ‘start’ times rather than earlier ‘stop’ times or a combination of both could well be the reason for the observed leftward shifts of peak times. In contrast, VH lesions produced a temporary rightward shift of peak times, which could be explained exclusively by later ‘stop’ times. Moreover, when peak times and peak rates were modulated by reversal learning, pre-DH lesions appear to have dramatic effects on the adaptability of temporal associations, whereas VH lesions only have effects on response levels. These data suggest that the DH is more closely related to the core timing mechanisms involved in duration encoding [13,14,16,28,29] and the VH is more closely related to motivation and context-dependent modulation of timing performance [10,61].
(b). Implications for hippocampal–striatal interactions
Do the effects observed here indicate a role for the hippocampus in timing per se or suggest an indirect effect by its modification of striatal function? The hippocampus is thought to be involved in navigation and memory in both spatial and temporal space [71–75]. In addition, both hippocampal ‘time cells’ that integrate episodic information across events [19,76] and a hippocampal neuronal coding mechanism that represents the recency of an experience over extended intervals [77] have been reported. These constructs demonstrate the importance of the hippocampus to interval timing per se; however, from an evolutionary standpoint, it would be hard to believe that each brain area works in isolation. Indeed, although knowledge about the action–outcome contingency can be regarded as a type of declarative memory, few reports have suggested a role for the hippocampus to be directly involved in striatal-mediated functions, such as goal-directed exploration and habit formation [78]. Corbit & Balleine [51] showed that electrolytic lesions of the DH impair contingency degradation, but not outcome devaluation—two forms of behavioural tests designed to evaluate the boundaries between goal-directed learning and habit formation. In contrast, Corbit et al. [79] showed that damage to the entorhinal efferents, rather than the DH itself, accounts for the effect on delayed match-to-sample performance and goal-directed behaviour. Interestingly, our work demonstrates that DH lesions enhance reversal learning in the bi-peak timing procedure, a form of contingency reversal. A similar case was recently demonstrated [80] in which lesions of ventromedial prefrontal cortex, known to inhibit dorsolateral striatum-mediated learning, enhance reversal learning. Therefore, these data suggest that DH lesions may cause substantial changes in the DS, as dorsomedial striatum and dorsolateral striatum are believed to be the mediators for goal-directed learning and habit formation, respectively [26,78]. The route of influences could be through the prefrontal cortex [81] or by way of the NAc and mid-brain dopamine systems [82–85], though the long-range GABAergic projections from the DH to the striatum have also been considered [86]. Interestingly, MacDonald et al. [60] have demonstrated that the acquisition of ‘start’ and ‘stop’ response thresholds in peak-interval timing procedures is differentially sensitive to protein synthesis inhibition in the DS and VS. Disruption of the DS resulted in altered ‘start’ times whereas disruption of the VS resulted in altered ‘stop’ times. Combined with the results from the current study, we propose that the DH typically expresses an inhibitory influence on the DS, whereas the VH has an excitatory influence on the VS.
Buhusi et al. [87] recently demonstrated that mice deficient in a close homologue to L1 (CHL1) cell adhesion molecules related to the immunoglobulin superfamily exhibit distortions in temporal memory such that they consistently under-reproduce the target duration when trained on a peak-interval timing procedure. These CHL1-deficient mice have morphological changes in hippocampal and thalamocortical pathways, and display abnormalities in exploratory behaviour [88] and sensorimotor gating [89]. As a consequence, they have been used as a model of schizophrenia and other types of intellectual disabilities in humans. Because the behavioural phenotype of these CHL1-deficient mice displays some of the qualitative (e.g. under-reproduction) changes observed in interval timing following DH lesions [25,87], we considered it important to establish additional parallels between alterations in gene expression and selective changes in hippocampal–striatal interactions that might alter interval timing in the multi-seconds range.
Oprd1−/− mice have been studied in order to identify potential molecular and cellular mechanisms underlying impaired hippocampal and facilitated striatal function. Le Merrer et al. [1] examined the expression of genes in the DH and the DS. Genes were selected to represent key actors of GABA, glutamate and monoamine signalling pathways (transporters, receptors, receptor subunits and enzymes), or were known neuronal markers. The expression of these genes in the VS (NAc) was also analysed in order to test for regional specificity. In the DH, Oprd1−/− mice showed altered expression of genes coding for GABA and glutamate transporters, receptors or receptor subunits. Within the DS, gene expression related to glutamate function was altered in a fashion likely to result in an imbalance between the nigrostriatal and pallidal–striatal pathways. In contrast, the VS was left largely unchanged in this respect, although some changes in neuronal markers were noted. Moreover, pharmacological tests using dopamine D1/D5 and D2/D3 receptor agonists show that the lack of δ-opioid receptors in the Oprd1−/− mice modifies the D1/D5-nigral/D2/D3-pallidal balance in the striatum in favour of the nigral output [1]. Hence, the facilitated acquisition of striatal-dependent tasks observed in Oprd1−/− mice is likely the result of potentiated dopaminergic/glutaminergic activity in striatonigral pathways—possibly involving striatal cholinergic interneurons [13,84,90,91]. As a consequence, the inclusion of the Oprd1−/− mice in this study strengthens the argument for hippocampal–striatal interactions as the source of the proportional leftward shifts produced by DH lesions.
Facilitated striatal activity resulting from the loss of hippocampal inhibition and/or internal changes in the balance between nigro- and pallidal–striatal pathways might contribute to an alteration in the memory translation constant (K*) and provide an explanation for maintained leftward or rightward shifts in timing functions. Previous work has shown that speeding up or facilitating memory storage results in a K* < 1.0 and slowing down or impairing memory storage results in a K* > 1.0. Changes in K* can either lead to under-reproductions (K* < 1.0) or over-reproductions (K* > 1.0). Moreover, changes in timing behaviour that reflect the memory translation constant are uncorrected by feedback and are maintained throughout extended training [28,92–96]. Such changes in K* have been observed following fimbria–fornix (K* < 1.0) or frontal cortex (K* > 1.0) lesions, or the administration of cholinergic agonists (K* < 1.0) or antagonists (K* > 1.0) [9–11,13,93,94,97]. Here, we report changes in K* (leftward shifts) following both pre- and post-DH lesions, but not VH lesions, and also as a function of δ-opioid receptor gene deletion.
In summary, the data reported here establish that mice can be trained to perform in a bi-peak timing procedure with a degree of accuracy and precision similar to previous studies [5]. Moreover, control mice exhibited the scalar property of interval timing as previously demonstrated [62], a hallmark of short-interval timing shared by numerous species [4,98]. The underestimation of target durations, i.e. leftward shifts in psychometric functions as a result of earlier ‘start’ (and possibly ‘stop’) times, produced by hippocampal lesions was demonstrated to be a result of DH damage (as opposed to VH damage) which leads to a broadening of the timing functions as a result of increases in the ‘stop’ times and failure of the scalar property [99]. In contrast, the changes in K* produced by the DH lesions were proportional to the target durations and scaled in a normal manner when the bi-peak functions were plotted on a relative timescale. These results strongly support a change in temporal memory resulting from hippocampal–striatal interactions such that the coincidence detection mechanism of the striatal beat-frequency model of interval timing [14,100] is biased towards shorter durations by the increased dorsal striatal activity resulting from the inhibition of DH function [12–15,101–104]. Although speculative, this proposed mechanism receives support from the proportional leftward shifts (decreases in K*) observed in Oprd1−/− mice which have also been shown to exhibit impaired hippocampal-dependent and facilitated striatal-dependent learning in a manner similar to rodents with DH lesions [36,105–111].
All experiments were conducted under a protocol approved by the Duke University Institutional Animal Care and Use Committee in accordance with the National Institutes of Health guidelines for the care and use of animals.
References
- 1.Allman MJ, Teki S, Griffiths TD, Meck WH. In press Properties of the internal clock: first- and second-order principles of subjective time. Annu. Rev. Psychol. 65 ( 10.1146/annurev-psych-010213-115117) [DOI] [PubMed] [Google Scholar]
- 2.Buhusi CV, Meck WH. 2005. What makes us tick? Functional and neural mechanisms of interval timing. Nat. Rev. Neurosci. 6, 755–765. ( 10.1038/nrn1764) [DOI] [PubMed] [Google Scholar]
- 3.Buhusi CV, Meck WH. 2009. Relative time sharing: new findings and an extension of the resource allocation model of temporal processing. Phil. Trans. R. Soc. B 364, 1875–1885. ( 10.1098/rstb.2009.0022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meck WH. 2003. Functional and neural mechanisms of interval timing. Boca Raton, FL: CRC Press. [Google Scholar]
- 5.Agostino PV, Cheng RK, Williams CL, West AE, Meck WH. 2013. Acquisition of response thresholds for timed performance is regulated by a calcium-responsive transcription factor, CaRF. Genes Brain Behav. 12, 633–644. ( 10.1111/gbb.12059) [DOI] [PubMed] [Google Scholar]
- 6.Coull JT, Vidal F, Nazarian B, Macar F. 2004. Functional anatomy of the attentional modulation of time estimation. Science 303, 1506–1508. ( 10.1126/science.1091573) [DOI] [PubMed] [Google Scholar]
- 7.Hinton SC, Meck WH. 2004. Frontal-striatal circuitry activated by human peak-interval timing in the supra-seconds range. Cogn. Brain Res. 21, 171–182. ( 10.1016/j.cogbrainres.2004.08.005) [DOI] [PubMed] [Google Scholar]
- 8.Matell MS, Meck WH, Nicolelis MAL. 2003. Interval timing and the encoding of signal duration by ensembles of cortical and striatal neurons. Behav. Neurosci. 117, 760–773. ( 10.1037/0735-7044.117.4.760) [DOI] [PubMed] [Google Scholar]
- 9.Meck WH. 2006. Frontal cortex lesions eliminate the clock speed effect of dopaminergic drugs on interval timing. Brain Res. 1108, 157–167. ( 10.1016/j.brainres.2006.06.046) [DOI] [PubMed] [Google Scholar]
- 10.Meck WH. 2006. Neuroanatomical localization of an internal clock: a functional link between mesolimbic, nigrostriatal, and mesocortical dopaminergic systems. Brain Res. 1109, 93–107. ( 10.1016/j.brainres.2006.06.031) [DOI] [PubMed] [Google Scholar]
- 11.Meck WH. 2006. Temporal memory in mature and aged rats is sensitive to choline acetyltransferase inhibition. Brain Res. 1108, 168–175. ( 10.1016/j.brainres.2006.06.047) [DOI] [PubMed] [Google Scholar]
- 12.Allman MJ, Meck WH. 2012. Pathophysiological distortions in time perception and timed performance. Brain 135, 656–677. ( 10.1093/brain/awr210) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coull JT, Cheng R-K, Meck WH. 2011. Neuroanatomical and neurochemical substrates of timing. Neuropsychopharmacology 36, 3–25. ( 10.1038/npp.2010.113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matell MS, Meck WH. 2004. Cortico-striatal circuits and interval timing: coincidence detection of oscillatory processes. Cogn. Brain Res. 21, 139–170. ( 10.1016/j.cogbrainres.2004.06.012) [DOI] [PubMed] [Google Scholar]
- 15.Meck WH, Penney TB, Pouthas V. 2008. Cortico-striatal representation of time in animals and humans 2005. Curr. Opin. Neurobiol. 18, 145–152. ( 10.1016/j.conb.2008.08.002) [DOI] [PubMed] [Google Scholar]
- 16.Merchant H, Harrington DL, Meck WH. 2013. Neural basis of the perception and estimation of time. Annu. Rev. Neurosci. 36, 313–336. ( 10.1146/annurev-neuro-062012-170349) [DOI] [PubMed] [Google Scholar]
- 17.Van Rijn H, Gu B-M, Meck WH. In press Dedicated clock/timing-circuit theories of interval timing. In Neurobiology of interval timing (eds Merchant H, de Lafuente V.). New York, NY: Springer. [Google Scholar]
- 18.Dickerson BC, Eichenbaum H. 2010. The episodic memory system: neurocircuitry and disorders. Neuropsychopharmacology 35, 86–104. ( 10.1038/npp.2009.126) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.MacDonald CJ. 2013. Prospective and retrospective duration memory in the hippocampus: is time in the foreground or background? Phil. Trans. R. Soc. B. 369, 20120463 ( 10.1098/rstb.2012.0463) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meck WH, Church RM, Olton DS. 1984. Hippocampus, time, and memory. Behav. Neurosci. 98, 3–22. ( 10.1037/0735-7044.98.1.3) [DOI] [PubMed] [Google Scholar]
- 21.Melgire M, Ragot R, Samson S, Penney TB, Meck WH, Pouthas V. 2005. Auditory/visual duration bisection in patients with left or right medial-temporal lobe resection. Brain Cogn. 58, 119–124. ( 10.1016/j.bandc.2005.12.003) [DOI] [PubMed] [Google Scholar]
- 22.Tam SKE, Bonardi C. 2012. Dorsal hippocampal involvement in appetitive trace conditioning and interval timing. Behav. Neurosci. 126, 258–269. ( 10.1037/a0027164) [DOI] [PubMed] [Google Scholar]
- 23.Tam SK, Jennings DJ, Bonardi C. 2013. Dorsal hippocampal involvement in conditioned-response timing and maintenance of temporal information in the absence of the CS. Exp. Brain Res. 227, 547–559. ( 10.1007/s00221-013-3530-4) [DOI] [PubMed] [Google Scholar]
- 24.Vidalaki VN, Ho MY, Bradshaw CM, Szabadi E. 1999. Interval timing performance in temporal lobe epilepsy: differences between patients with left and right hemisphere foci. Neuropsychologia 37, 1061–1070. ( 10.1016/S0028-3932(98)00155-9) [DOI] [PubMed] [Google Scholar]
- 25.Meck WH, Church RM, Matell MS. 2013. Hippocampus, time, and memory—a retrospective analysis. Behav. Neurosci. 127, 642–654. ( 10.1037/a0034201) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yin B, Troger AB. 2011. Exploring the 4th dimension: hippocampus, time, and memory revisited. Front. Integr. Neurosci. 5, 36 ( 10.3389/fnint.2011.00036) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gu B-M, Jurkowski AJ, Lake JI, Malapani C, Meck WH. In press Bayesian models of interval timing and distortions in temporal memory as a function of Parkinson's disease and dopamine-related error processing. In Time distortions in mind: temporal processing in clinical populations (eds Vatakis A, Allman MJ.). Boston, MA: Brill Academic Publishers. [Google Scholar]
- 28.Meck WH. 2002. Choline uptake in the frontal cortex is proportional to the absolute error of a temporal memory translation constant in mature and aged rats. Learn. Motiv. 33, 88–104. ( 10.1006/lmot.2001.1101) [DOI] [Google Scholar]
- 29.Meck WH. 2002. Distortions in the content of temporal memory: neurobiological correlates. In Animal cognition and sequential behavior: behavioral, biological, and computational perspectives (eds Fountain SB, Bunsey MD, Danks JH, McBeath MK.), pp. 175–200. Boston, MA: Kluwer Academic Press. [Google Scholar]
- 30.Fanselow MS, Dong H-W. 2010. Are the dorsal and ventral hippocampus functionally distinct structures? Neuron 65, 7–19. ( 10.1016/j.neuron.2009.11.031) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nadel L. 1968. Dorsal and ventral hippocampal lesions and behavior. Physiol. Behav. 3, 891–900. ( 10.1016/0031-9384(68)90174-1) [DOI] [Google Scholar]
- 32.Pennartz CMA, Ito R, Verschure PFMJ, Battaglia FP, Robbins TW. 2011. The hippocampal–striatal axis in learning, prediction and goal-directed behavior. Trends Neurosci. 34, 548–559. ( 10.1016/j.tins.2011.08.001) [DOI] [PubMed] [Google Scholar]
- 33.Moser MB, Moser EI, Forrest E, Andersen P, Morris RG. 1995. Spatial learning with a minislab in the dorsal hippocampus. Proc. Natl Acad. Sci. USA 92, 9697–9701. ( 10.1073/pnas.92.21.9697) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ghiglieri V, Sgobio C, Costa C, Picconi B, Calabresi P. 2011. Striatum-hippocampus balance: from physiological behavior to interneuronal pathology. Progr. Neurobiol. 94, 102–114. ( 10.1016/j.pneurobio.2011.04.005) [DOI] [PubMed] [Google Scholar]
- 35.Poldrack RA, Packard MG. 2003. Competition among multiple memory systems: converging evidence from animal and human brain studies. Neuropsychologia 41, 245–251. ( 10.1016/S0028-3932(02)00157-4) [DOI] [PubMed] [Google Scholar]
- 36.Le Merrer J, Rezai X, Scherrer G, Becker JAJ, Kieffer B. 2013. Impaired hippocampus-dependent and facilitated striatum-dependent behaviors in mice lacking the delta opioid receptor. Neuropsychopharmacology 38, 1050–1059. ( 10.1038/npp.2013.1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Le Merrer J, Plaza-Zabala A, Del Boca C, Matifas A, Maldonado R, Kieffer BL. 2011. Deletion of the δ opioid receptor gene impairs place conditioning but preserves morphine reinforcement. Biol. Psychiatry 69, 700–703. ( 10.1016/j.biopsych.2010.10.021) [DOI] [PubMed] [Google Scholar]
- 38.Olmstead MC, Ouagazzal AM, Kieffer BL. 2009. Mu and delta opioid receptors oppositely regulate motor impulsivity in the signaled nose poke task. PLoS ONE 4, e4410 ( 10.1371/journal.pone.0004410) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Erbs E, Faget L, Scherrer G, Kessler P, Hentsch D, Vonesch JL, Matifas A, Kieffer BL, Massotte D. 2012. Distribution of delta opioid receptor-expressing neurons in the mouse hippocampus. Neuroscience 221, 203–213. ( 10.1016/j.neuroscience.2012.06.023) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bramham CR, Milgram NW, Srebro B. 1991. Delta opioid receptor activation is required to induce LTP of synaptic transmission in the lateral perforant path in vivo. Brain Res. 567, 42–50. ( 10.1016/0006-8993(91)91433-2) [DOI] [PubMed] [Google Scholar]
- 41.Chavkin C, Shoemaker WJ, McGinty JF, Bayon A, Bloom FE. 1985. Characterization of the prodynorphin and proenkephalin neuropeptide systems in rat hippocampus. J. Neurosci. 5, 808–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fouquet C, Babayan BM, Watilliaux A, Bontempi B, Tobin C, Rondi-Reig L. 2013. Complementary roles of the hippocampus and the dorsomedial striatum during spatial and sequence-based navigation behavior . PLoS ONE 8, e67232 ( 10.1371/journal.pone.0067232) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Packard MG, McGaugh JL. 1996. Inactivation of hippocampus or caudate nucleus with lidocaine differentially affects expression of place and response learning. Neurobiol. Learn. Mem. 65, 65–72. ( 10.1006/nlme.1996.0007) [DOI] [PubMed] [Google Scholar]
- 44.Packard MG, Gabriele A. 2009. Peripheral anxiogenic drug injections differentially affect cognitive and habit memory: role of basolateral amygdala. Neuroscience 164, 457–462. ( 10.1016/j.neuroscience.2009.07.054) [DOI] [PubMed] [Google Scholar]
- 45.Schroeder JP, Wingard JC, Packard MG. 2002. Post-training reversible inactivation of hippocampus reveals interference between memory systems. Hippocampus 12, 280–284. ( 10.1002/hipo.10024) [DOI] [PubMed] [Google Scholar]
- 46.Ciamei A, Morton AJ. 2009. Progressive imbalance in the interaction between spatial and procedural memory systems in the R6/2 mouse model of Huntington's disease. Neurobiol. Learn. Mem. 92, 417–428. ( 10.1016/j.nlm.2009.06.002) [DOI] [PubMed] [Google Scholar]
- 47.Cheng R-K, Meck WH. 2007. Prenatal choline supplementation increases sensitivity to time by reducing non-scalar sources of variance in adult temporal processing. Brain Res. 1186, 242–254. ( 10.1016/j.brainres.2007.10.025) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Meck WH, Cheng RK, MacDonald CJ, Gainetdinov RR, Caron MG, Çevik MÖ. 2012. Gene-dose dependent effects of methamphetamine on interval timing in dopamine-transporter knockout mice. Neuropharmacology 62, 1221–1229. ( 10.1016/j.neuropharm.2011.01.042) [DOI] [PubMed] [Google Scholar]
- 49.Cheng R-K, Etchegaray M, Meck WH. 2007. Impairments in timing, temporal memory, and reversal learning linked to neurotoxic regimens of methamphetamine intoxication. Brain Res. 1186, 255–266. ( 10.1016/j.brainres.2007.10.002) [DOI] [PubMed] [Google Scholar]
- 50.Cheung TH, Cardinal RN. 2005. Hippocampal lesions facilitate instrumental learning with delayed reinforcement but induce impulsive choice in rats. BMC Neurosci. 6, 36 ( 10.1186/1471-2202-6-36) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Corbit LH, Balleine BW. 2000. The role of the hippocampus in instrumental conditioning. J. Neurosci. 20, 4233–4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Meck WH. 1988. Hippocampal function is required for feedback control of an internal clock's criterion. Behav. Neurosci. 102, 54–60. ( 10.1037/0735-7044.102.1.54) [DOI] [PubMed] [Google Scholar]
- 53.Meck WH, Church RM, Wenk GL, Olton DS. 1987. Nucleus basalis magnocellularis and medial septal area lesions differentially impair temporal memory. J. Neurosci. 7, 3505–3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Olton DS, Meck WH, Church RM. 1987. Separation of hippocampal and amygdaloid involvement in temporal memory dysfunctions. Brain Res. 404, 180–188. ( 10.1016/0006-8993(87)91369-2) [DOI] [PubMed] [Google Scholar]
- 55.Olton DS, Wenk GL, Church RM, Meck WH. 1988. Attention and the frontal cortex as examined by simultaneous temporal processing. Neuropsychologia 26, 307–318. ( 10.1016/0028-3932(88)90083-8) [DOI] [PubMed] [Google Scholar]
- 56.Paxinos G, Franklin KBJ. 2008. The mouse brain in stereotaxic coordinates, 3rd edn San Diego, CA: Academic Press. [Google Scholar]
- 57.Church RM, Meck WH, Gibbon J. 1994. Application of scalar timing theory to individual trials. J. Exp. Psychol. Anim. B. 20, 135–155. ( 10.1037/0097-7403.20.2.135) [DOI] [PubMed] [Google Scholar]
- 58.Matell MS, Bateson M, Meck WH. 2006. Single-trials analyses demonstrate that increases in clock speed contribute to the methamphetamine-induced horizontal shifts in peak-interval timing functions. Psychopharmacology (Berl.) 188, 201–212. ( 10.1007/s00213-006-0489-x) [DOI] [PubMed] [Google Scholar]
- 59.Buhusi CV, Meck WH. 2009. Relativity theory and time perception: single or multiple clocks? PLoS ONE 4, e6268 ( 10.1371/journal.pone.0006268) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.MacDonald CJ, Cheng R-K, Meck WH. 2012. Acquisition of ‘start’ and ‘stop’ response thresholds in peak-interval timing is differentially sensitive to protein synthesis inhibition in the dorsal and ventral striatum. Front. Integr. Neurosci. 6, 10 ( 10.3389/fnint.2012.00010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Drew MR, Simpson EH, Kellendonk C, Herzberg WG, Lipatova O, Fairhurst S, Kandel ER, Malapani C, Balsam PD. 2007. Transient overexpression of striatal D2 receptors impairs operant motivation and interval timing. J. Neurosci. 27, 7731–7739. ( 10.1523/JNEUROSCI.1736-07.2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Buhusi CV, Aziz D, Winslow D, Carter RE, Swearingten JE, Buhusi MC. 2009. Interval timing accuracy and scalar timing in C57BL/6 mice. Behav. Neurosci. 123, 1102–1113. ( 10.1037/a0017106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gibbon J, Church RM, Meck WH. 1984. Scalar timing in memory. Ann. NY Acad. Sci. 423, 52–77. ( 10.1111/j.1749-6632.1984.tb23417.x) [DOI] [PubMed] [Google Scholar]
- 64.Malapani C, Rakitin B, Levy R, Meck WH, Deweer B, Dubois B, Gibbon J. 1998. Coupled temporal memories in Parkinson's disease: a dopamine-related dysfunction. J. Cogn. Neurosci. 10, 316–331. ( 10.1162/089892998562762) [DOI] [PubMed] [Google Scholar]
- 65.Gallistel CR, King AP, McDonald RV. 2004. Sources of variability and systematic error in mouse timing behavior. J. Exp. Psychol. Anim. Behav. Process. 30, 3–16. ( 10.1037/0097-7403.30.1.3) [DOI] [PubMed] [Google Scholar]
- 66.Cheng RK, Hakak OL, Meck WH. 2007. Habit formation and the loss of control of an internal clock: inverse relationship between the level of baseline training and the clock-speed enhancing effects of methamphetamine. Psychopharmacology 193, 351–362. ( 10.1007/s00213-007-0783-2) [DOI] [PubMed] [Google Scholar]
- 67.Matell MS, Portugal GS. 2007. Impulsive responding on the peak-interval procedure. Behav. Process. 74, 198–208. ( 10.1016/j.beproc.2006.08.009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Buhusi CV, Meck WH. 2013. Scalar time contraction following neurotoxic hippocampal lesions is reversed by the D2R antagonist raclopride.
- 69.Bannerman DM, Yee BK, Good MA, Heupel MJ, Iversen SD, Rawlins JN. 1999. Double dissociation of function within the hippocampus: a comparison of dorsal, ventral, and complete hippocampal cytotoxic lesions. Behav. Neurosci. 113, 1170–1188. ( 10.1037/0735-7044.113.6.1170) [DOI] [PubMed] [Google Scholar]
- 70.Balci F, Meck WH, Moore H, Brunner D. 2009. Timing deficits in aging and neuropathology. In Animal models of human cognitive aging (eds Bizon JL, Woods A.), pp. 161–201. Totowa, NJ: Humana Press. [Google Scholar]
- 71.Eichenbaum H. 2013. Hippocampus: remembering the choices. Neuron 77, 999–1001. ( 10.1016/j.neuron.2013.02.034) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Foerde K, Race E, Verfaellie M, Shohamy D. 2013. A role for the medial temporal lobe in feedback-driven learning: evidence from amnesia. J. Neurosci. 33, 5698–5704. ( 10.1523/JNEUROSCI.5217-12.2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gorchetchnikov A, Grossberg S. 2007. Space, time and learning in the hippocampus: how fine spatial and temporal scales are expanded into population codes for behavioral control. Neural Netw. 20, 182–193. ( 10.1016/j.neunet.2006.11.007) [DOI] [PubMed] [Google Scholar]
- 74.Hirel J, Gaussier P, Quoy M, Banquet JP, Save E, Poucet B. 2013. The hippocampo-cortical loop: spatio-temporal learning and goal-oriented planning in navigation. Neural Netw. 43, 8–21. ( 10.1016/j.neunet.2013.01.023) [DOI] [PubMed] [Google Scholar]
- 75.Teki S, Kumar S, von Kriegstein K, Stewart L, Lyness CR, Moore BCJ, Capleton B, Griffiths TD. 2012. Navigating the auditory scene: an expert role for the hippocampus. J. Neurosci. 32, 12 251–12 257. ( 10.1523/JNEUROSCI.0082-12.2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.MacDonald CJ, Lepage KQ, Eden UT, Eichenbaum H. 2011. Hippocampal ‘time cells’ bridge the gap in memory for discontiguous events. Neuron 71, 737–749. ( 10.1016/j.neuron.2011.07.012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mankin EA, Sparks FT, Slayyeh B, Sutherland RJ, Leutgeb S, Leutgeb JK. 2012. Neuronal code for extended time in the hippocampus. Proc. Natl Acad. Sci. USA 109, 19 462–19 467. ( 10.1073/pnas.1214107109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yin HH, Knowlton BJ. 2006. The role of the basal ganglia in habit formation. Nat. Rev. Neurosci. 7, 464–476. ( 10.1038/nrn1919) [DOI] [PubMed] [Google Scholar]
- 79.Corbit LH, Ostlund SB, Balleine BW. 2002. Sensitivity to instrumental contingency degradation is mediated by the entorhinal cortex and its efferents via the dorsal hippocampus. J. Neurosci. 22, 10 976–10 984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Graybeal C, Feyder M, Schulman E, Saksida LM, Bussey TJ, Brigman JL, Holmes A. 2011. Paradoxical reversal learning enhancement by stress or prefrontal cortical damage: rescue with BDNF. Nat. Neurosci. 14, 1507–1509. ( 10.1038/nn.2954) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dégenètais E, Thierry A-M, Glowinski J, Gioanni Y. 2003. Synaptic influence of hippocampus on pyramidal cells of the rat prefrontal cortex: an in vivo intracellular recording study. Cereb. Cortex 13, 782–792. ( 10.1093/cercor/13.7.782) [DOI] [PubMed] [Google Scholar]
- 82.Luo AH, Tahsili-Fahadan P, Wise RA, Lupica CR, Aston-Jones G. 2011. Linking context with reward: a functional circuit from hippocampal CA3 to ventral tegmental area. Science 333, 353–357. ( 10.1126/science.1204622) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Grace AA. 2012. Dopamine system dysregulation by the hippocampus: implications for the pathophysiology and treatment of schizophrenia. Neuropharmacology 62, 1342–1348. ( 10.1016/j.neuropharm.2011.05.011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gu BM, Cheng RK, Yin B, Meck WH. 2011. Quinpirole-induced sensitization to noisy/sparse periodic input: temporal synchronization as a component of obsessive-compulsive disorder. Neuroscience 179, 143–150. ( 10.1016/j.neuroscience.2011.01.048) [DOI] [PubMed] [Google Scholar]
- 85.Voorn P, Vanderschuren LJMJ, Groenewegen HJ, Robbins TW, Pennartz CMA. 2004. Putting a spin on the dorsal-ventral divide of the striatum. Trends Neurosci. 27, 468–474. ( 10.1016/j.tins.2004.06.006) [DOI] [PubMed] [Google Scholar]
- 86.Melzer S, Michael M, Caputi A, Eliava M, Fuchs EC, Whittington MA, Monyer H. 2012. Projecting GABAergic neurons modulate inhibition in hippocampus and entorhinal cortex. Science 335, 1506–1510. ( 10.1126/science.1217139) [DOI] [PubMed] [Google Scholar]
- 87.Buhusi M, Scripa I, Williams CL, Buhusi CV. 2013. Impaired interval timing and spatial-temporal integration in mice deficient in CHL1, a gene associated with schizophrenia. Timing Time Percept. 1, 21–38. ( 10.1163/22134468-00002003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Montag-Sallaz M, Baarke A, Montag D. 2003. Aberrant neuronal connectivity in CHL1-deficient mice is associated with altered information processing-related immediate early gene expression. J. Neurobiol. 57, 67–80. ( 10.1002/neu.10254) [DOI] [PubMed] [Google Scholar]
- 89.Morellini F, Lepsveridze E, Kahler B, Dityatev A, Schachner M. 2007. Reduced reactivity to novelty, impaired social behavior, and enhanced basal synaptic excitatory activity in perforant path projections to the dentate gyrus in young adult mice deficient in the neural cell adhesion molecule CHL1. Mol. Cell. Neurosci. 34, 121–136. ( 10.1016/j.mcn.2006.10.006) [DOI] [PubMed] [Google Scholar]
- 90.Cheng RK, Ali YM, Meck WH. 2007. Ketamine ‘unlocks’ the reduced clock-speed effect of cocaine following extended training: evidence for dopamine-glutamate interactions in timing and time perception. Neurobiol. Learn. Mem. 88, 149–159. ( 10.1016/j.nlm.2007.04.005) [DOI] [PubMed] [Google Scholar]
- 91.Cheng RK, MacDonald CJ, Meck WH. 2006. Differential effects of cocaine and ketamine on time estimation: implications for neurobiological models of interval timing. Pharm. Biochem. Behav. 85, 114–122. ( 10.1016/j.pbb.2006.07.019) [DOI] [PubMed] [Google Scholar]
- 92.Buhusi CV, Oprisan SA. 2013. Time-scale invariance as an emergent property in a perceptron with realistic, noisy neurons. Behav. Process. 95, 60–70. ( 10.1016/j.beproc.2013.02.015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Meck WH. 1983. Selective adjustment of the speed of internal clock and memory processes. J. Exp. Psychol. Anim. Behav. Process. 9, 171–201. ( 10.1037/0097-7403.9.2.171) [DOI] [PubMed] [Google Scholar]
- 94.Meck WH. 1996. Neuropharmacology of timing and time perception. Cogn. Brain Res. 3, 227–242. ( 10.1016/0926-6410(96)00009-2) [DOI] [PubMed] [Google Scholar]
- 95.Oprisan SA, Buhusi CV. 2011. Modeling pharmacological clock and memory patterns of interval timing in a striatal beat-frequency model with realistic, noisy neurons. Front. Integr. Neurosci. 5, 52 ( 10.3389/fnint.2011.00052) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Oprisan SA, Buhusi CV. 2013. How noise contributes to time-scale invariance of interval timing. Phys. Rev. E 87, 052717 ( 10.1103/PhysRevE.87.052717) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Meck WH, Church RM. 1987. Cholinergic modulation of the content of temporal memory. Behav. Neurosci. 101, 457–464. ( 10.1037/0735-7044.101.4.457) [DOI] [PubMed] [Google Scholar]
- 98.Gibbon J, Malapani C, Dale CL, Gallistel C. 1997. Toward a neurobiology of temporal cognition: advances and challenges. Curr. Opin. Neurobiol. 7, 170–184. ( 10.1016/S0959-4388(97)80005-0) [DOI] [PubMed] [Google Scholar]
- 99.Abela AR, Dougherty SD, Fagen ED, Hill CJ, Chudasama Y. 2013. Inhibitory control deficits in rats with ventral hippocampal lesions. Cereb. Cortex 23, 1396–1409. ( 10.1093/cercor/bhs121) [DOI] [PubMed] [Google Scholar]
- 100.Agostino PV, Golombek DA, Meck WH. 2011. Unwinding the molecular basis of interval and circadian timing. Front. Integr. Neurosci. 5, 64 ( 10.3389/fnint.2011.00064) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dalla Barba G, La Corte V. 2013. The hippocampus, a time machine that makes errors. Trends Cogn. Sci. 17, 102–104. ( 10.1016/j.tics.2013.01.005) [DOI] [PubMed] [Google Scholar]
- 102.Eichenbaum H. 2013. Memory on time. Trends Cogn. Sci. 17, 81–88. ( 10.1016/j.tics.2013.04.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.MacDonald CJ, Meck WH. 2004. Systems-level integration of interval timing and reaction time. Neurosci. Biobehav. Rev. 28, 747–769. ( 10.1016/j.neubiorev.2004.09.007) [DOI] [PubMed] [Google Scholar]
- 104.Meck WH, MacDonald CJ. 2007. Amygdala inactivation reverses fear's ability to impair divided attention and make time stand still. Behav. Neurosci. 121, 707–720. ( 10.1037/0735-7044.121.4.707) [DOI] [PubMed] [Google Scholar]
- 105.Eckart MT, Huelse-Matia MC, Schwarting RKW. 2012. Dorsal hippocampal lesions boost performance in the rat sequential reaction time task. Hippocampus 22, 1202–1214. ( 10.1002/hipo.20965) [DOI] [PubMed] [Google Scholar]
- 106.Hinton SC, Meck WH. 1997. How time flies: function and neural mechanisms of interval timing. Adv. Psychol. 120, 409–457. ( 10.1016/S0166-4115(97)80062-3) [DOI] [Google Scholar]
- 107.Jacobs NS, Allen TA, Nguyen N, Fortin NJ. 2013. Critical role of the hippocampus in memory for elapsed time. J. Neurosci. 33, 13 888–13 893. ( 10.1523/JNEUROSCI.1733-13.2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.MacDonald CJ, Carrow S, Place R, Eichenbaum H. 2013. Distinct hippocampal time cell sequences represent odor memories in immobilized rats. J. Neurosci. 33, 14 607–14 616. ( 10.1523/JNEUROSCI.1537-13.2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Goncalves J, Baptista S, Olesen MV, Fontes-Ribeiro C, Malva JO, Woldbye DP, Silva AP. 2012. Methamphetamine-induced changes in the mice hippocampal neuropeptide Y system: implications for memory impairment. J. Neurochem. 123, 1041–1053. ( 10.1111/jnc.12052) [DOI] [PubMed] [Google Scholar]
- 110.McDonald RJ, Jones J, Richards B, Hong NS. 2006. A double dissociation of dorsal and ventral hippocampal function on a learning and memory task mediated by the dorso-lateral striatum. Eur. J. Neurosci. 24, 1789–1801. ( 10.1111/j.1460-9568.2006.05064.x) [DOI] [PubMed] [Google Scholar]
- 111.Meck WH. 2001. Interval timing and genomics: what makes mutant mice tick? Int. J. Comp. Psychol. 14, 211–231. [Google Scholar]