

Abstract
Sleep homoeostasis refers to a process in which the propensity to sleep increases as wakefulness progresses and decreases as sleep progresses. Sleep is tightly organized around the circadian clock and is regulated by genetic and epigenetic mechanisms. The homoeostatic response of sleep, which is classically triggered by sleep deprivation, is generally measured as a rebound effect of electrophysiological measures, for example delta sleep. However, more recently, gene expression changes following sleep loss have been investigated as biomarkers of sleep homoeostasis. The genetic background of an individual may affect this sleep-dependent gene expression phenotype. In this study, we investigated whether parental genetic background differentially modulates the expression of genes following sleep loss. We tested the progeny of reciprocal crosses of AKR/J and DBA/2J mouse strains and we show a parent-of-origin effect on the expression of circadian, sleep and neuronal plasticity genes following sleep deprivation. Thus, we further explored, by in silico, specific functions or upstream mechanisms of regulation and we observed that several upstream mechanisms involving signalling pathways (i.e. DICER1, PKA), growth factors (CSF3 and BDNF) and transcriptional regulators (EGR2 and ELK4) may be differentially modulated by parental effects. This is the first report showing that a behavioural manipulation (e.g. sleep deprivation) in adult animals triggers specific gene expression responses according to parent-of-origin genomic mechanisms. Our study suggests that the same mechanism may be extended to other behavioural domains and that the investigation of gene expression following experimental manipulations should take seriously into account parent-of-origin effects.
Keywords: parent-of-origin effects, sleep, gene, expression
1. Introduction
Sleep is a genetically and epigenetically regulated phenomenon that is subjected to two fundamental processes: a homoeostatic process and a circadian process [1]. The homoeostatic process of sleep depends on previous wakefulness, representing the pressure for sleep according to the time of day. The circadian process dictates the timing of sleep; it is a self-sustained periodic mechanism that develops with approximately 24 h, cell-autonomous, oscillations. The molecular machinery that sets the circadian clock is composed of positive and negative feedback loops, which involve transcriptional and translational core elements within the cell (reviewed in [2]). Alternative translational and post-translational components participate in the fundamental modulatory mechanisms that maintain circadian timing. These activities involve several epigenetic changes, such as histone modification, acetylation and methylation [3,4], that modulate the dynamic on/off switches of physiological circadian sleep–wake processes [4]. Sleep homoeostasis is biologically related to the circadian clock, and several clock gene mutations result in significant alterations to the electrophysiological measures of sleep [5–7].
Pioneering studies by Franken et al. [8] have shown that the genetics of mouse strains influence electrophysiological measures of sleep, for example slow oscillations in the delta frequency range (1–4 Hz), a fundamental measure of sleep intensity. Several studies in mice have shown that sleep deprivation induces changes in gene expression and it has been reported that the genetic backgrounds of mouse strains affect the transcriptional changes that follow sleep loss [9]. However, different studies have identified different classes of genes that depend on sleep [10]. Transcriptome analysis in three mice strains (AKR/J, C57BL/6J and DBA/2J) [9] described the Homer1a gene as an ideal sleep-dependent target that is rapidly and strongly induced by sleep deprivation in all strains.
In our study, we have tested for the first time whether the effect of genetic background on sleep-dependent gene expression is determined in a parent-of-origin manner. Parent-of-origin effects (i.e. genomic imprinting) have been suggested to modulate fundamental aspects of sleep. Clinical observations of neurodevelopmental sleep disorders suggest that genomic imprinting plays a pivotal role in the architecture of both rapid eye movement (REM) and non-REM (NREM) sleep [11–13]. Interestingly, diverse sleep deficits occur in diseases, such as Prader–Willi syndrome (PWS) and Angelman syndrome (AS), which are classically characterized by opposing imprinting profiles. PWS is caused by maternal duplications/paternal deletions of alleles on chromosome 15q11–13, whereas AS is associated with paternal duplications/maternal deletions on the same region, 15q11–13. The former is characterized by REM sleep abnormalities, excessive sleepiness and core temperature abnormalities [14–17], while the latter is characterized by reductions in sleep. Sleep abnormalities associated with the PWS/AS imprinting region may be linked to the UBE3A gene. Indeed, the lack of the maternal allele of Ube3a in mice results in reduced NREM sleep, deterioration in REM sleep and an increased frequency of waking during the dark-to-light transition [18]. Moreover, serotonin (5-HT) 2A receptors, mediating aminergic inhibition of REM-on cells [19], are primarily expressed by maternal alleles [20]. The importance of studying the link between parental genomic background and sleep has been emphasized by our recent study in mice [21]. We have shown that loss of imprinting of the maternally imprinted gene Gnas dramatically affects REM and NREM physiology in mice [21]. To test the hypothesis that parent-of-origin genetic background affects the expression of specific genes, determining the presence or the absence of a homoeostatic response to sleep loss, we studied reciprocal crosses of two mouse strains that differ in their homoeostatic responses to sleep deprivation: AKR/J and DBA/2J. These two strains have distinct delta-power profiles [8] and different gene expression responses [9,22] after sleep deprivation. While AKR/J mice exhibit dramatic increases in delta power after 6 h of sleep deprivation, DBA/2J mice present a milder response following the same deprivation protocol [8]. Furthermore, AKR/J mice show a greater increase in mRNA levels of core circadian clock genes, such as Bmal1, Clock, Cry1, Cry2, Per1 and Per2, after 6 h of sleep deprivation than do DBA/2J mice [22].
The rationale for our study involves the phenotypic expression patterns of reciprocal heterozygous F1 mice. A parent-of-origin effect would lead to a differential phenotype (i.e. different gene expression) between two reciprocal F1s (hereafter referred to as F1 and F1r). For the purpose of this study, we screened a large list of genes that are involved in circadian, sleep, genomic imprinting and neuronal plasticity regulation in the prefrontal cortex (PFC), as this brain area has been closely linked to sleep function in mammals [23,24]. Remarkably, we detected a sleep-dependent modulation of certain genes that depends on an individual's parental background. This proves, for the first time, that parent-of-origin effects regulate specific sleep-dependent genetic mechanisms.
2. Material and methods
(a). Animals and procedures
The initial AKR/J mouse strain was obtained from Jackson Laboratories (Bar Harbour, USA) and the DBA/2J strain was obtained from Charles River (Wilmington, USA). The mice were kept in an IIT (Istituto Italiano di Tecnologia) animal facility and bred in reciprocal crosses to obtain two different experimental cohorts, AKR/JxDBA/2J F1 mice (the maternal strain is reported first) and DBA/2JxAKR/J F1r mice. Each cohort included a total of six males (13 weeks old) that were equally subdivided into a sleep-deprived (SD) group and a control group (figure 1). All mice were group-housed a week before the experiment, with food and water ad libitum, under a 12 L : 12 D cycle (lights on from 7.00 to 19.00). On the day of the experiment, SD mice underwent 6 h of sleep deprivation starting at 7.00. At 13.00, SD was interrupted, and the mice were left undisturbed for 1 h before they were sacrificed and their PFC tissue was collected. Tissue from the control group was collected at the same time, but control group mice were not subjected to sleep deprivation. All sleep experiments were conducted in the home-cage environment and all procedures were performed under the guidance of the Italian Policy (licence number 039, expires on 15 June 2015).
Figure 1.

The creation of reciprocal cohorts permits the investigation of the role of parental epigenetic controls after SD. A schematic of experimental design and reciprocal crossing mating is presented. The F1 generation was obtained from an AKR/JXDBA/2J crossing, and the F1r generation was obtained from a DBA/2JXAKR/J breeding. Males (13 weeks old) from both F1 and F1r were used as follows: three were maintained under sleep deprivation (SD), and three were subjected to a standard sleep pattern protocol as controls.
(b). Quantitative real-time PCR
Total RNA was extracted from approximately 1 g of snap-frozen PFC using the Rneasy Microarray tissue mini kit (Qiagen, Hilden, Germany). RNA samples were quantified with an ND1000 Nanodrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). Reverse transcription of 1 μg of RNA was performed using the RT2 First Strand Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. RT-qPCR was conducted using a custom RT2 Profiler PCR array for 234 imprinted, circadian and epigenetic-related genes, based on a 384-well plate format developed by Sabioscience Qiagen technical service (Carlsbad, USA; electronic supplementary material, tables S1 and S2). Reconfirmation experiments for the genes of interest were performed using a different set of primers (table 1). RT-qPCR was performed on a ViiA 7 Real-time System machine (Applied Biosystem, Foster City, CA, USA) using the following conditions: 10 min at 95°C, 40 cycles of denaturation at 95°C for 15 s and an annealing and extension step at 60°C for 1 min. Each sample was run to obtain average Ct values according to the manufacturer's specifications. All samples were normalized against a panel of four different housekeeping genes: Gapdh, GusB, β-actin and B2M. Expression levels relative to these housekeeping genes were determined by the calculation of ΔCt, and the data are expressed as 2−ΔΔCt, where ΔΔCt is the difference between the SD and not-SD cohorts.
Table 1.
Primers used for the confirmation of the presence of the genes of interest.
| primer | sequence |
|---|---|
| Rian forward | AGGATTGATTGTGCTGTTAGAGT |
| Rian reverse | CCTCACTGTCTTCCATTCCAA |
| Dlk1 forward | ACAATGGAACTTGCGTGGA |
| Dlk1 reverse | CTTGTGCTGGCAGTCCTT |
| Mtrnr2 forward | AAAGGAGGGTTCAACTGTCT |
| Mtrnr2 reverse | CCAAGGGTCTTCTCGTCTT |
| Prok2 forward | TGCTGTGCTGTCAGTATCT |
| Prok2 reverse | TCTTCTTTCCTGCCTTCCA |
| Per2 forward | AGCTACACCACCCCTTACAAGCT |
| Per2 reverse | GACACGGCAGAAAAAAGATTTCTC |
| Egr1 forward | CCTATGAGCACCTGACCACAGAGT |
| Egr1 reverse | CTCGTCTCCACCATCGCCTTCT |
| Fos forward | ACAGCCTTTCCTACTACCAT |
| Fos reverse | GCACTAGAGACGGACAGA |
| Gapdh forward | GAACATCATCCCTGCATCCA |
| Gapdh reverse | CCAGTGAGCTTCCCGTTCA |
| β-Actin forward | AAGTGGTTACAGGAAGTCC |
| β-Actin reverse | ATAATTTACACAGAAGCAATGC |
(c). Statistical analysis
Statistically significant gene expression differences between SD and not-SD mice were visualized by pooling together the two normal sleep cohorts as a control group (F1 and F1r) (figure 2c). Statistically significant parent-of-origin-regulated genes were identified by splitting the controls of these sleep-modulated genes and comparing the fold change of F1 SD/F1 versus F1r SD/F1r (figure 2d). In both cases, a two-way ANOVA with Bonferroni's multiple comparison test analysis was performed (*p < 0.05; **p < 0.01; ***p < 0.001). All average data are presented as the mean ± s.e.m.
Figure 2.

Gene expression of 230 genes in F1 and F1r mice cohorts after sleep deprivation. (a) Representation of the statistical analysis of the gene expression of 183 detectable targets (Ct ≤ 30) among the F1 and F1r cohorts. (b) Visual classification of the gene expression profiles of F1 and F1r animals after SD. Sleep-regulated genes compose 8.2% and 7.1% of the total genes of the F1 and F1r groups, respectively. (c) Gene expression levels were quantified using Qiagen RT-PCR custom plates. Bars represent the mean + s.e.m. of three different samples for F1 SD and F1r SD mice. *p < 0.05; **p < 0.01; ***p < 0.001 by two-way ANOVA plus Bonferroni's post-test. (d) Parent-of-origin regulation of sleep-dependent genes among F1 SD/F1 and F1r SD/F1r. Bars represent the mean + s.e.m. of three different samples for F1 SD and F1r SD mice related to their control groups (F1 or F1r, respectively). *p < 0.05; **p < 0.01; ***p < 0.001 by two-way ANOVA plus Bonferroni's post-test.
(d). Ingenuity pathway analysis
Significantly enriched functional classes and upstream regulators (figure 4 and electronic supplementary material, figure S1) were identified through Ingenuity Pathway Analysis (Ingenuity Systems, www.ingenuity.com) running a core-analysis using as input the 22 regulated genes detected in PFC samples of F1 SD/ F1r SD reciprocal crosses and the initial set of 230 genes as background. A complete list of all the identified significant classes is reported in the electronic supplementary material, table S3.
Figure 4.

The functional analysis and networks of F1 SD- and F1r SD-regulated genes. (a) A heat map shows the significantly enriched upstream mechanisms of regulation of F1 SD-regulated or F1r SD-regulated genes according to Ingenuity Pathway Analysis (see the electronic supplementary material, table S3 and figure S1 for the complete list). (b) A number of highly significant classes in F1 SD were further selected and shown as individual networks; p-values of overlap are also reported. (Online version in colour.)
We distinguished the genes that were regulated in F1 SD from those regulated in F1r SD to calculate the enrichment values in the Upstream analysis. Some representative classes are shown in figure 4b.
3. Results and discussion
Of the 230 genes selected across genomic imprinting, circadian clock and neural plasticity domains (see electronic supplementary material, tables S1 and S2), 20% (47 genes) presented very low expression levels (Ct ≥ 30) in our PFC samples. We compared the gene expression values of the other 80% (183 genes) between F1 and F1r control mice and no differences, with the exception of the expression of the Rian gene, were observed between the two control groups (figure 2a). The Rian gene was retested in a subsequent RT-PCR experiment with the second set of primers (table 1), and non-statistically significant differences were found between the two F1 progenies (data not shown). These results suggest that, under a normal sleep regimen, we observed no parental effects on gene expression.
In order to maximize the effect of sleep deprivation, we pooled together the two normal sleep groups into one group (referred to as a unique ‘control’ group). Two-way ANOVA statistical analysis with Bonferroni's multiple comparisons of the 182 detectable genes revealed minimal gene expression changes in both SD progenies with respect to control. Specifically, 15 (8.2%) and 13 (7.1%) genes were sleep modulated in F1 and F1r, respectively, with six genes in common between the two cohorts (figure 2b,c). Among the transcripts that were overexpressed after sleep deprivation in both F1 and F1r progenies, we found Homer1a and Per2, which confirms the findings of previous studies that these genes have fundamental roles in sleep homoeostatic mechanisms [9,25,26]. Homer1a upregulation was lessened in DBA/2J mice when compared with AKR/J mice [27] immediately after sleep deprivation. Per2 has also been described as differentially upregulated between the two strains [26] in the same conditions. One hour after sleep deprivation, we observed differences in the fold changes of both Homer1a and Per2 gene expression levels in the reciprocal crosses of the two strains (figure 2c). The other four genes modulated by sleep depletion in both progenies were Dlk1, Peg10, Prok2 and Egr1 (figure 2c). We also found nine genes that were differentially regulated in AKR/JxDBA/2J F1 SD (Egr3, Fos, Airn, Mtrnr2, Nptx2, Plagl1, Synj1, Dbp and Igf2r) and seven genes that were differentially regulated in DBA/2JxAKR/J F1r SD (Pde10a, Atp10a, Drd1a, Sfmbt2, Stat5a, Zim1 and L3mbtl; figure 2b,c).
By splitting the control group in F1 and F1r, we studied whether these sleep-deprivation-regulated genes are controlled in a parent-of-origin manner. Comparing the fold change of F1 SD/F1 versus F1r SD/F1r, we found that Dlk1, Per2, Prok2, Egr1, Fos and Mtrnr2 regulation 1 h after sleep deprivation was significantly different among reciprocal crosses (figure 2d); therefore, these genes are subjected to a parent-of-origin regulation after sleep deprivation.
Furthermore, we tried to confirm the differential regulation of these six genes between the reciprocal crosses by repeating the RT-PCR with a different set of primers (figure 3 and table 1). In this second analysis, non-statistical differences were found between offspring after sleep deprivation in Dlk1 and Mtrnr2 expression (figure 3), whereas Prok2 was not detected (data not shown). On the other hand, the rest of genes tested were consistent with the previous observation. Figure 3 shows that Per2, Egr1 and Fos are upregulated after 1 h of sleep rebound following sleep deprivation in the F1 progeny (F1 SD/F1) but not in the reciprocal cohort (F1r SD/F1r). This result demonstrates that Per2, Egr1 and Fos are genes subjected to a parent-of-origin regulation following sleep loss.
Figure 3.
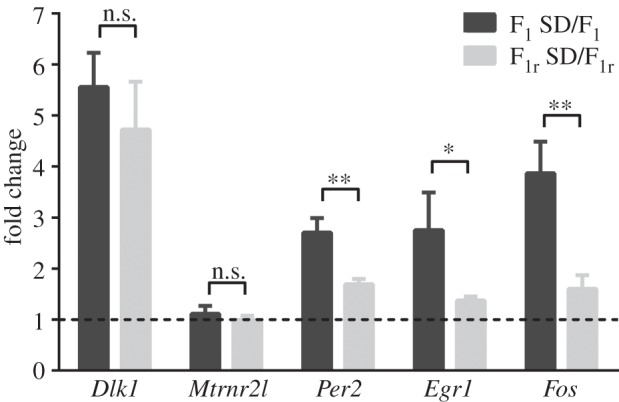
Parent-of-origin regulation of sleep-dependent gene expression. The genes shown were quantified by RT-PCR with the second set of primers (table 1). Bars represent the mean + s.e.m. of three different samples for F1 SD and F1r SD mice related to their control groups (F1 or F1r, respectively). *p < 0.05; **p < 0.01; ***p < 0.001 by Bonferroni's test. n.s., Non-significant.
Per2 is a core regulator of the circadian clock machinery and has been described as differentially regulated between the two parental strains used in this study. Specifically, Per2 upregulation was lessened in AKR/J mice when compared with DBA/2J mice [26]. This effect was detected both after sleep deprivation and 2 h from the end of deprivation. In our study, we showed that Per2 is modulated by sleep deprivation in both reciprocal crosses of these strains; however, this regulation is genotype dependent. This confirms the existence of a parent-of-origin regulation of this gene under sleep deprivation. Moreover, Fos and Egr1 are immediate early genes (IEGs) that were previously reported to respond to sleep deprivation according to an individual's specific genetic background [9]. Following sleep deprivation, expression levels of Fos, Egr1 and Egr3 were reported to significantly increase in AKR/J mice but not in DBA/2J mice [9]. Egr1 and Egr3 exhibit circadian oscillations with differential regulation between light and dark periods in the suprachiasmatic nucleus of the hypothalamus, the master clock of the body [28,29]. Both Egr1 and Fos are reported to respond to the photic phase shift of the circadian clock [30]. The light intensity required to induce Egr1 expression can be 10 times less than the amount necessary to produce a circadian phase shift [30]. Egr1 and Egr3 are characterized by peculiarly timed regulatory mechanisms. Egr1 and Egr3 mRNA levels peak 30–60 min after seizure activity in hippocampus granule cells [31], while their protein levels peak at different time scales: EGR1 protein levels peak 1 h after treatment and return to background levels in 3–4 h, while EGR3 protein levels peak after 4–6 h and basal levels are restored after 24 h [31]. The different temporal patterns of the molecular circuits of Egr1 and Egr3 could represent a common genetic mechanism that, at a cellular level, acts at different timescales.
Our study demonstrates for the first time that the involvement of certain genes in sleep homoeostatic mechanisms is parent-of-origin dependent. Thus, we further explored whether the list of parent-of-origin-regulated genes observed in our study might be implicated in specific functions or upstream mechanisms of regulation. We performed a functional analysis searching for classes that are significantly enriched for either F1 SD- or F1r SD-regulated genes (see electronic supplementary material, table S3). We observed that several differentially enriched upstream mechanisms (see the electronic supplementary material, figure S1 for the complete list) involving these IEGs in the F1 SD include chemicals (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, H89, leukotriene C4, phorbol myristate acetate, U0126, apomorphine, clozapine, haloperidol, kainic acid, N-methyl-d-aspartate (NMDA) and Ca2+), signalling pathways (CHRM1, DICER1, PKA and PSEN1), growth factors (CSF3 and brain-derived neurotrophic factor (BDNF)) and transcriptional regulators (EGR2 and ELK4) (figure 4a). Relevant gene networks for the identified upstream regulators are depicted in figure 4b. Most of these gene networks are related to synaptic transduction, which suggests that specific parent-of-origin mechanisms can modulate sleep-dependent synaptic plasticity mechanisms in the brain. For example, NMDA is the agonist for the ionotropic glutamate receptor NMDAR, which is a pivotal ion channel implicated in the regulation of synaptic functions in the central nervous system [32]. The G protein-coupled receptor CHRM1 (cholinergic receptor muscarinic 1) can also modulate neuronal excitability and synaptic transmission [33] by interacting with glutamatergic neurotransmitter systems, for example NMDARs [34,35], and by potentiating or inhibiting NMDARs in a cell-dependent manner [36].
Ca2+ influx through NMDARs is essential for long-lasting changes in synaptic efficacy, such as long-term potentiation (LTP) and long-term depression [37]. Calcium is widely known to be a major player in neuronal intracellular communication and signalling processes capable of activating and promoting gene transcription in the nucleus. Indeed, Ca2+ influx in the postsynaptic terminal via NMDARs and L-type voltage activates calcium channels and stimulates the production of the second messenger 3′-5′-cyclic adenosine monophosphate (cAMP) by adenylyl cyclase. cAMP activates PKA (protein kinase A), among other important targets in memory processing. Once activated, PKA and other plasticity-associated kinases can phosphorylate and activate the cAMP response element-binding protein (CREB), which regulates the transcription of genes involved in synaptic plasticity, memory and cell survival [38–41]. BDNF is largely expressed in the nervous system [42]. BDNF regulates several aspects of neurodevelopment and synaptic plasticity (see [43] for review), and BDNF-mediated signalling induces synaptic potentiation and plasticity in cortical networks during wakefulness, playing a crucial role in the synaptic homoeostasis regulation of sleep [44]. Egr1 and Fos are tightly linked to neuronal plasticity and memory [45,46]. Egr1 is implicated in the maintenance of synaptic plasticity and is necessary for the persistence of LTP [47] and the consolidation of different forms of long-term memory as well as during the transition from short- to long-term memories [48,49]. Interestingly, sleep is significantly involved in the regulation of brain plasticity and cognition (see [50] for review). A series of studies have shown that PKA and CREB signalling pathways promote wakefulness [51]. The same results were obtained in mice lacking two of the three isoforms of CREB: the loss of alpha and delta causes a reduction in CREB activity and a reduction in wakefulness during the light-off period, with respect to controls [52–54].
Although our study focused only on few genes, our functional analysis identified specific domains such as behaviour, neurological diseases and cell death and survival as enriched processes involving these IEGs (see electronic supplementary material, table S3). Our study suggests that the role of sleep in neuroprotection can be determined by parental epigenetic mechanisms. Indeed, Homer1a is implicated in intracellular calcium homoeostasis and sleep restorative mechanisms [9], and Egr1 and Fos are involved in molecular neuroprotective responses, for example those triggered by ischaemia [55]. Altogether, these data indicate that sleep restriction activates molecular pathways associated with the preservation of neuronal integrity [56].
In our study, we concentrated on the PFC, one of the main targets of the restorative effects of sleep on cognition [57]. The PFC is pivotal in coordinating high-level cognitive processes, such as response inhibition, higher order attention processes, working memory and episodic learning memory [58–62]. Different phases of sleep were reported to be associated with specific activation and deactivation modes of PFC regions [63,64]. Moreover, the disruption or the alteration of normal sleep–wake cycles or circadian rhythms delayed the time required by the PFC to achieve the attention levels of other brain cortical regions [65]. In addition, sleep deprivation alters the neuronal functionality and gene expression profile in the PFC [66–68]. The evidence that neuronal plasticity genes (and possibly many neuronal plasticity pathways) are differently regulated in the PFC according to parent-of-origin mechanisms casts a new light on the epigenetic regulation of these genes in sleep and sleep-related functions.
Acknowledgements
We thank Raquel Garcia Garcia for graphical support. We also thank Glenda Lassi for reading and discussion of the manuscript, Riccardo Navone and Daniela Cantatore for assistance with the management of the mouse colonies.
References
- 1.Borbely AA. 1982. A two process model of sleep regulation. Hum. Neurobiol. 1, 195–204. [PubMed] [Google Scholar]
- 2.Tucci V. 2012. Sleep, circadian rhythms, and interval timing: evolutionary strategies to time information. Front. Integr. Neurosci. 5, 92 ( 10.3389/fnint.2011.00092) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aguilar-Arnal L, Sassone-Corsi P. 2013. The circadian epigenome: how metabolism talks to chromatin remodeling. Curr. Opin. Cell Biol. 25, 170–176. ( 10.1016/j.ceb.2013.01.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borrelli E, Nestler EJ, Allis CD, Sassone-Corsi P. 2008. Decoding the epigenetic language of neuronal plasticity. Neuron 60, 961–974. ( 10.1016/j.neuron.2008.10.012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Laposky A, Easton A, Dugovic C, Walisser J, Bradfield C, Turek F. 2005. Deletion of the mammalian circadian clock gene BMAL1/Mop3 alters baseline sleep architecture and the response to sleep deprivation. Sleep 28, 395–409. [DOI] [PubMed] [Google Scholar]
- 6.Wisor JP, O'Hara BF, Terao A, Selby CP, Kilduff TS, Sancar A, Edgar DM, Franken P. 2002. A role for cryptochromes in sleep regulation. BMC Neurosci. 3, 20 ( 10.1186/1471-2202-3-20) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Naylor E, Bergmann BM, Krauski K, Zee PC, Takahashi JS, Vitaterna MH, Turek RW. 2000. The circadian clock mutation alters sleep homeostasis in the mouse. J. Neurosci. 20, 8138–8143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Franken P, Chollet D, Tafti M. 2001. The homeostatic regulation of sleep need is under genetic control. J. Neurosci. 21, 2610–2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maret S, et al. 2007. Homer1a is a core brain molecular correlate of sleep loss. Proc. Natl Acad. Sci. USA 104, 20 090–20 905. ( 10.1073/pnas.0710131104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mackiewicz M, Zimmerman JE, Shockley KR, Churchill GA, Pack AI. 2009. What are microarrays teaching us about sleep? Trends Mol. Med. 15, 79–87. ( 10.1016/j.molmed.2008.12.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McNamara P. 2004. Genomic imprinting and neurodevelopmental disorders of sleep. Sleep Hypn. 6, 82–90. [Google Scholar]
- 12.McNamara P, Dowdall J, Auerbach S. 2002. REM sleep, early experience, and the development of reproductive strategies. Hum. Nat. 13, 405–435. ( 10.1007/s12110-002-1001-x) [DOI] [PubMed] [Google Scholar]
- 13.Tucci V, Nolan PM. 2009. Toward an understanding of the function of sleep: new insights from mouse genetics. In Evolution of sleep phylogenetic functional perspectives (eds McNamara P, Barton RA, Nunn CL.), pp. 218–237. Cambridge, UK: Cambridge University Press. [Google Scholar]
- 14.Hertz G, et al. 1993. Sleep and breathing patterns in patients with Prader Willi syndrome (PWS): effects of age and gender. Sleep 16, 366–371. [DOI] [PubMed] [Google Scholar]
- 15.Vela-Bueno A, et al. 1984. Sleep in the Prader–Willi syndrome. Clinical and polygraphic findings. Arch. Neurol. 41, 294–296. ( 10.1001/archneur.1984.04050150072020) [DOI] [PubMed] [Google Scholar]
- 16.Vgontzas AN, Bixler EO, Kales A, Centurione A, Rogan PK, Mascari M, Vela-Bueno A. 1996. Daytime sleepiness and REM abnormalities in Prader–Willi syndrome: evidence of generalized hypoarousal. Int. J. Neurosci. 87, 127–139. ( 10.3109/00207459609070832) [DOI] [PubMed] [Google Scholar]
- 17.Vgontzas AN, et al. 1996. Relationship of sleep abnormalities to patient genotypes in Prader–Willi syndrome. Am. J. Med. Genet. 67, 478–482. () [DOI] [PubMed] [Google Scholar]
- 18.Colas D, Wagstaff J, Fort P, Salvert D, Sarda N. 2005. Sleep disturbances in Ube3a maternal-deficient mice modeling Angelman syndrome. Neurobiol. Dis. 20, 471–478. ( 10.1016/j.nbd.2005.04.003) [DOI] [PubMed] [Google Scholar]
- 19.Amici R, Sanford LD, Kearney K, McInerney B, Ross RJ, Horner RL, Morrison AR. 2004. A serotonergic (5-HT2) receptor mechanism in the laterodorsal tegmental nucleus participates in regulating the pattern of rapid-eye-movement sleep occurrence in the rat. Brain Res. 996, 9–18. ( 10.1016/j.brainres.2003.09.026) [DOI] [PubMed] [Google Scholar]
- 20.Kato MV, et al. 1996. Genomic imprinting of the human serotonin-receptor (HTR2) gene involved in development of retinoblastoma. Am. J. Hum. Genet. 59, 1084–1090. [PMC free article] [PubMed] [Google Scholar]
- 21.Lassi G, et al. 2012. Loss of Gnas imprinting differentially affects REM/NREM sleep and cognition in mice. PLoS Genet. 8, e1002706 ( 10.1371/journal.pgen.1002706) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wisor JP, Pasumarthi RK, Gerashchenko D, Thompson CL, Pathak S, Sancar A, Franken P, Lein ES, Kilduff TS. 2008. Sleep deprivation effects on circadian clock gene expression in the cerebral cortex parallel electroencephalographic differences among mouse strains. J. Neurosci. 28, 7193–7201. ( 10.1523/JNEUROSCI.1150-08.2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krueger JM, Rector DM, Roy S, Van Dongen HPA, Belenky G, Panksepp J. 2008. Sleep as a fundamental property of neuronal assemblies. Nat. Rev. Neurosci. 9, 910–919. ( 10.1038/nrn2521) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tononi G, Cirelli C. 2006. Sleep function and synaptic homeostasis. Sleep Med. Rev. 10, 49–62. ( 10.1016/j.smrv.2005.05.002) [DOI] [PubMed] [Google Scholar]
- 25.Mackiewicz M, et al. 2007. Macromolecule biosynthesis: a key function of sleep. Physiol. Genomics 31, 441–457. ( 10.1152/physiolgenomics.00275.2006) [DOI] [PubMed] [Google Scholar]
- 26.Franken P, Thomason R, Heller HC, O'Hara BF. 2007. A non-circadian role for clock-genes in sleep homeostasis: a strain comparison. BMC Neurosci. 8, 87 ( 10.1186/1471-2202-8-87) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Andretic R, Franken P, Tafti M. 2008. Genetics of sleep. Annu. Rev. Genet. 42, 361–388. ( 10.1146/annurev.genet.42.110807.091541) [DOI] [PubMed] [Google Scholar]
- 28.Lin JT, Kornhauser JM, Singh NP, Mayo KE, Takahashi JS. 1997. Visual sensitivities of nur77 (NGFI-B) and zif268 (NGFI-A) induction in the suprachiasmatic nucleus are dissociated from c-fos induction and behavioral phase-shifting responses. Brain Res. Mol. Brain Res. 46, 303–310. ( 10.1016/S0169-328X(97)00005-3) [DOI] [PubMed] [Google Scholar]
- 29.Morris ME, et al. 1998. A screen for genes induced in the suprachiasmatic nucleus by light. Science 279, 1544–1547. ( 10.1126/science.279.5356.1544) [DOI] [PubMed] [Google Scholar]
- 30.O'Donovan KJ, Tourtellotte WG, Millbrandt J, Baraban JM. 1999. The EGR family of transcription-regulatory factors: progress at the interface of molecular and systems neuroscience. Trends Neurosci. 22, 167–173. ( 10.1016/S0166-2236(98)01343-5) [DOI] [PubMed] [Google Scholar]
- 31.O'Donovan KJ, Wilkens EP, Baraban JM. 1998. Sequential expression of Egr-1 and Egr-3 in hippocampal granule cells following electroconvulsive stimulation. J. Neurochem. 70, 1241–1248. ( 10.1046/j.1471-4159.1998.70031241.x) [DOI] [PubMed] [Google Scholar]
- 32.Lau CG, Zukin RS. 2007. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat. Rev. Neurosci. 8, 413–426. ( 10.1038/nrn2153) [DOI] [PubMed] [Google Scholar]
- 33.Picciotto MR, Higley MJ, Mineur YS. 2012. Acetylcholine as a neuromodulator: cholinergic signaling shapes nervous system function and behavior. Neuron 76, 116–129. ( 10.1016/j.neuron.2012.08.036) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu WY, Lu W-Y, Xiong Z-G, Lei S, Orser BA, Dudek E, Browning MD. 1999. G-protein-coupled receptors act via protein kinase C and Src to regulate NMDA receptors. Nat. Neurosci. 2, 331–338. ( 10.1038/7243) [DOI] [PubMed] [Google Scholar]
- 35.Marino MJ, Rouse ST, Levey AI, Potter LT, Conn PJ. 1998. Activation of the genetically defined m1 muscarinic receptor potentiates N-methyl-d-aspartate (NMDA) receptor currents in hippocampal pyramidal cells. Proc. Natl Acad. Sci. USA 95, 11 465–11 470. ( 10.1073/pnas.95.19.11465) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grishin AA, Benquet P, Gerber U. 2005. Muscarinic receptor stimulation reduces NMDA responses in CA3 hippocampal pyramidal cells via Ca2+-dependent activation of tyrosine phosphatase. Neuropharmacology 49, 328–337. ( 10.1016/j.neuropharm.2005.03.019) [DOI] [PubMed] [Google Scholar]
- 37.Collingridge GL, Isaac JT, Wang YT. 2004. Receptor trafficking and synaptic plasticity. Nat. Rev. Neurosci. 5, 952–962. ( 10.1038/nrn1556) [DOI] [PubMed] [Google Scholar]
- 38.Cohen S, Greenberg ME. 2008. Communication between the synapse and the nucleus in neuronal development, plasticity, and disease. Annu. Rev. Cell Dev. Biol. 24, 183–209. ( 10.1146/annurev.cellbio.24.110707.175235) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Greer PL, Greenberg ME. 2008. From synapse to nucleus: calcium-dependent gene transcription in the control of synapse development and function. Neuron 59, 846–860. ( 10.1016/j.neuron.2008.09.002) [DOI] [PubMed] [Google Scholar]
- 40.Saha RN, Dudek SM. 2008. Action potentials: to the nucleus and beyond. Exp. Biol. Med. 233, 385–393. ( 10.3181/0709-MR-241) [DOI] [PubMed] [Google Scholar]
- 41.Wiegert JS, Bading H. 2011. Activity-dependent calcium signaling and ERK-MAP kinases in neurons: a link to structural plasticity of the nucleus and gene transcription regulation. Cell Calcium 49, 296–305. ( 10.1016/j.ceca.2010.11.009) [DOI] [PubMed] [Google Scholar]
- 42.West AE, Chen EG, Dalva MB, Dolmetsch RE, Kornhauser JM, Shaywitz AJ, Takasu MA, Tao X, Greenberg ME. 2001. Calcium regulation of neuronal gene expression. Proc. Natl Acad. Sci. USA 98, 11 024–11 031. ( 10.1073/pnas.191352298) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Park H, Poo MM. 2013. Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 14, 7–23. ( 10.1038/nrn3379) [DOI] [PubMed] [Google Scholar]
- 44.Tononi G, Cirelli C. 2003. Sleep and synaptic homeostasis: a hypothesis. Brain Res. Bull. 62, 143–150. ( 10.1016/j.brainresbull.2003.09.004) [DOI] [PubMed] [Google Scholar]
- 45.Guzowski JF, Setlow B, Wagner EK, McGaugh JL. 2001. Experience-dependent gene expression in the rat hippocampus after spatial learning: a comparison of the immediate-early genes Arc, c-fos, and zif268. J. Neurosci. 21, 5089–5098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Poirier R, et al. 2008. Distinct functions of Egr gene family members in cognitive processes. Front. Neurosci. 2, 47–55. ( 10.3389/neuro.01.002.2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jones MW, et al. 2001. A requirement for the immediate early gene Zif268 in the expression of late LTP and long-term memories. Nat. Neurosci. 4, 289–296. ( 10.1038/85138) [DOI] [PubMed] [Google Scholar]
- 48.Bozon B, Davis S, Laroche S. 2002. Regulated transcription of the immediate-early gene Zif268: mechanisms and gene dosage-dependent function in synaptic plasticity and memory formation. Hippocampus 12, 570–577. ( 10.1002/hipo.10100) [DOI] [PubMed] [Google Scholar]
- 49.Bozon B, Kelly A, Josselyn SA, Silva AJ, Davis S, Laroche S. 2003. MAPK, CREB and zif268 are all required for the consolidation of recognition memory. Phil. Trans. R. Soc. Lond. B 358, 805–814. ( 10.1098/rstb.2002.1224) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang G, Grone B, Colas D, Appelbaum L, Mourrain P. 2011. Synaptic plasticity in sleep: learning, homeostasis and disease. Trends Neurosci. 34, 452–463. ( 10.1016/j.tins.2011.07.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hendricks JC, Williams JA, Panckeri K, Kirk D, Tello M, Yin JC-P, Sehgal A. 2001. A non-circadian role for cAMP signaling and CREB activity in Drosophila rest homeostasis. Nat. Neurosci. 4, 1108–1115. ( 10.1038/nn743) [DOI] [PubMed] [Google Scholar]
- 52.Hummler E, Cole TJ, Blendy JA, Ganss R, Aguzzi A, Schmid W, Beermann F, Schutz G. 1994. Targeted mutation of the CREB gene: compensation within the CREB/ATF family of transcription factors. Proc. Natl Acad. Sci. USA 91, 5647–5651. ( 10.1073/pnas.91.12.5647) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Graves LA, et al. 2003. Genetic evidence for a role of CREB in sustained cortical arousal. J. Neurophysiol. 90, 1152–1159. ( 10.1152/jn.00882.2002) [DOI] [PubMed] [Google Scholar]
- 54.Graves L, et al. 2002. Behavioral analysis of CREB alphadelta mutation on a B6/129 F1 hybrid background. Hippocampus 12, 18–26. ( 10.1002/hipo.10003) [DOI] [PubMed] [Google Scholar]
- 55.Kamphuis W, et al. 2007. Global gene expression profiling of ischemic preconditioning in the rat retina. Mol. Vis. 13, 1020–1030. [PMC free article] [PubMed] [Google Scholar]
- 56.Mongrain V, et al. 2010. Separating the contribution of glucocorticoids and wakefulness to the molecular and electrophysiological correlates of sleep homeostasis. Sleep 33, 1147–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maquet P. 1995. Sleep function(s) and cerebral metabolism. Behav. Brain Res. 69, 75–83. ( 10.1016/0166-4328(95)00017-N) [DOI] [PubMed] [Google Scholar]
- 58.Couyoumdjian A, Sdoia S, Tempesta D, Curcio G, Rastellini E, De Gennaro L, Ferrara M. 2010. The effects of sleep and sleep deprivation on task-switching performance. J. Sleep Res. 19, 64–70. ( 10.1111/j.1365-2869.2009.00774.x) [DOI] [PubMed] [Google Scholar]
- 59.Harrison Y, Horne JA. 1998. Sleep loss impairs short and novel language tasks having a prefrontal focus. J. Sleep Res. 7, 95–100. ( 10.1046/j.1365-2869.1998.00104.x) [DOI] [PubMed] [Google Scholar]
- 60.Killgore WD, Kahn-Greene ET, Lipizzi EL, Newman RA, Kamimori GH, Balkin TJ. 2008. Sleep deprivation reduces perceived emotional intelligence and constructive thinking skills. Sleep Med. 9, 517–526. ( 10.1016/j.sleep.2007.07.003) [DOI] [PubMed] [Google Scholar]
- 61.Granon S, Floresco S. 2009. Functional neuroanatomy of flexible behaviors in mice and rats. Endophenotypes of psychiatric and neurodegenerative disorders in rodent models, pp. 83–103. Kerala: Transworld Research Network. [Google Scholar]
- 62.Dalley JW, Cardinal RN, Robbins TW. 2004. Prefrontal executive and cognitive functions in rodents: neural and neurochemical substrates. Neurosci. Biobehav. Rev. 28, 771–784. ( 10.1016/j.neubiorev.2004.09.006) [DOI] [PubMed] [Google Scholar]
- 63.Maquet P, Dive D, Salmon E, Sadzot B, Franco G, Poirrier R, von Frenckell R, Franck G. 1990. Cerebral glucose utilization during sleep-wake cycle in man determined by positron emission tomography and [18F]2-fluoro-2-deoxy-D-glucose method. Brain Res. 513, 136–143. ( 10.1016/0006-8993(90)91099-3) [DOI] [PubMed] [Google Scholar]
- 64.Maquet P, et al. 2000. Experience-dependent changes in cerebral activation during human REM sleep. Nat. Neurosci. 3, 831–836. ( 10.1038/77744) [DOI] [PubMed] [Google Scholar]
- 65.Nofzinger EA, Mintun MA, Wiseman M, Kupfer DJ, Moore RY. 1997. Forebrain activation in REM sleep: an FDG PET study. Brain Res. 770, 192–201. ( 10.1016/S0006-8993(97)00807-X) [DOI] [PubMed] [Google Scholar]
- 66.Winters BD, Huang YH, Dong Y, Krueger JM. 2011. Sleep loss alters synaptic and intrinsic neuronal properties in mouse prefrontal cortex. Brain Res. 1420, 1–7. ( 10.1016/j.brainres.2011.08.078) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu ZW, Faraguna U, Cirelli C, Tononi G, Gao X-B. 2010. Direct evidence for wake-related increases and sleep-related decreases in synaptic strength in rodent cortex. J. Neurosci. 30, 8671–8675. ( 10.1523/JNEUROSCI.1409-10.2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cirelli C, Faraguna U, Tononi G. 2006. Changes in brain gene expression after long-term sleep deprivation. J. Neurochem. 98, 1632–1645. ( 10.1111/j.1471-4159.2006.04058.x) [DOI] [PubMed] [Google Scholar]