

Abstract
Statistical species delimitation usually relies on singular data, primarily genetic, for detecting putative species and individual assignment to putative species. Given the variety of speciation mechanisms, singular data may not adequately represent the genetic, morphological and ecological diversity relevant to species delimitation. We describe a methodological framework combining multivariate and clustering techniques that uses genetic, morphological and ecological data to detect and assign individuals to putative species. Our approach recovers a similar number of species recognized using traditional, qualitative taxonomic approaches that are not detected when using purely genetic methods. Furthermore, our approach detects groupings that traditional, qualitative taxonomic approaches do not. This empirical test suggests that our approach to detecting and assigning individuals to putative species could be useful in species delimitation despite varying levels of differentiation across genetic, phenotypic and ecological axes. This work highlights a critical, and often overlooked, aspect of the process of statistical species delimitation—species detection and individual assignment. Irrespective of the species delimitation approach used, all downstream processing relies on how individuals are initially assigned, and the practices and statistical issues surrounding individual assignment warrant careful consideration.
Keywords: ecology, individual assignment, morphology, multilocus genetics, species delimitation, species detection
1. Introduction
Increased efficacy in species delimitation is critically important in biology given the pending biodiversity crisis [1]. Statistical developments in species delimitation, often reliant only on genetic data and not the intrinsic characters distinguishing species, have been increasingly adopted as an approach to meet this need [2–9]. Under traditional taxonomic practices, the discovery, delineation and description of species often involves qualitative decisions on what a species should be and are thus subjected to the implementation of various philosophical species concepts [10,11]. Alternatively, statistical species delimitation treats species as hypotheses in a statistical framework using these objective tests to delineate evolutionarily independent lineages as species, and therefore satisfying numerous species concepts (see [12] for review). However, the sole use of genetic data in these methods has brought about questions regarding the utility of these approaches to the taxonomic description of species [13], which is the focus of ongoing debate [12–14]. Given the importance of distinguishing intrinsic characters, attempts have been made to ensure that approaches remain integrative in empirical studies that employ statistical species delimitation [15–17]. However, a truly integrative approach for statistically identifying species, classifying individuals and estimating the probability that putative species represent evolutionary species remains elusive—limiting cohesiveness between species discovery and description [12,18].
To date, statistical species delimitation methods have focused on four areas: detecting putative species, assigning individuals to species, estimating species relationships and estimating the probability that species are evolutionarily distinct in a coalescent-based framework. The latter two have been the focus of methodological advancement, involving the adoption of coalescent-based tests of species' evolutionary independence [7,9,19–22], and we do not focus on these here. We instead focus on individual assignment and species detection, which must be defined a priori in coalescent-based species delimitation and have received nominal attention for methodological advancement. Methods to detect species and assign individuals prior to coalescent-based species delimitation include clustering using genetic allele frequency data [23], genetic distances [4] and morphological data [24]. These statistical, rather than qualitative approaches, provide much-needed objectivity. Furthermore, the widespread availability of genetic data has created a paradigm that only genetic data should be used for individual assignment and species detection (e.g. [3–5,7,9]) in statistical species delimitation frameworks. Nevertheless, before a purely genetic approach to species detection and individual assignment is adopted, we need to understand: (i) what is being sacrificed by the failure to consider multiple data-types and (ii) if genetic data are sufficient for species detection and individual assignment. Could accuracy be improved by the addition of multiple data-types?
Taking a single line of evidence in delimiting species may result in undetected species or too many species [25,26]. Coalescent-based tests of evolutionary independence may be effective at rejecting overly split hypothesized species [22], however, given that a priori species detection and individual assignment are typically prerequisites (excepting the heuristic approach of [6]), a considerably more problematic issue arises when putative species fail to be detected. Species may go undetected with a purely genetic approach when species show divergent morphologies and/or ecological niches, which is often the case in adaptive radiations [27,28], where vast amounts of genetic data are required to recover species [29]. This is largely because the processes of incomplete lineage sorting and hybridization in recent radiations interfere with individual assignment using genetic approaches [4,23], as would convergent morphological evolution when using purely morphological approaches [24].
We propose a statistical approach, through modification of existing Gaussian clustering methods, by which multiple data-types can be used to detect putative species in a group and then assign individuals without prior knowledge of the number of species in a group. This approach can then be combined with coalescent-based tests of species boundaries [12], but specifically focuses on improving: (i) species detection and individual assignment and (ii) the cohesiveness between species discovery and description. We compare this method using various amounts of integrated data for species detection and individual assignment using: (i) a genetic clustering approach [23] and (ii) a qualitative, integrative traditional taxonomic framework [8]. We test these comparisons in an empirical system comprising 11 Australian amphibolurine lizard species (Ctenophorus maculatus complex (Squamata: Agamidae)). This empirical system was chosen considering that purely genetic data, as commonly applied in statistical species delimitation [12], failed to recognize the morphological and ecological diversity in this group. We predict that by directly incorporating data on morphological and environmental components with multilocus genetic data, we can identify putative species and assign individuals in a statistically integrative manner, even though the level of divergence in different data-types varies.
2. Material and methods
(a). Empirical system and taxon definition
Multilocus genetic, morphological and environmental data were collected from 153 individuals across 11 species within the C. maculatus species complex (see electronic supplementary material, table S1). Species in the C. maculatus complex differentiate variably across genetic, morphological and environmental axes. The complex represents a recent, rapid radiation of species occupying distinct ecological niches [30]. Many species are morphologically distinct (see electronic supplementary material, figure S1)—satisfying morphological approaches—however, shallow multilocus genetic divergences (Sp.7–10—electronic supplementary material, figure S2) could lead to unrecognized species lineages based solely on genetic criteria. Alternatively, some species are morphologically indistinguishable (Sp.3 and 11—electronic supplementary material, figure S1), yet show deep genetic divergences (see electronic supplementary material, figure S2) that would not be recognized using morphological methods. Variance is exemplified in non-metric multidimensional scaling (NMDS) plots between species pairs using different data-types (see electronic supplementary material, figure S3). A taxonomic approach was used to a priori define species whereby genetic (mtDNA lineages), geographical, morphological and ecological data are qualitatively combined to distinguish species [8]. This approach is hereafter termed traditional taxonomic criteria (TTC), as it is a common integrative approach used to delimit and formally describe species. Details of the empirical morphological, ecological and genetic data used are given in the electronic supplementary material. All analyses were conducted using add-on packages within the R statistical package (http://www.r-project.org/), unless otherwise stated.
(b). Gaussian clustering
Gaussian clustering methods, in combination with multivariate approaches, allow incorporation of information from integrated data in a statistical framework. Individual pairwise Gower distances [31] were calculated for environmental and morphological data (daisy, cluster package [32]). Individual pairwise genetic distances were calculated using MEGA v. 5.0 [33] with a Jukes Cantor correction [34] to account for multiple substitutions. Genetic distances for individual loci were divided by mean pairwise distances to account for differences in substitution rates among loci. Distances across phased haplotypes within individuals were averaged and individual distances were averaged across loci. Distance matrices for each data-type were standardized using NMDS (isoMDS, MASS package [35]) prior to further analysis. The recommendations of Hausdorf & Hennig [4] were followed such that four dimensions for each data-type were retained, ensuring that no more than five individuals were required to recognize a cluster. NMDS stress values were generally below 10%, with an absolute maximum of 20%. Combined data consisted of combining two (environmental and genetic, E + G; morphological and genetic, M + G) and all (morphological, environmental and genetic—M + E + G) data-types, by concatenating the four NMDS dimensions for each dataset.
(c). Clustering of individuals using allele frequency data
Clustering using allele frequency data was undertaken using the program STRUCTURAMA [23], a commonly used approach for detecting species clusters and assigning individuals in species delimitation [12]. Multilocus genetic data were converted to haplotypic frequencies, and STRUCTURAMA v. 1.0 [23] was run for 10 million generations, sampling every 100th cycle with 10 000 samples discarded as burn-in. A range of prior values was used for the shape and scale values of the gamma distribution of k (shape : scale—2.5 : 0.5, 2.5 : 1, 5 : 2, 10 : 1). The number of populations (k) was treated as a random variable and the k with the highest probability was chosen with probabilities of cluster assignment used to assign individuals to a species cluster.
(d). Bayesian species delimitation
Bayesian species delimitation has been shown to perform well in delimiting species in broad range of divergence scenarios [36]. We used Bayesian species delimitation here (BPP v. 2.1 [22]) to estimate the posterior probabilities of species clusters using multilocus genetic data. Guide tree topologies were obtained from *BEAST [21]; the results from three individual species-tree analyses were combined (50 million generations/run, sampling every 1000 with a 10% burnin) after removing the burnin using LogCombiner [37]. Log files were checked for stationarity, high effective sample size (ESS) values confirmed convergence, and species trees were summarized using TreeAnnotator [37]. BPP v. 2.1 [22] was run for 2.5 million reversible jump Markov chain Monte Carlo (rjMCMC) generations sampling every five generations, using algorithm 1 with ɛ = 15. Values for θ (ancestral population size) and τ (root age) impact the posterior probabilities and therefore number of species delimited using BPP [22], therefore, we employed a range of prior distributions for θ and τ (θ and τ = 1 : 10, 2 : 400, 2 : 2000 and θ = 1 : 10 and τ = 2 : 2000). Low values for priors (i.e. 1 : 10) generally infer large population sizes and deep divergences, whereas higher values infer small population sizes and shallow divergences for θ and τ, respectively.
(e). Comparing singular and integrative data in distinguishing traditional taxonomic criteria species
To compare the ability of the singular and integrative datasets to detect species, we used linear discriminant function analyses (DFA) to assess the information content of each dataset in distinguishing among TTC-defined species. While testing informativeness using TTC delineation appears arbitrary, it is nevertheless unavoidable when testing new methods using empirical datasets [4,5,22]. Individuals were categorized as being TTC assigned (posterior probability (PP) > 0.95; assigned to their TTC species), differently assigned (PP > 0.95; assigned to a species it was not designated to under TTC), or unassigned (PP < 0.95; not assigned to any species).
(f). Comparing singular and integrative data in detecting species clusters
We compared the species detection and individual assignment of Gaussian clustering, using singular and integrated data, to allele frequency clustering using TTC assignment as a benchmark. In using TTC species as a benchmark, we potentially misclassified species/individuals, however the comparison is unavoidable when using empirical datasets as historically taxonomies have inevitably been determined under qualitative frameworks [4,5,22]. Gaussian clustering of NMDS dimensions (mclust package [38]) was used to detect species clusters, with the number of clusters determined using the Bayesian Information Criterion (BIC). Noise in the standardized dimensional data was accounted for using ‘noise’ estimation (NNclean, prabclus package [4]). The choice of a k tuning constant for noise estimation was four, enabling cluster detection with few individuals [4].
Gaussian clustering was carried out on genetic, environmental and morphological singular data in addition to three integrated datasets. To assess the ability of each data-type to recover TTC-defined species, individuals were categorized as: assigned to TTC species (PP > 0.95), different assignment (assigned to a species it not designated under TTC; PP > 0.95), unassigned (unassigned to any species; PP ≥ 0.95) or unrecognized (where species delimited using TTC were unrecognized). Clusters and subsequent individual assignment were then used to assess the probabilities of species boundaries using BPP v. 2.1 [22] implementing a *BEAST [21] guide topology. Analyses were based on male complete integrative data (M + E + G) given the limitations of using morphological data in sexually dimorphic species, and considering that secondary sexual characteristics largely distinguish species [39]. Unsupported nodes (PP < 0.95 for 2/4 θ and τ priors) were collapsed.
3. Results
NMDS stress values for each data-type were below 10%, excepting male environmental data, which did not exceed the maximal limit of 20% (see electronic supplementary material, table S2). DFA results indicate that singular data could not distinguish TTC species in comparison with integrated data (figure 1). In general, M + E + G data were more effective at recovering TTC assigned males than other integrated data (M + G and E + G). M + G data were slightly more effective at recovering TTC assignment for females than M + E + G data (figure 1). For males, M + E + G data minimize differently classified and unassigned individuals.
Figure 1.
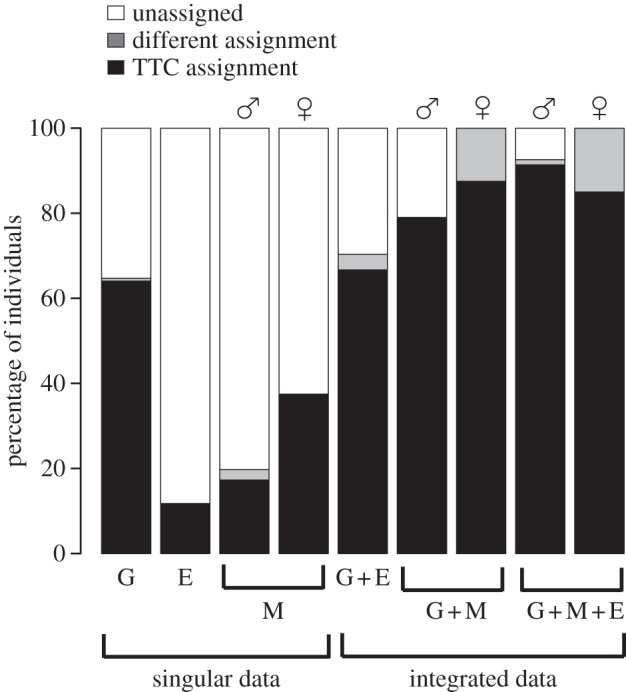
DFA showing the power of singular and integrative data in distinguishing among TTC species. The proportions of individuals assigned to their TTC species, unassigned or assigned to a species other than their TTC species, are shown. Male (♂) and female (♀) morphological data were analysed separately. G, genetic data; E, environmental data; M, morphological data.
Clusters detected generally fell below the number of TTC species (figure 2a). Genetic allele frequency clustering indicated six clusters (see electronic supplementary material, table S4) with stable individual assignment across prior sets. Gaussian clustering of environmental data, male M + G data, and female and male M + E + G data were either above or close to a similar number of clusters to TTC assignment. High levels of unassigned, differently assigned individuals and unrecognized TTC species were prevalent using singular data (figure 2b). Integrated data minimized unassigned individuals, and more often statistically assigned individuals to their TTC species compared to singular data and allele frequency clustering (figure 2b). For M + E + G data, males showed higher levels of TTC assignment than females, while the highest proportional TTC assignment was for female M + G data. Given the limited sampling for females, we used clustering of male M + E + G data for subsequent analyses. Individual assignment differed from TTC assignment using male M + E + G data in a small number of cases, specifically where samples from different TTC species were geographically proximate, in parapatry (e.g. Sp.4 and 5) or where samples were separated by a sampling gap (see electronic supplementary material, figure S4). To explore the effect of alternative groupings, we analysed the data using both strict assignment to cluster, as well as the removal of individuals with a different assignment to that under TTC.
Figure 2.
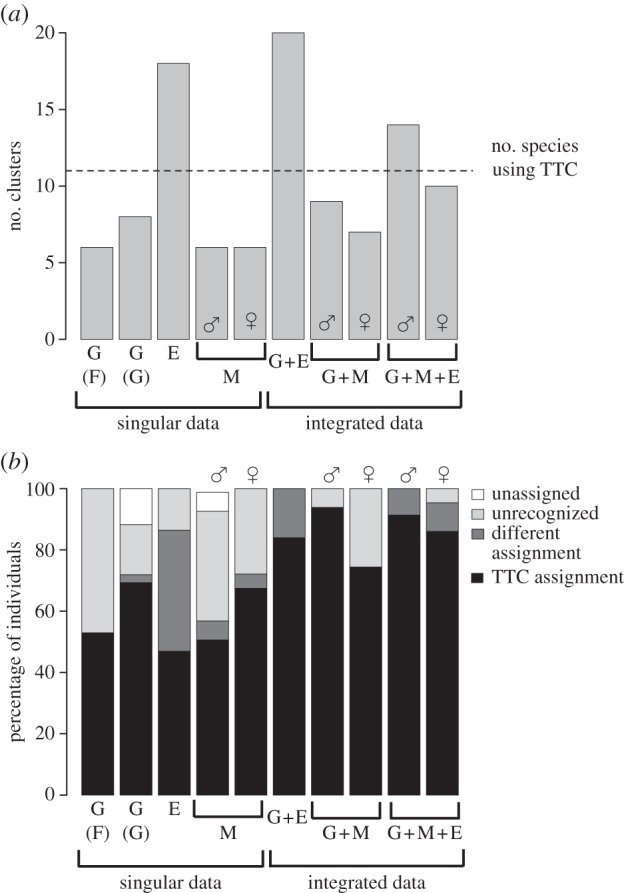
Clusters inferred using different methods and data (a) relative to TTC species (dotted line). Shown are individuals unassigned to a species, in a TTC species unrecognized by clustering, assigned correctly to TTC species, and assigned to a different species than specified by TTC (b). Male (♂) and female (♀) morphological data were analysed separately. G, genetic data; E, environmental data; M, morphological data; (F), allele frequency clustering; (G), Gaussian clustering genetic data.
TTC species were well supported for all θ and τ priors (figure 3a). Bayesian species delimitation of allele frequency clustering inferred six putative well-supported species with θ and τ = 1 : 10, and θ = 1 : 10 and τ = 2 : 2000 (figure 3b), but no support for Sp.4–10 when θ and τ = 2 : 400 and 2 : 2000, respectively. These analyses did not mix well despite a range of starting and automatic tuning of fine-tune parameters, probably related to errors in individual assignment and the resulting guide tree used in coalescent-based species delimitation [20]. One collapsed clade potentially encompasses four TTC species, while several others represent two distinct TTC species (figure 3b). However, if priors for θ and τ = 2 : 400 and 2 : 2000 were assumed, up to seven species may be collapsed.
Figure 3.

Species delimitation for TTC delimited (a), allele frequency species (b), along with species inferred from Gaussian clustering of male data strictly (c), and with misclassified individuals removed (d). Unsupported nodes (PP > 0.95 for 2/4 θ and τ prior combinations) were collapsed. Priors for θ and τ are shown in the following order for θ and τ = 1 : 10, 2 : 400 and 2 : 2000, and then θ—1 : 10 and τ = 2 : 2000. Asterisk (*)—marks nodes supported by only two combinations of θ and τ.
The topology of the species trees obtained using TTC (figure 3a) and integrative clustering techniques with (figure 3c) and without potentially misclassified individuals removed (figure 3d) shows only slight topological differences among some closely related species, namely between Sp.6 and Sp.9. Removing individuals with a different TTC assignment from integrative clustering resulted in the statistical support for the species split between Sp.4 and Sp.5 (figure 3c,d), which is equivalent to the support for the split between Sp.4 and Sp.5 under TTC (figure 3a). Differences between TTC and integrative assignment highlight a relationship between some populations of Sp.3 (Sp.3a—figure 3) and Sp.2 that was not recognized under TTC (figure 3a).
4. Discussion
Despite the widespread use of genetic data for species detection and individual assignment [2–9], we were unable to recover TTC species utilizing genetic data alone, irrespective of the method used (figures 1 and 2). While this may indicate low genetic signal, we find genetic support for more finely split species using TTC and integrative clustering (figure 3). Purely genetic methods can inadequately describe diversity [28] and require significant amounts of genetic data (well beyond that typically used in species delimitation [40]) to delimit rapid radiations diverging along ecological/morphological axes [29]. This suggests that the evolutionary processes underlying the speciation of diverse groups, often the subject of species delimitation studies, may impact the ability to detect species and assign individuals using a single data-type alone—in this case genetic data, but also using morphological data alone [17].
The data used for detecting species and assigning individuals are likely to have broad impacts on efficacy. Hybridization and/or incomplete lineage sorting, often associated with rapid radiation, could cause reduced resolution of species detection using genetic data [41], even though species trees can be reconstructed under these scenarios [42,43]. A tiered sampling strategy involving microsatellite/SNP data may enable hybrid detection and exclusion [44]. Indeed, these methods may be more suited in detecting species and assigning individuals compared with the current practice of using lower resolution sequence data, but are often intractable for large delimitation studies. Broad-scale environmental variables, used here, may not adequately distinguish between sympatric species occupying differential microhabitats for which finer-scale microhabitat data may be more appropriate [45]. Morphological data, as demonstrated in our study (see also [24]), represent a valuable resource for species detection and individual assignment but should be informative with respect to phenotypic divergences among species. Collectively, these results highlight the importance of consistent sampling strategies and informative data in species delimitation studies.
While integrative data recovered species and assigned individuals similar to the qualitative taxonomic framework (TTC), many species were not detected using singular data-types, including when allele frequency clustering methods were used (figure 2). While caution should be taken in interpretation of a single dataset, the incorporation of a statistically integrative approach to detecting species is likely to have wide utility for species delimitation, especially when divergence varies across phenotypic, genetic and ecological axes. While debate over species concepts is ongoing [10,11], there is consensus regarding the importance of the evolutionary independence of species [10,12] and the need for an integrative taxonomy. Using an integrative statistical framework for species discovery and delineation, as proposed herein, allows for the statistical inclusion of variance in intrinsic characters in species discovery—which may then be used to describe species. Furthermore, using an objective statistical approach can also circumvent taxonomic inflation related to the adoption of different species concepts in qualitative taxonomic frameworks [46].
While the proposed integrative approach has the potential to improve the detection of species, it did not assign individuals to the correct group 100% of the time, if TTC assignment is taken as a benchmark (figure 2). We concede that in using TTC as a benchmark we potentially misclassify species/individuals, however the comparison is unavoidable when using empirical datasets, as taxonomies historically have been determined under qualitative frameworks [4,22]. Nevertheless, this comparison can provide valuable insights into the limitations of our statistically integrative method. For example, individuals close to species boundaries or geographically separated were often assigned to a different species compared with TTC assignment. Similarly, individuals in areas of parapatry and potential hybridization have dubious individual assignment (figure 3 and electronic supplementary material, S4). Our method may be sensitive to gaps in geographical sampling and outliers (e.g. hybrids) that may skew results. Outlier individuals are likely to impact species detection and individual assignment regardless of the method employed, and the sensitivity of our method in comparison to other delimitation methods [2,3,6] requires further attention.
There were only minor differences in the species delimited and topological relationships inferred between integrative and qualitative TTC methods of species detection and individual assignment (figure 3). Differences in the topological relationships among the different methods (figure 3) could be driven by differences in individual assignment [21], or by differences in the number of individuals included for each species [47]. By excluding individuals that had a different assignment in integrative clustering compared with their TTC assignment, the split between Sp.4 and 5 was recovered. These species are recently diverged with only secondary sexual characteristics distinguishing the two [30], which emphasizes the impact of recent divergence and potential hybridization on species detection and individual assignment. Additionally, integrative clustering detected a cluster within Sp.3 that is possibly more closely related to Sp.2 and unrecognized by TTC. This may indicate that our integrative framework is sensitive to over-splitting as opposed to under-detection, or that the current taxonomic criteria for the group are inadequate and cryptic taxa were undetected.
Despite attempts to ensure that species delimitation remains integrative [15–17] and considerable work on statistical methods [2–9], the paradigm in species identification and individual assignment in statistical species delimitation relies mostly upon genetic data. Genetic species discovery may be useful for identifying groups needing appraisal and for higher level taxonomic resolution in diverse groups [48]. However, with shallow genetic divergence, our results show that species numbers might be dramatically underestimated using purely genetic methods (figure 2), and recovered topologies may misrepresent the evolutionary history if individual assignment is not correct (figure 3). These situations are often those where statistical species delimitation methods are deployed empirically, and we would urge extreme caution in using singular data-types for putative species detection and individual assignment to putative species.
Biodiversity crises have led to a push for more efficient ways to delimit species and the prevalent use of genetic data in statistical approaches has reduced consideration of morphological and ecological diversity, often implicit in sampling. Methodological advances have brought necessary statistical rigor to species delimitation, however, the use of singular data may misrepresent diversity. Our integrative method narrows the parameter space where species are undetected in species groups differentially diverging along multiple axes, by allowing a statistically integrative approach to species detection and individual assignment that can be validated using statistical species delimitation. The use of integrative data also provides cohesiveness between species detection and description in an integrative taxonomy. Furthermore, our method requires the user to carefully consider sampling design and which data-types are used by considering the axes of divergence within the group being assessed, therefore allowing for variance in the potential mechanisms driving speciation.
Acknowledgements
We thank M. J. Sistrom for discussion, A. Caccone for support, J. Melville, J. S. Keogh, L Joseph and M. N. Hutchinson for assistance with empirical data.
Data accessibility
Data deposited in Dryad: http://dx.doi.org/10.5061/dryad.mm11q.
Funding statement
Funding was to D.L.E. (Australian National Wildlife Collection) and to L.L.K. (National Science Foundation (DEB-07-15487)).
References
- 1.Wheeler QD, Raven PH, Wilson EO. 2004. Taxonomy: impediment or expedient? Science 303, 285 (doi:10.1126/science.303.5656.285) [DOI] [PubMed] [Google Scholar]
- 2.Knowles LL, Carstens BC. 2007. Delimiting species without monophyletic gene trees. Syst. Biol. 56, 887–895 (doi:10.1080/10635150701701091) [DOI] [PubMed] [Google Scholar]
- 3.Carstens BC, Dewey TA. 2010. Species delimitation using a combined coalescent and information-theoretic approach: an example from North American Myotis bats. Syst. Biol. 59, 400–414 (doi:10.1093/sysbio/syq024) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hausdorf B, Hennig C. 2010. Species delimitation using dominant and codominant multilocus markers. Syst. Biol. 59, 491–503 (doi:10.1093/sysbio/syq039) [DOI] [PubMed] [Google Scholar]
- 5.Leaché AD, Fujita MK. 2010. Bayesian species delimitation in West African forest geckos (Hemidactylus fasciatus). Proc. R. Soc. B 277, 3071–3077 (doi:10.1098/rspb.2010.0662) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O'Meara BC. 2010. New heuristic methods for joint species delimitation and species tree inference. Syst. Biol. 59, 59–73 (doi:10.1093/sysbio/syp077) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ence DD, Carstens BC. 2011. SpedeSTEM: a rapid and accurate method for species delimitation. Mol. Ecol. Res. 11, 473–480 (doi:10.1111/j.1755-0998.2010.02947.x) [DOI] [PubMed] [Google Scholar]
- 8.Yeates DK, Seago A, Nelson L, Cameron SL, Joseph L, Trueman JWH. 2010. Integrative taxonomy, or iterative taxonomy? Syst. Entomol. 36, 209–217 (doi:10.1111/j.1365-3113.2010.00558.x) [Google Scholar]
- 9.Camargo A, Morando M, Avila LJ, Sites JW., Jr 2012. Species delimitation with ABC and other methods: a test of accuracy with simulations and an empirical example with lizards of the Liolaemus darwinii complex (Squamata: Liolaemidae). Syst. Biol. 66, 2834–3849 [DOI] [PubMed] [Google Scholar]
- 10.de Queiroz K. 2007. Species concepts and species delimitation. Syst. Biol. 56, 879–886 (doi:10.1080/10635150701701083) [DOI] [PubMed] [Google Scholar]
- 11.Hausdorf B. 2011. Progress toward a general species concept. Evolution 65, 923–931 (doi:10.1111/j.1558-5646.2011.01231.x) [DOI] [PubMed] [Google Scholar]
- 12.Fujita MK, Leaché AD, Burbrink FT, McGuire JA, Moritz C. 2012. Coalescent-based species delimitation in an integrative taxonomy. Trends Ecol. Evol. 27, 480–488 (doi:10.1016/j.tree.2012.04.012) [DOI] [PubMed] [Google Scholar]
- 13.Bauer AM, et al. 2011. Availability of new Bayesian-delimited gecko names and the importance of character-based species descriptions. Proc. R. Soc. B 278, 490–492 (doi:10.1098/rspb.2010.1330) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fujita MK, Leaché AD. 2011. A coalescent perspective on delimiting and naming species: a reply to Bauer et al. Proc. R. Soc. B 278, 493–495 (doi:10.1098/rspb.2010.1864) [Google Scholar]
- 15.Wiens JJ, Penkrot TA. 2002. Delimiting species using DNA and morphological variation and discordant species limits in spiny lizards (Sceloperus). Syst. Biol. 51, 69–91 (doi:10.1080/106351502753475880) [DOI] [PubMed] [Google Scholar]
- 16.Rissler LJ, Apodaca JJ. 2007. Adding more ecology into species delimitation: Ecological niche models and phylogeography help define cryptic species in the Black Salamander (Aneides flavipunctatus). Syst. Biol. 56, 924–942 (doi:10.1080/10635150701703063) [DOI] [PubMed] [Google Scholar]
- 17.Sistrom MJ, Donnellan SC, Hutchinson MN. 2013. Delimiting species in recent radiations with low levels of morphological divergence: a case study in Australian Gehyra geckos. Mol. Phyl. Evol. 68, 135–143 (doi:10.1016/j.ympev.2013.03.007) [DOI] [PubMed] [Google Scholar]
- 18.Pardial JM, Miralles A, De la Riva I, Vences M. 2010. The integrative future of taxonomy. Front. Zool. 7, 16 (doi:10.1186/1742-9994-7-16) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu L. 2008. BEST: Bayesian estimation of species trees under the coalescent model. Bioinformatics 24, 2542–2543 (doi:10.1093/bioinformatics/btn484) [DOI] [PubMed] [Google Scholar]
- 20.Kubatko LS, Carstens BC, Knowles LL. 2009. STEM: species tree estimation using maximum likelihood for gene trees under coalescence. Bioinformatics 25, 971–973 (doi:10.1093/bioinformatics/btp079) [DOI] [PubMed] [Google Scholar]
- 21.Heled J, Drummond AJ. 2010. Bayesian inference of species trees from multilocus data. Mol. Biol. Evol. 27, 570–580 (doi:10.1093/molbev/msp274) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang Z, Rannala B. 2010. Bayesian species delimitation using multilocus sequence data. Proc. Natl Acad. Sci. USA 107, 9264–9269 (doi:10.1073/pnas.0913022107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huelsenbeck JP, Andolfatto P. 2007. Inference of population structure under a Dirichlet process model. Genetics 175, 1787–1802 (doi:10.1534/genetics.106.061317) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zapata F, Jiménez I. 2012. Species delimitation: inferring gaps in morphology across geography. Syst. Biol. 61, 179–194 (doi:10.1093/sysbio/syr084) [DOI] [PubMed] [Google Scholar]
- 25.Sobel JM, Chen GF, Watt LR, Schemske DW. 2010. The biology of speciation. Evolution 64, 295–315 (doi:10.1111/j.1558-5646.2009.00877.x) [DOI] [PubMed] [Google Scholar]
- 26.Carstens BC, Pelletier TA, Reid NM, Salter JD. 2013. How to fail at species delimitation. Mol. Ecol. 22, 4369–4383 (doi:10.1111/mec.12413) [DOI] [PubMed] [Google Scholar]
- 27.Elmer KR, Lehtonen TK, Kautt AF, Harrod C, Meyer A. 2010. Rapid sympatric ecological differentiation of crater lake cichlid fishes within historic times. BMC Biol. 8, 60 (doi:10.1186/1741-7007-8-60) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harrington RC, Near TJ. 2012. Phylogenetic and coalescent strategies of species delimitation in Snubnose Darters (Percidae: Etheostoma). Syst. Biol. 61, 63–79 (doi:10.1093/sysbio/syr077) [DOI] [PubMed] [Google Scholar]
- 29.Wagner CE, Keller I, Wittwer S, Selz OM, Mwaiko S, Greuter L, Sivasundar A, Seehausen O. 2013. Genome-wide RAD sequence data provide unprecedented resolution of species boundaries and relationships in the Lake Victoria cichlid adaptive radiation. Mol. Ecol. 22, 787–798 (doi:10.1111/mec.12023) [DOI] [PubMed] [Google Scholar]
- 30.Edwards DL, Melville J, Joseph L, Keogh JS. In revision. Ecological divergence, adaptive radiation and the evolution of sexual signaling traits in a complex of Australian agamid lizards. Evolution. [DOI] [PubMed] [Google Scholar]
- 31.Gower JC. 1971. A general coefficient of similarity and some of its properties. Biometrics 27, 857–871 (doi:10.2307/2528823) [Google Scholar]
- 32.Maechler M. 2010. Cluster 1.13.2 See http://cran.r-project.org/web/packages/cluster/index.html
- 33.Tamura K, Peterson D, Peterson N, Stecher G, Neo M, Kumar S. 2011. Mega5: Molecular evolutionary genetic analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739 (doi:10.1093/molbev/msr121) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jukes TH, Cantor CR. 1969. Evolution of protein molecules. In Mammalian protein metabolism (ed. Munro HN.), pp. 21–123 New York, NY: Academic Press [Google Scholar]
- 35.Venables WN, Ripley BD. 2002. Modern applied statistics with S, 4th edn New York, NY: Springer [Google Scholar]
- 36.Zhang C, Zhang D-X, Zhu T, Yang Z. 2011. Evaluation of a Bayesian coalescent method of species delimitation. Syst. Biol. 60, 747–761 (doi:10.1093/sysbio/syr071) [DOI] [PubMed] [Google Scholar]
- 37.Drummond AJ, Rambaut A. 2007. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7, 214 (doi:10.1186/1471-2148-7-214) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fraley C, Raftery AE. 2002. Model-based clustering, discriminant analysis, and density estimation. J. Am. Stat. Assoc. 97, 611–631 (doi:10.1198/016214502760047131) [Google Scholar]
- 39.Storr GM. 1965. The Amphibolurus maculatus species group (Lacertilia, Agamidae) in Western Australia. J. Roy. Soc. West. Aust. 48, 45–54 [Google Scholar]
- 40.Olave M, Solà E, Knowles LL. 2014 Upstream analyses create problems with DNA-based species delimitation. Syst. Biol. In press. [DOI] [PubMed] [Google Scholar]
- 41.Petit RJ, Excoffier L. 2009. Gene flow and species delimitation. Trends Ecol. Evol. 24, 386–393 (doi:10.1016/j.tree.2009.02.011) [DOI] [PubMed] [Google Scholar]
- 42.Degnan JH, Rosenberg NA. 2009. Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends Ecol. Evol. 24, 332–340 (doi:10.1016/j.tree.2009.01.009) [DOI] [PubMed] [Google Scholar]
- 43.Knowles LL. 2009. Estimating species trees: methods of phylogenetic analysis when there is incongruence across genes. Syst. Biol. 58, 463–467 (doi:10.1093/sysbio/syp061) [DOI] [PubMed] [Google Scholar]
- 44.Vähä J-P, Primmer CR. 2006. Efficiency of model-based Bayesian methods for detecting hybrid individuals under different hybridization scenarios and with different numbers of loci. Mol. Ecol. 15, 63–72 (doi:10.1111/j.1365-294X.2005.02773.x) [DOI] [PubMed] [Google Scholar]
- 45.Kerr JT, Ostrovsky M. 2003. From space to species: ecological applications for remote sensing. Trends Ecol. Evol. 18, 299–305 (doi:10.1016/S0169-5347(03)00071-5) [Google Scholar]
- 46.Isaac NJ, Mallet J, Mace GM. 2004. Taxonomic inflation: its influence on macroecology and conservation. Trends Ecol. Evol. 19, 464–469 (doi:10.1016/j.tree.2004.06.004) [DOI] [PubMed] [Google Scholar]
- 47.Huang H, He Q, Kubatko LS, Knowles LL. 2010. Sources of error inherent in species-tree estimation: impact of mutational and coalescent effects on accuracy and implications for choosing different methods. Syst. Biol. 59, 573–583 (doi:10.1093/sysbio/syq047) [DOI] [PubMed] [Google Scholar]
- 48.Tänzler R, Sagata K, Surbakti S, Balke M, Reidel A. 2012. DNA barcoding for community ecology – How to tackle a hyperdiverse, mostly undescribed Melanesian fauna. PLoS ONE 7, e28832 (doi:10.1371/journal.pone.0028832) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data deposited in Dryad: http://dx.doi.org/10.5061/dryad.mm11q.