

Abstract
A simultaneous action of several pro-fibrotic mediators appears relevant in the development of fibrosis. There are evidences that transforming growth factor-β (TGF-β)/Smad3 pathway forms with αvβ6 integrin, mammalian target of Rapamycin (mTOR) and peroxisome proliferator-activated receptor-γ (PPARγ) a complex signalling network with extensive crosstalk and strong effects on fibrosis development. The present study evaluated the expression of TGFβ, Smad3, αvβ6 integrin, mTOR and PPARγ in 2, 4, 6-trinitrobenzenesulphonic acid (TNBS)-induced colorectal fibrosis in Smad3 wild-type (WT) and null mice. Smad3 WT mice treated with TNBS developed a marked colorectal fibrosis and showed a concomitant up-regulation of TGFβ, Smad3, αvβ6 and mTOR and a reduction of PPARγ expression. On the other hand, Smad3 Null mice similarly treated with TNBS did not develop fibrosis and showed a very low or even absent expression of TGFβ, Smad3, αvβ6 and mTOR and a marked over-expression of PPARγ. At the same time the expression of α-smooth muscle actin (a marker of activated myofibroblasts), collagen I-III and connective tissue growth factor (a downstream effector of TGFβ/Smad3-induced extracellular matrix proteins) were up-regulated in Smad3 WT mice treated with TNBS compared to Null TNBS-treated mice. These preliminary results suggest a possible interaction between these pro-fibrotic molecules in the development of intestinal fibrosis.
Key words: intestinal fibrosis, integrins, TGF-β, SMAD, mTOR, PPAR, IBD
Introduction
In inflammatory bowel disease (IBD), as well as in other enteropathies, the chronic transmural damage elicits an excessive wound-healing response that may lead to fibrosis, strictures, stenosis and obstruction.1-3 Intestinal fibrosis results from an abnormal response to a chronic local injury and is characterized by abnormal production and deposition of extracellular matrix (ECM) proteins produced by activated myofibroblasts, which are also called ECM-producing cells.4-7 These cells are derived not only from resident mesenchymal cells (fibroblasts, sub-epithelial myofibroblasts and smooth muscle cells), but also from epithelial and endothelial cells (by a process known as epithelial/endothelial-mesenchymal transition), stellate cells, pericytes, as well as intestinal or bone marrow stem cells.3,6,7 The most important soluble factors that regulate the activation of ECM-producing cells include cytokines, chemokines, growth factors, components of the renin-angiotensin system (RAS), angiogenic factors, peroxisome proliferator-activated receptors (PPARs), mammalian target of Rapamycin (mTOR), and products of oxidative stress.8,9 Other molecules, such as matrix metalloproteinases (MMPs) and specific tissue inhibitors of metalloproteinases (TIMPs), are also involved in regulating ECM turnover. Timing, concentration and sources of the main pro-fibrotic mediators might affect their individual contribution to tissue remodelling and fibrosis. Furthermore, a simultaneous action of some pro-fibrotic mediators appears relevant in the development of fibrosis.
Transforming growth factor-β (TGF-β) appears to play a central role in regulating the development, proliferation, differentiation and activation of intestinal mesenchymal cells, as well as in fibrosis.3-7, 10
All three mammalian isoforms of TGF-β (β1, β2, β3) are expressed in the intestine that are synthesized and secreted in the latent form and must be activated before they can bind to their receptors and induce TGFβ-mediated effects. TGF-β1 is the most extensively studied and is considered the primary pro-fibrotic factor. Latency-associated protein (LAP) forms a non-covalent complex with TGF-β, termed latent complex which retains TGF-β in its inactive state until released.11 Sequestration and regulated release of active TGF-β from LAP in this complex provide a mechanism by which the biologic function of TGF-β is controlled at the cellular level. Latent TGF-β1 can be activated by both proteolytic and non-proteolytic mechanisms.11,12 Several proteases can release TGF-β1 from LAP-β1 including plasmin, urokinase-type and plasmin activators, tissue-type plasminogen activators, matrix metalloproteinases 2 and 9, and cathepsin.12 Non-proteolytic activation involves the interaction of LAP-β1 with another protein and induction of a conformational change thereby activating TGF-β1. LAP-β1 can bind to any of the αv-containing integrins, but not all integrins that bind LAP-β1 activate latent TGF-β1.11 Integrins are heterodimeric transmembrane proteins made up of α and βsubunits. Six, of the 24 currently described integrins are able to bind the RGD motif in the LAP of TGF-β. Four of these integrins (αvβ3, αvβ5, αvβ6 and αvβ8) are thought to be able to activate TGF-β. The role of integrin-mediated TGF-β activation in vivo has only been confirmed for the αvβ6 and αvβ8 integrins.11
Once activated, TGF-β1 binds to specific membrane receptors (TGF-βRI, TGF-βRII, TGF-β1RIII) leading to activation of intracellular transduction pathways. The canonical pathway is represented by Smad proteins.13,14 The activation of TGF-β receptors phosphorylates Smad2 and Smad3 which bind with the common mediated Smad4. The Smad2/3-Smad4 complex translocates into the nucleus where it regulates specific TGF-β target genes. TGF-beta signalling is negatively regulated by inhibitory Smad7. Besides Smads downstream pathways, TGF-β can also modulate, in a Smad/independent manner, other signal transduction pathways such ERK/cJUN/p38 MAP kinases and the phosphoinositide-3 kinase (PI3-K) and its downstream target Akt, also known as protein kinase B (PKB).13 Of the several fibrogenic molecules, αvβ6 integrin, mTOR and PPAR-γ appear to interact directly with TGF-β/Smad pathway.
Integrins regulate cell-cell and cell-extracellular matrix interactions, thus influencing growth, differentiation, and development, as well wound healing and development of fibrosis.15,16 αvβ6 is not expressed in normal condition, but it is up-regulated after tissue injury, in woung healing, in some types of epithelial cancers and in many human fibrotic diseases of various organs (skin, lung, kidney and liver).16-23 αvβ6 colocalizes with TGFβ. αvβ6 ligands include fibronectin, tenascin, vitronectin and LAP. Interaction with LAP activates latent TGF-β and promotes fibrosis. αvβ6 integrin inhibitors significantly reduce tissue levels of profibrogenic transcripts, such as procollagen α1(I), αSMA, TGFβ1, TGFβ2, connective tissue growth factor (CTGF), TIMP-1 and αvβ6 integrin itself. Inhibition of the αvβ6 integrin, a key activator of TGF-β, could be an attractive therapeutic strategy for fibrosis, as it may be possible to inhibit TGF-β at sites of αvβ6 integrin up-regulation without affecting other vital homeostatic roles of TGF-β.
mTOR, a phosphatidylinositol 3-kinase-related kinase (PIKK), forms at least two distinct complexes.24 The mTOR complex 1 (mTORC1) which is composed of mTOR, G protein beta subunit-like (GβL) and regulatory associated protein of TOR (Raptor) and control protein synthesis and cell growth and proliferation, as well as autophagy, angiogenesis and fibrosis. The mTOR complex 2 (mTORC2) consists of mTOR, GβL and Rapamycin-insensitive companion of TOR (Rictor) and is involved in the cell proliferation and survival, metabolic regulation and actin cytosckeleton organization. mTOR signalling is activated by hormones, growth factors, amino acid levels, stress and alterations in cellular energy status.24 mTOR inhibitors (mTORis) exerts direct antifibrotic activities both by reducing the number of fibroblast and myofibroblasts and by down-regulating the production of fibrogenic cytokines, such as IL-4, IL-6, IL-13, IL-17, and TGFβ1, and the synthesis of type I and III collagen.25-27 Their antifibrotic effectiveness has been reported in fibrotic diseases of various organs including skin, lung, kidney, liver and intestine.28-33
PPARs are nuclear receptors, which regulate gene transcription by binding to retinoid X receptors (RXR) as functional heterodimers in response to a variety of endogenous and exogenous ligands.9,34 Three different isoforms of PPARs have been identified, termed PPARα, PPARγ and PPARδ, each one encoded by specific genes. In particular the PPAR-γ isoform, identified mainly in the colorectal mucosa, but also in adipocytes, liver, vascular tissue and several inflammatory cells (monocytes and macrophages, dendritic cells, B and T cells) seems to be involved in several physiological processes, such as differentiation of adipocytes, glucose homeostasis, lipid metabolism, inflammatory and immune processes, as well as fibrosis.9,34 PPAR-γ activation seems to be strongly related to the TGFβ/Smads pathway. The stimulation of PPAR-γ with specific ligands interferes with the Smad3 pathway by directly antagonizing Smad3 or down-regulating CTGF expression (a downstream effector of TGFβ/Smad3-induced extracellular matrix proteins).9,35,36 There are evidences, therefore, that αvβ6, mTOR and PPARγ form with TGFβ/Smad3 pathway a complex signalling network with extensive crosstalk and strong effects on fibrosis development.
The aim of the present study was to evaluate the expression of TGβF, Smad3, αvβ6 integrin, mTOR and PPARγ in 2, 4, 6-trinitrobenzenesulphonic acid (TNBS) induced colorectal fibrosis in Smad3 wild-type (WT) and null mice.
Materials and Methods
Animals
Twenty healthy adult mice, (Black Swiss × 129SVJ strain) 5 weeks of age, were included in the study: 10 Smad3 wild-type (5 controls, 5 receiving TNBS) and 10 Smad3 null mice (5 controls, 5 receiving TNBS). All mice were maintained in a specific pathogen-free facility and routinely monitored. Mice were kept in microisolator cages and allowed free access to food and water.
The study protocol was approved by the Animal Research Committee of the University of L’Aquila, Italy.
Induction of colitis
Chronic colonic inflammation and fibrosis was induced in 5 Smad3 wild-type and 5 null mice, by weekly intrarectal administration of TNBS (Sigma Aldrich, Milan, Italy) under light anaesthesia according to the method previously reported.37 Each mouse received an incremental dose of TNBS over a 6-week period. At weeks I and II, mice received 0.5 mg of TNBS in 30% ethanol; at weeks III and IV, mice received 0.75 mg of TNBS in 45% ethanol; at weeks V and VI, mice received 1.0 mg of TNBS in 45% ethanol. The solution of TNBS-ethanol was administered in a total volume of 100 μL through a medical-grade polyurethane tube (diameter, 1 mm) the tip of which was positioned at 3 cm beyond the anus. Animals in the control groups (5 Smad3 wild-type and 5 null mice) received 100 μL of 0.9% saline instead of TNBS by enema. Animals were monitored daily for food and fluid intake and examined for signs of colitis including weight loss, diarrhoea, rectal bleeding and prolapse as well as signs of systemic inflammation such as piloerection, lethargy, and periorbital exudates.37
Sample recovery and preparation
Laparotomy was performed under anaesthesia; the entire large bowel was rapidly excised and placed in a Petri dish containing sterile saline solution. The presence of adhesions between the colon and adjacent organs was scored on a 0-2 scale.37 The colon was then opened longitudinally, rinsed with sterile saline solution, weighed, measured, and then attached to a wooden tongue depressor for the assessment of macroscopic lesions. The colonic tissue samples were then fixed in 4% buffered formaldehyde and embedded in paraffin for histological and immunohistochemistry studies.
Assessment of macroscopic and microscopic colonic lesions
The sum of the scores of colonic lesions including adhesions, strictures, dilation, thickness, oedema/hyperaemia and ulcers was expressed as total macroscopic score (maximum score possible = 12).37 Specimens obtained from the large bowel of all animals were washed and immediately immersed in 10% buffered formalin in phosphate buffer saline (PBS; pH 7·4) for 3 h at room temperature followed by the standard procedure for paraffin embedding. Serial 3-μm sections were stained with haematoxylin and eosin (H&E) to assess the degree of inflammation and with Masson’s Trichrome to detect connective tissue and fibrosis. Stained sections were then observed under an Olympus BX51 Light Microscope (Olympus, Optical Co. Ltd, Tokyo, Japan). Intestinal fibrosis was scored as absent, mild, or severe, depending on the density and extent of Trichrome-positive connective tissue staining and disruption of tissue architecture.37
Immunohistochemistry analysis
Colorectal specimens were promptly fixed with 10% buffered formalin in PBS (pH 7·4) for 3 h, dehydrated in graded ethanol, and embedded in a low-temperature fusion paraffin. Serial 3-μm sections were incubated for 40 min in methanol and 3% hydrogen peroxide solution and then rinsed in PBS. Thereafter, sections were incubated overnight at 4 °C with polyclonal antibodies (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) to α-SMA (sc-32251), Collagen I-III (sc-8784; sc-8781), CTGF (sc-14939), TGFβ (sc-146), Smad3 (sc-6202), and PPARγ (sc-7273), mTOR (Epitomics Inc., Burlingame, CA, USA; catalog 1612-1) used at a dilution of 1:100, 1:400, 1:200, 1:250, 1:100, 1:100 and 1:100, respectively, in PBS. Anti-αvβ6 integrin antibody (kindly provided by Biogen Idec, Cambridge, MA, USA) was used at a dilution of 1:100 in PBS.
Samples were then rinsed with PBS for 5 min and incubated with a labelled streptavidin-biotin-peroxidase conjugate kit (Dako LSAB, cod. K0675, Dako-Cytomation, Milan, Italy). After rinsing in PBS, for 10 min, the sections were incubated with 3,3-diaminobenzidinetetrahydrochloride (Sigma Aldrich) for 1-3 min. To control for specificity of the immune reaction, sections were incubated omitting the primary antibody (i.e. incubated only with the secondary antibody alone). Finally, samples were counterstained with Mayer’s Haematoxylin and observed under a photomicroscope (Olympus BX51 Light Microscopy; Olympus, Optical Co. Ltd.).
Statistical analysis
Statistical analyses were performed using the Kruskal-Wallis non-parametric ANOVA system. Post-hoc comparisons between pairs of groups were assessed by using Wilcoxon rank sum test. Results were expressed as means ±SD; a P-value <0.05 was considered statistically significant.
Results
Macroscopic findings
After TNBS treatment, the colons of Smad3 wild-type mice appeared, at macroscopic examination, significantly harder, thicker and shorter than those of the Smad3 null mice. The total macroscopic score was significantly higher in Smad3 WT mice treated with TNBS compared to that of Null TNBS-treated mice (5.40 0.89 vs 0.80 0.84, respectively, P<0.05).
Microscopic findings
In the untreated control mice, histological assessment showed normal morphological pattern and a similar connective tissue distribution both in Smad3 WT and null mice. In the TNBS-treated mice, marked changes were observed in the structure of the colon from Smad3 WT mice. The total microscopic score was significantly higher in Smad3 WT mice treated with TNBS compared to that of Null TNBS-treated mice (5.60 1.82 vs 1.80 0.84, respectively, P<0.05). A marked increase in connective tissue in the submucosa and serosa was found in Smad3 WT mice treated with TNBS compared to that in Null TNBS-treated mice (Figure 1). The degree of colonic fibrosis was significantly higher in WT treated with TNBS compared to that of Null TNBS-treated mice (1.60 0.55 vs 0.20 0.44, respectively, P<0.05).
Figure 1.
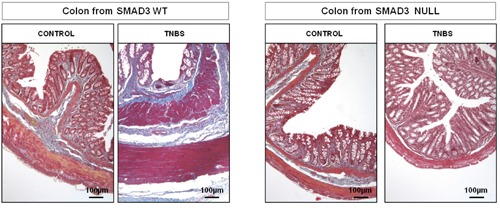
Masson’s Trichromic staining. Connective tissue distribution is similar in the two groups of control mice; in WT TNBS-treated mice a marked changes in colonic wall architecture due to abnormal deposition of connective tissue in lamina propria, submucosa and serosa were present, whereas the colonic wall of Null TNBS-treated mice is similar to that of untreated mice. Magnification: 10x.
Immunohistochemical evaluation
In WT control mice and in Null untreated and treated mice, α-SMA immunostaining, a marker of activated myofibroblasts, was localized in typical layers, while in the WT TNBS-treated mice α-SMA was more evident in muscolaris mucosae, muscularis externa and it was also present in submucosa and serosa layers (Figure 2).
Figure 2.
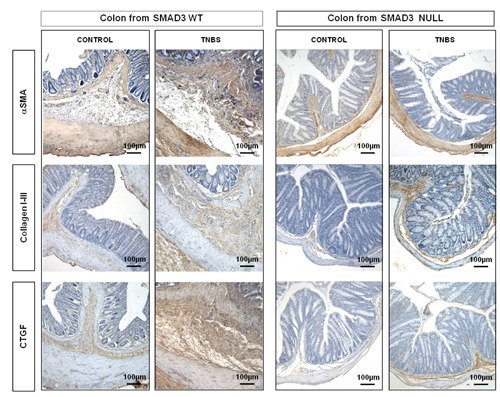
The αSMA expression is located in the typical areas (muscolaris mucosae and muscolaris propria) of Smad3 WT and Null control mice and in Smad3 Null TNBS-treated mice. Its expression is markedly increased in the colonic submucosa and serosa of Smad3 WT TNBS-treated mice. In TNBS-treated mice, collagen I-III and CTGF staining is markedly increased in lamina propria, submucosa and serosa layers from Smad3 WT mice compared to Null mice. Magnification: 10x.
In the untreated control mice, both WT and Null, collagen I-III and CTGF staining did not differ between the two groups and these were localized in the typical sides. In the WT TNBS-treated mice, collagen I-III and CTGF stainings were markedly increased in the lamina propria, submucosa and serosa layers compared to those of Null TNBS-treated mice (Figure 2). TGFβ1 and Smad3 stainings, absent in WT and Null control mice and in Null TNBS-treated mice, were present in the submucosa and serosa of WT TNBS-treated mice (Figure 3). In the untreated control mice, both WT and Null, αvβ6 staining was absent. In Smad3 WT mice treated with TNBS, αvβ6 immunostaining was increased in submucosa and in serosa whereas it was absent in Smad3 Null TNBS-treated mice (Figure 3). In WT and control mice, mTOR was expressed in epithelial cells and in the apical portion of lamina propria, while it was absent in Smad3 Null control mice. mTOR staining was slightly increased in the epithelium and in submucosa and serosa layers of WT mice treated with TNBS compared to that of Null TNBS-treated mice (Figure 4). In Null mice treated with TNBS, PPARγ staining was increased, both in the mucosa and in the submucosa layers, compared to WT TNBS-treated mice; its positivity was also more evident in Null control mice compared to WT controls (Figure 4).
Figure 3.
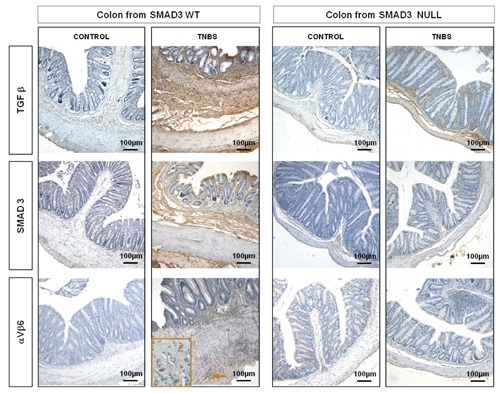
TGFβ1 and Smad3 stainings, absent in WT and Null control mice are markedly increased in lamina propria, submucosa and serosa layers in colon of Smad3 WT mice treated with TNBS compared to Null TNBS-treated mice. αVβ6 immunostaining is increased in submucosa in Smad3 WT TNBS-treated mice, whereas it is absent in Smad3 Null TNBS-treated mice. Magnification: 10x.
Figure 4.
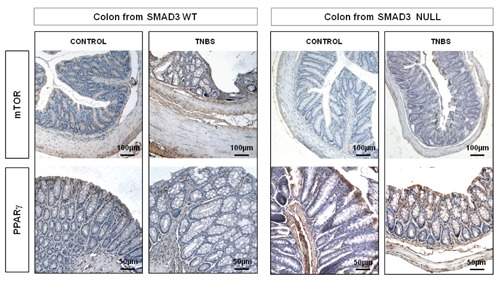
mTOR immunostainin: in the untreated WT and Null control mice mTOR is expressed in the epithelium and in the apical portion of lamina propria; mTOR staining is slightly increased in the epithelium, submucosa and serosa layers of Smad3 WT mice treated with TNBS compared to that of Smad3 Null TNBS-treated mice. Magnification: 10x. PPARγ immunostaining: overall, PPARγ staining is more marked in Smad3 Null mice, both in treated and untreated mice; it increased, both in the mucosa and submucosa layers, in Smad3 Null mice treated with TNBS compared to that of Smad3 WT TNBS-treated mice. Magnification: 20x.
Discussion
Intestinal fibrosis is a chronic and progressive process mediated by complex cell/matrix/cytokine and growth factor interaction,1,6-9 and TGFβ has long emerged as a prominent regulator of fibrogenesis determining onset and progression of fibrosis in many chronic diseases. TGFβ intracellular Smads transduction pathways appear to be crucial for development of fibrosis. Several studies have demonstrated that disruption of the TGFβ/Smad3 signalling pathway by the loss of Smad3 confers resistant to tissues fibrosis in several organs including skin, kidney, lung and liver.38-42 In a previous study we demonstrated that Smad3 Null mice are resistant to the development of experimental intestinal fibrosis induced by TNBS.37 Histological and morphometric evaluation revealed a significantly higher degree of colonic fibrosis and accumulation of collagen in the Smad3 wild-type compared to null mice. Immunohistochemical evaluation showed a marked increase in αSMA, collagen I-III, CTGF, TGFβ and Smad3 staining in the colon of Smad3 wild-type compared to null mice. All these findings have been confirmed by the present study.
In colonic fibrosis Smad3 could induce an abnormal activation of a fibrogenic phenotype of mesenchymal cells which turns into an increase of local deposition and accumulation of ECM proteins.1,5-7,9,10,37,43 The Smad2/3-Smad4 complex, by translocating into the nucleus, regulates specific pro-fibrogenic genes. Specifically, the target genes known to contain Smad-responsive regions and that are directly or indirectly involved in fibrogenesis, include several fibrillar ECM proteins (collagen, fibronectin), matrix-degrading enzymes (MMPs) and some specific inhibitors (TIMPs), as well as genes regulating epithelial-mesenchymal cell transition, proliferation (cyclin-dependent kinase inhibitor p21) and apoptosis (caspases).14,44
In this study we have evaluated whether TGFβ/Smad3 pathway and αvβ6 integrin, mTOR and PPARγ may interact in TNBS-induced colorectal fibrosis in Smad3 WT and Null mice. Smad3 WT mice treated with TNBS developed a marked colorectal fibrosis and showed a concomitant up-regulation of TGFβ, Smad3, αvβ6 and mTOR an a reduction of PPARγ expression. On the other hand, Smad3 Null mice similarly treated with TNBS did not develop fibrosis and showed a very low or even absent expression of TGFβ, Smad3, αvβ6 and mTOR and a marked overexpression of PPARγ. At the same time the expression of αSMA (a marker of activated myofibroblasts), collagen I-III and CTGF (a downstream effector of TGFβ/Smad3-induced ECM) were up-regulated in Smad3 WT mice treated with TNBS compared to Null TNBS-treated mice. These preliminary data suggest a possible interaction between the above-mentioned molecules in the development of intestinal fibrosis, findings that need to be confirmed by in vitro studies using human intestinal fibroblasts or myofibroblasts cultures. To the best of our knowledge, this is the first report underlining the role of avb6 in the intestinal fibrosis, finding that may have important clinical implications in all human fibrogenic enteropathies, specially in the IBD.
The specific interaction between TGFβ/Smad3 pathway and αvβ6 integrin, mTOR and PPARγ is still unclear. TGFβ isoforms are synthesized as latent molecules, consisting of mature TGFβ that is covalently bound to the LAP.13,14 This latent complex associates with a family member of the latent TGFβ binding proteins (LTBPs) that facilitates TGFβ storage in the ECM. To be functional, TGF-β must be activated. There are several activators of TGFβ that can dissociate the mature TGFβ from LAP, allowing it to interact with its cell surface signalling receptors.11,12,15-17 Integrin-mediated activation seems to be the main mechanism of TGFβ activation in vivo. αvβ6 integrin can activate fibrogenic TGFβ1 through a mechanism that requires LTBP-1. αvβ6 is not expressed in normal condition, but it is up-regulated after tissue injury in epithelial cells of skin, kidney, lung, liver and intestine, as well as in many human fibrotic diseases of various organs including skin, kidney, lung and liver.11,12,15-23,45-47 αvβ6 can lead to local activation of TGFβ1 generating new active growth factor and then maintaining the TGFβ mediated fibrotic process. Various genetic and pharmacologic interventions targeting the αvβ6 integrin have been shown to reduce the activation of TGFβ1 and fibrosis. Therefore, the αvβ6 blockade, could provide a new mechanism for injury specific attenuation of TGFβ activity and fibrosis.
Given its pleiotropic effects, TGFβ inhibition using strategies targeted to specific regions involved in fibrosis might be a better alternative.18 Most other approaches are currently under consideration for targeting TGFβ block either TGFβ receptors or TGFβ itself. These approaches might lead to unwanted side effects by interfering with important homeostatic effects of TGFβ at sites outside the organs affected by tissue fibrosis. Mice deficient in TGFβ1 exhibit uncontrolled tissue inflammation, autoimmunity, and premature death, demonstrating a critical role for TGFβ1 in immune homeostasis. Although mice lacking αvβ6 do have mild inflammation in kidney, lung and skin, these effects are much less severe than those seen in mice lacking even a single TGFβ isoform.18 Additionally, the αvβ6 integrin is highly upregulated in diseased tissue providing a mechanism for injury-induced TGFβ activation as compared to homeostatic control of TGFβ activity. By inhibiting a subset only of TGFβ activation, particularly in injured epithelial organs, targeting αvβ6 could allow treatment of tissue fibrosis with substantially reduced risk of disrupting beneficial homeostatic control of inflammation and immunity.18
Extensive interaction also exists between mTOR and TGFβ/Smads pathway which contributes to the proliferation of fibroblasts in many fibrotic disorders.25 The activation of TGF-β receptors promote the phosphorylation of the PI3-K, which is a branch point for the activation of Akt. Once activated, Akt phosphorylates the tuberous sclerosis complex (TSC) that negatively regulates mTORC1.48-50 Thus active TSC is an inhibitor of mTORC1 and loss of TSC activity increase mTORC1 activity which induces fibroblast and myofibroblast proliferation.25,28 mTOR inhibitors reduce the myofibroblasts and down-regulate the production of TGFβ1, and the synthesis of type I and III collagen.25-27 Their antifibrotic properties have been reported in fibrotic diseases of several organs including skin, lung, kidney, liver and intestine.28-33,51,52
PPAR-γ and TGFβ/Smads pathway activities seems to be strongly related. PPAR-γ ligands may directly antagonize Smad3 or down-regulate CTGF expression that promotes the TGF-induced synthesis of collagen.9,35,36 PPARγ agonists inhibit the fibroblast migration and proliferation53 as well as the transdifferentiation of epithelial and mesenchymal cells in activated myofibroblasts,54 one of the key points in fibrosis development. PPAR-γ ligands repress TGFβ-induced myofibroblast differentiation and activation by targeting the PI3K/Akt and Smad3 pathways, respectively.55,56 Overexpression of PPARγ prevents the development of tissue fibrosis, whereas its loss increases susceptibility to fibrosis.57,58 All these findings could explain the ability of PPARγ to interfere in multiple phases of the tissue fibrotic processes. Therefore, PPARγ should be regarded as an innate protection from excessive fibrogenesis and a potential new target for the development of novel compounds with antifibrotic properties.59 Several PPARγ ligands with selective activity are under development. Experimental studies have shown that PPARγ agonists attenuate fibrosis in various organs including lung, kidney, pancreas, liver and intestine, antifibrotic effects that are abolished by the use of a PPARγ selective antagonists.60-65
Given all this, the data obtained suggest that the development of intestinal fibrosis could be influenced not only by TGFβ-Smads signalling but also by αvβ6 integrin, mTOR and PPARγ in a crosstalk integrated system. αvβ6 integrin may act by stimulating TGFβ canonical (mediated by Smads) and non-canonical (mediated by mTOR) intracellular pathways. Increased expression of αvβ6 integrin, TGFβ, Smad3 and mTOR is associated to the development of fibrosis, whereas up-regulation of PPARγ appears to be protective towards fibrosis. Selective Smad3 disruption affects the expression of all these molecules and their effects on TNBS-induced colorectal fibrosis.
References
- 1.Rieder F, Brenmoehl J, Leeb S, Scholmerich J, Rogler G. Wound healing and fibrosis in intestinal disease. Gut. 2007;56:130- 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burke JP, Mulsow JJ, O’Keane C, Docherty NG, Watson RW, O’Connell PR. Fibrogenesis in Crohn’s disease. Am J Gastroenterol. 2007;102:439-48 [DOI] [PubMed] [Google Scholar]
- 3.Rieder F, Fiocchi C. Intestinal fibrosis in inflammatory bowel disease-Current knowledge and future perspectives. J Crohns Colitis. 2008;2:279-90 [DOI] [PubMed] [Google Scholar]
- 4.Powell DW, Mifflin RC, Valentich JD, Crowe SE, Saada JI, West AB., Myofibroblasts I. Paracrine cells important in health and disease. Am J Physiol Cell Physiol. 1999;277:C1-19 [DOI] [PubMed] [Google Scholar]
- 5.Pucilowska JB, Williams KL, Lund PK. Fibrogenesis. IV. Fibrosis and inflammatory bowel disease: cellular mediators and animal models. Am J Physiol Gastrointest Liver Physiol. 2000;279:G653-9 [DOI] [PubMed] [Google Scholar]
- 6.Fiocchi C, Lund PK. Themes in fibrosis and gastrointestinal inflammation. Am J Physiol Gastrointest Liver Physiol. 2011; 300:G677-83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rieder F, Fiocchi C. Intestinal fibrosis in IBD-a dynamic, multifactorial process. Nat Rev Gastroenterol Hepatol. 2009;6:228-35 [DOI] [PubMed] [Google Scholar]
- 8.Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199-210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Speca S, Giusti I, Rieder R, Latella G. Cellular and molecular mechanisms of intestinal fibrosis. World J Gastroenterol. 2012;18:3635-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mifflin RC, Pinchuk IV, Saada JI, Powell DW. Intestinal myofibroblasts: targets for stem cell therapy. Am J Physiol Gastrointest Liver Physiol. 2011;300:G684-96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wipff PJ, Hinz B. Integrins and the activation of latent transforming growth factor beta1 – An intimate relationship. Eur J Cell Biol. 2008;87:601-15 [DOI] [PubMed] [Google Scholar]
- 12.Jenkins G. The role of proteases in transforming growth factor-beta activation. Int J Biochem Cell Biol. 2008;40:1068-78 [DOI] [PubMed] [Google Scholar]
- 13.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature. 2003;425:577-84 [DOI] [PubMed] [Google Scholar]
- 14.Roberts AB, Russo A, Felici A, Flanders KC. Smad3: a key player in pathogenetic mechanisms dependent on TGF-beta. Ann NY Acad Sci. 2003;995:1-10 [DOI] [PubMed] [Google Scholar]
- 15.van der Flier A, Sonnenberg A. Function and interactions of integrins. Cell Tissue Res. 2001;305:285-98 [DOI] [PubMed] [Google Scholar]
- 16.Margadant C, Sonnenberg A. Integrin-TGF-beta crosstalk in fibrosis, cancer and wound healing. EMBO Rep. 2010;11:97-105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Margadant C, Monsuur HN, Norman JC, Sonnenberg A. Mechanisms of integrin activation and trafficking. Curr Opin Cell Biol. 2011;23:607-14 [DOI] [PubMed] [Google Scholar]
- 18.Katsumoto TR, Violette SM, Sheppard D. Blocking TGFβ via inhibition of the αvβ6 integrin: a possible therapy for systemic sclerosis interstitial lung disease. Int J Rheumatol. 2011;2011:208219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Puthawala K, Hadjiangelis N, Jacoby SC, Bayongan E, Zhao Z, Yang Z, et al. Inhibition of integrin alpha(v)beta6, an activator of latent transforming growth factor-beta, prevents radiation-induced lung fibrosis. Am J Respir Crit Care Med. 2008;177:82-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horan GS, Wood S, Ona V, Li DJ, Lukashev ME, Weinreb PH, et al. Partial inhibition of integrin alpha(v)beta6 prevents pulmonary fibrosis without exacerbating inflammation. Am J Respir Crit Care Med. 2008;177:56-65 [DOI] [PubMed] [Google Scholar]
- 21.Hahm K, Lukashev ME, Luo Y, Yang WJ, Dolinski BM, Weinreb PH, et al. Alphav beta6 integrin regulates renal fibrosis and inflammation in Alport mouse. Am J Pathol. 2007;170:110-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nadler EP, Patterson D, Violette S, Weinreb P, Lewis M, Magid MS, et al. Integrin alphavbeta6 and mediators of extracellular matrix deposition are up-regulated in experimental biliary atresia. J Surg Res. 2009;154:21-9 [DOI] [PubMed] [Google Scholar]
- 23.Sullivan BP, Weinreb PH, Violette SM, Luyendyk JP. The coagulation system contributes to alphaVbeta6 integrin expression and liver fibrosis induced by cholestasis. Am J Pathol. 2010;177:2837-49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsang CK, Qi H, Liu LF, Zheng XF. Targeting mammalian target of rapamycin (mTOR) for health and diseases. Drug Discov Today. 2007;12:112-24 [DOI] [PubMed] [Google Scholar]
- 25.Wang S, Wilkes MC, Leof EB, Hirschberg R. Noncanonical TGF-beta pathways, mTORC1 and Abl, in renal interstitial fibrogenesis. Am J Physiol Renal Physiol. 2010;298:F142-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Poulalhon N, Farge D, Roos N, Tacheau C, Neuzillet C, Michel L, et al. Modulation of collagen and MMP-1 gene expression in fibroblasts by the immunosuppressive drug rapamycin. A direct role as an antifibrotic agent? J Biol Chem. 2006;281:33045-52 [DOI] [PubMed] [Google Scholar]
- 27.Osman B, Akool el-S, Doller A, Müller R, Pfeilschifter J, Eberhardt W. Differential modulation of the cytokine-induced MMP-9/TIMP-1 protease-antiprotease system by the mTOR inhibitor rapamycin. Biochem Pharmacol. 2011;81:134-43 [DOI] [PubMed] [Google Scholar]
- 28.Lieberthal W, Levine JS. The role of the mammalian target of rapamycin (mTOR) in renal disease. J Am Soc Nephrol. 2009;20:2493-502 [DOI] [PubMed] [Google Scholar]
- 29.Korfhagen TR, Le Cras TD, Davidson CR, Schmidt SM, Ikegami M, Whitsett JA, et al. Rapamycin prevents transforming growth factor-alpha-induced pulmonary fibrosis. Am J Respir Cell Mol Biol. 2009;41:562-72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Whaley-Connell A, Habibi J, Panfili Z, Hayden MR, Bagree S, Nistala R, et al. Angiotensin II activation of mTOR results in tubulointerstitial fibrosis through loss of N-cadherin. Am J Nephrol. 2011;34:115-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Neef M, Ledermann M, Saegesser H, Schneider V, Reichen J. Low-dose oral rapamycin treatment reduces fibrogenesis, improves liver function, and prolongs survival in rats with established liver cirrhosis. J Hepatol. 2006;45:786-96 [DOI] [PubMed] [Google Scholar]
- 32.Patsenker E, Schneider V, Ledermann M, Saegesser H, Dorn C, Hellerbrand C, et al. Potent antifibrotic activity of mTOR inhibitors sirolimus and everolimus but not of cyclosporine A and tacrolimus in experimental liver fibrosis. J Hepatol. 2011;55:388-98 [DOI] [PubMed] [Google Scholar]
- 33.Yoshizaki A, Yanaba K, Yoshizaki A, Iwata Y, Komura K, Ogawa F, et al. Treatment with rapamycin prevents fibrosis in tight-skin and bleomycin-induced mouse models of systemic sclerosis. Arthritis Rheum. 2010;62:2476-87 [DOI] [PubMed] [Google Scholar]
- 34.Rousseaux C, Desreumaux P. The peroxisome-proliferator-activated gamma receptor and chronic inflammatory bowel disease (PPARgamma and IBD). J Soc Biol. 2006;200:121-31 [DOI] [PubMed] [Google Scholar]
- 35.Zhao C, Chen W, Yang L, Chen L, Stimpson SA, Diehl AM. PPARgamma agonists prevent TGFbeta1/Smad3-signaling in human hepatic stellate cells. Biochem Biophys Res Commun. 2006;350:385-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang GY, Cheng T, Zheng MH, Yi CG, Pan H, Li ZJ, et al. Activation of peroxisome proliferator-activated receptor-gamma inhibits transforming growth factor-beta1 induction of connective tissue growth factor and extracellular matrix in hypertrophic scar fibroblasts in vitro. Arch Dermatol Res. 2009;301:515-22 [DOI] [PubMed] [Google Scholar]
- 37.Latella G, Vetuschi A, Sferra R, Zanninelli G, D’Angelo A, Catitti V, et al. Smad3 loss confers resistance to the development of trinitrobenzene sulfonic acid-induced colorectal fibrosis. Eur J Clin Invest. 2009;39:145-56 [DOI] [PubMed] [Google Scholar]
- 38.Lakos G, Tacagawa S, Chen SJ, Ferreira AM, Han G, Wang XJ, et al. Targeted disruption of TGF-beta/Smad3 signaling modulates skin fibrosis in a mouse model of scleroderma. Am J Pathol. 2004;165:203-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao J, Shi W, Wang YL, Chen H, Bringas P, Datto MB, et al. Smad3 deficiency attenuates bleomycin-induced pulmonary fibrosis in mice. Am J Physiol Lung Cell Mol Physiol. 2002;282:L585-93 [DOI] [PubMed] [Google Scholar]
- 40.Inazaki K, Kanamaru Y, Kojima Y, Sueyoshi N, Okumura K, Kaneko K, et al. Smad3 deficiency attenuates renal fibrosis, inflammation, and apoptosis after unilateral ureteral obstruction. Kidney Int. 2004;66:597-604 [DOI] [PubMed] [Google Scholar]
- 41.Latella G, Vetuschi A, Sferra R, Catitti V, D’Angelo A, Zanninelli G, et al. Targeted disruption of Smad3 confers resistance to the development of dimethylnitrosamine-induced hepatic fibrosis in mice. Liver Int. 2009;29:997-1009 [DOI] [PubMed] [Google Scholar]
- 42.Flanders KC. Smad3 as a mediator of the fibrotic response. Int J Exp Pathol. 2004;85:47-64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zanninelli G, Vetuschi A, Sferra R, D’Angelo A, Fratticci A, Continenza MA, et al. Smad3 knock-out mice as a useful model to study intestinal fibrogenesis. World J Gastroenterol. 2006;12:1211-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Verrecchia F, Chu ML, Mauviel A. Identification of novel TGF-b/smad gene targets in dermal fibroblasts using a combined cDNA microarray/promoter transactivation approach. J Biol Chem. 2001;276:17058-62 [DOI] [PubMed] [Google Scholar]
- 45.Tatler AL, Jenkins G. TGF-β activation and lung fibrosis. Proc Am Thorac Soc. 2012;9:130-6 [DOI] [PubMed] [Google Scholar]
- 46.Patsenker E, Popov Y, Stickel F, Jonczyk A, Goodman SL, Schuppan D. Inhibition of integrin alphavbeta6 on cholangiocytes blocks transforming growth factor-beta activation and retards biliary fibrosis progression. Gastroenterology. 2008;135:660-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen G, Zhang L, Chen L, Wang H, Zhang Y, Bie P.Role of integrin αvβ6 in the pathogenesis of ischemia-related biliary fibrosis after liver transplantation. Transplantation. 2013;95:1092-9 [DOI] [PubMed] [Google Scholar]
- 48.Huang J, Manning BD. The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem J. 2008;412:179-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Inoki K. Role of TSC-mTOR pathway in diabetic nephropathy. Diabetes Res Clin Pract. 2008;82:S59-62 [DOI] [PubMed] [Google Scholar]
- 50.Huang J, Manning BD. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem Soc Trans. 2009;37:217-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Massey DC, Bredin F, Parkes M. Case report Use of sirolimus (rapamycin) to treat refractory Crohn’s disease. Gut. 2008;57:1294-6 [DOI] [PubMed] [Google Scholar]
- 52.Dumortier J, Lapalus MG, Guillaud O, Poncet G, Gagnieu MC, Partensky C, et al. Everolimus for refractory Crohn’s disease: a case report. Inflamm Bowel Dis. 2008;14:874-7 [DOI] [PubMed] [Google Scholar]
- 53.Lin Q, Fang LP, Zhou WW, Liu XM. Rosiglitazone inhibits migration, proliferation, and phenotypic differentiation in cultured human lung fibroblasts. Exp Lung Res. 2010;36:120-8 [DOI] [PubMed] [Google Scholar]
- 54.Tan X, Dagher H, Hutton CA, Bourke JE. Effects of PPAR gamma ligands on TGF-beta1-induced epithelial-mesenchymal transition in alveolar epithelial cells. Respir Res. 2010;11:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kulkarni AA, Thatcher TH, Olsen KC, Maggirwar SB, Phipps RP, Sime PJ. PPAR-γ ligands repress TGFβ-induced myofibroblast differentiation by targeting the PI3K/Akt pathway: implications for therapy of fibrosis. PLoS One. 2011;6:e15909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhao C, Chen W, Yang L, Chen L, Stimpson SA, Diehl AM. PPARgamma agonists prevent TGFbeta1/Smad3-signaling in human hepatic stellate cells. Biochem Biophys Res Commun. 2006;350:385-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nan YM, Han F, Kong LB, Zhao SX, Wang RQ, Wu WJ, et al. Adenovirus-mediated peroxisome proliferator activated receptor gamma overexpression prevents nutritional fibrotic steatohepatitis in mice. Scand J Gastroenterol. 2011;46:358-69 [DOI] [PubMed] [Google Scholar]
- 58.Kapoor M, McCann M, Liu S, Huh K, Denton CP, Abraham DJ, et al. Loss of peroxisome proliferator-activated receptor gamma in mouse fibroblasts results in increased susceptibility to bleomycin-induced skin fibrosis. Arthritis Rheum. 2009;60:2822-9 [DOI] [PubMed] [Google Scholar]
- 59.Wei J, Ghosh AK, Sargent JL, Komra K, Wu M, Huang QQ, et al. PPARγ downregulation by TGFß in fibroblast and impaired expression and function in systemic sclerosis: a novel mechanism for progressive fibrogenesis. PLoS One. 2010;5:e13778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang L, Stimpson SA, Chen L, Wallace Harrington W, Rockey DC. Effectiveness of the PPARγ agonist, GW570, in liver fibrosis. Inflamm Res. 2010;59:1061-71 [DOI] [PubMed] [Google Scholar]
- 61.Wu M, Melichian DS, Chang E, Warner-Blankenship M, Ghosh AK, Varga J. Rosiglitazone abrogates bleomycin-induced scleroderma and blocks profibrotic responses through peroxisome proliferator-activated receptor-gamma. Am J Pathol. 2009;174:519-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kawai T, Masaki T, Doi S, Arakawa T, Yokoyama Y, Doi T, et al. PPAR-gamma agonist attenuates renal interstitial fibrosis and inflammation through reduction of TGF-beta. Lab Invest. 2009;89:47-58 [DOI] [PubMed] [Google Scholar]
- 63.Aoki Y, Maeno T, Aoyagi K, Ueno M, Aoki F, Aoki N, et al. Pioglitazone, a peroxisome proliferator-activated receptor gamma ligand, suppresses bleomycin-induced acute lung injury and fibrosis. Respiration. 2009;77:311-9 [DOI] [PubMed] [Google Scholar]
- 64.Talukdar R, Tandon RK. Pancreatic stellate cells: new target in the treatment of chronic pancreatitis. J Gastroenterol Hepatol. 2008;23:34-41 [DOI] [PubMed] [Google Scholar]
- 65.Chen H, He YW, Liu WQ, Zhang JH. Rosiglitazone prevents murine hepatic fibrosis induced by Schistosoma japonicum. World J Gastroenterol. 2008;14:2905-11 [DOI] [PMC free article] [PubMed] [Google Scholar]