

Abstract
The nephron number at birth is a quantitative trait that correlates inversely with the risk of hypertension and chronic kidney disease later in life. During kidney development, the nephron number is controlled by multiple factors including genetic, epige-netic, and environmental modifiers. Premature birth, which represents more than 12% of annual live births in the United States, has been linked to low nephron number and the development of hypertension later in life. In this report, we describe the development of a mouse model of prematurity-induced reduction of nephron number. Premature mice, delivered 1 and 2 days early, have 17.4 ± 2.3% (n = 6) and 23.6 ± 2% (n = 10) fewer nephrons, respectively, when compared with full-term animals (12,252 ± 571 nephrons/kidney, n = 10). After 5 weeks of age, the mice delivered 2 days premature show lower real-time glomerular filtration rate (GFR, 283 ± 13 vs 389 ± 26 μL/min). The premature mice also develop hypertension (mean arterial pressure (MAP), 134 ± 18 vs 120 ± 14 mm Hg) and albuminuria (286 ± 83 vs 176 ± 59 μg albumin/mg creatinine). This mouse model provides a proof of concept that prematurity leads to reduced nephron number and hypertension, and this model will be useful in studying the pathophysiology of prematurity-induced nephron number reductions and hypertension.
Premature birth before 37 weeks of gestation is a global health problem with immediate high infant mortality and morbidity, and often leads to an increased risk for adulthood diseases. In 2009, 12.1% of live births in the United States were preterm with 3.48% of infants born before 33 weeks of gestation.1,2 The incidence of preterm birth varies by race and ethnicity, and it is highest in African Americans (33%) compared to all other groups.3 Prematurity and low birth weight are independent, quantitative risk factors for the development of hypertension in adults.4–7 Premature females examined at 26 years of age had significantly higher systolic blood pressure compared to full-term but small-for-gestational-age controls.4,5 Cohorts from the Netherlands showed that prematurely born children have a higher blood pressure than full term but small-for-gestational-age children, and both groups had higher blood pressure than full-term, normal-birth-weight individuals.8
The fetal origin of adult disease was postulated more than 2 decades ago by David Barker and is known as the “Barker hypothesis.” The essence of the hypothesis is that intrauterine events alter organ programming leading to lifelong increased incidence of adult diseases including hypertension, metabolic syndrome, and coronary artery disease. This hypothesis is supported by multiple epidemiologic cohorts.9–11
A reduced nephron number is an important risk for development of hypertension and chronic kidney disease.12–14 The relationship between nephron number and hypertension was illustrated in a sentient report by Keller et al15 who determined the nephron number in autopsy samples and found that subjects who were hypertensive have, on average, half the number of nephrons of nonhypertensive individuals.15 No noninvasive method of counting nephrons in live individuals is available, and birth weight is used as a surrogate for the nephron number. Birth weight correlates directly to the nephron number, and this measurement was valid for both white and African American subjects.16 Moreover, birth weight showed a significant correlation to the odds of developing chronic kidney disease.17 Underdeveloped kidneys containing fewer nephrons frequently is adduced to be the link between adult hypertension and kidney disease as an outcome and intrauterine growth retardation (IGUR) with or without prematurity as risk factors.7,18
Multiple murine models of a reduced nephron number have been generated by genetically engineering mice to knockout genes critical in nephrogenesis (eg, Gdnf, Six2, Fgfr2, and Sall1). Surviving mice with mutations in these critical genes are born with fewer nephrons compared with the wild type and develop higher arterial blood pressure.19–22 Although a genetic mutation in these nephrogenesis critical genes is reported in human syndromic diseases with a reduced nephron number, for example, renal coloboma syndrome and Townes-Brock syndrome,23,24 the incidence of such genetic mutations in human is dwarfed by the incidence of prematurity. This makes premature birth, especially extreme prematurity, a prominent risk factor for adult hypertension and kidney disease. The mechanisms of prematurity-induced reduction of nephron number are not clear, and animal models can provide an excellent experimental system that might help elucidate this link. In this report, we developed a murine model for studying premature birth and its effect on kidney development and nephron number, and we characterized effect of prematurity-induced nephron number reduction on blood pressure and renal function.
MATERIALS AND METHODS
Animals, housing, and diet
The study was conducted in a 60,000 sq ft Association for Assessment and Accreditation of Laboratory Animal Care accredited animal facility devoted exclusively to housing rodents. All experimental procedures involving animals in the facility were reviewed and approved by the Medical College of Wisconsin Institutional Animal Care and Use Committee. Experimental procedures are performed in compliance with the Guide for the Care and Use of Laboratory Animals.25,26 Accordingly, relevant ethical guidelines for animal research were all enforced.
Animals were housed in ventilated racks (Model No. MS75JU70MVSPSHR-R; Allentown Inc, Allentown, NJ). All rodent food was purchased commercially and irradiated prior to receipt at the facility. All caging and cage accessories were autoclaved prior to use. Cage changing was performed in laminar flow hoods (NuAire Allegard Dual Access Small Animal Cage Changing and Transfer Stations; models 612 and 617; NuAire, Inc, Plymouth, Minn) present in each animal holding room. The animals were fed an autoclaved breeder diet (5K67, Purina LabDiet; PMI Nutrition International, LLC, Brentwood, Mo). The light cycle in the animal holding rooms was 14/10 at 4 AM and 6 PM.
Cesarean delivery (C-section)
Adult, timed-pregnant CD-1 mice (Charles River Laboratories, Wilmington, Mass) were purchased. The animals were ordered timed pregnant with the pregnancy staggered days apart to allow foster animals to deliver pups prior to use of experimental animals in which pups would be collected prematurely by C-section at specified days prior to anticipated parturition.
At the specified gestational date, the female was euthanized by CO2 exposure. As soon as CO2 anesthesia was observed, cervical dislocation was then performed to ensure humane euthanasia. To reduce bacterial contamination of the abdominal cavity, the abdominal skin was prepped by application of isopropyl alcohol and the abdominal skin was retracted. Additional preparation of the external tissue included use of Clidox-S (Pharmacal Research Labs, Inc, Naugatuck, Conn). The abdomen was incised using a clean scalpel or sharp scissors. The uterus was removed and placed on sterile gauze sponges (gauze sponge, 4 × 4 in, Pro Advantage; NDC, La Vergne, Tenn). To prevent hypothermia of the feti in the uterus, the gauze sponges were placed on clean drapes (Kendall Surgi-Drape; Tyco Healthcare Group LP, Mansfield, Mass). A heat therapy pump (Gaymar T/Pump, Gaymar Industries, Orchard Park, NY) was placed beneath the drapes to provide thermal support.
The individual feti were removed by cutting the gravid uterus gently with a sharp scissors. The premature pups were then expelled by gentle pressure, and the umbilical cord was cut approximately 1–3 mm distal to the umbilical attachment. Cotton swabs (6-in cotton-tipped applicators; Dukal Corporation, Hauppauge, NY) were used to tear the amniotic membrane and massage each pup gently until spontaneous breathing was noted. If a pup did not breathe spontaneously within 30–60 s after initiating physical stimulation, a cotton swab dipped in doxapram hydrochloride (doxapram injection, USP, 20 mg/mL; Baxter Healthcare Corporation, Deerfield, Ill) was applied gently to the oral mucous membrane of the affected animal for 1–3 s, and physical stimulation was continued until spontaneous respiration was observed. It has to be mentioned that the need for Doxapram was not constant, and at times, it was not required at all for the whole litter. Resuscitating these mice requires practice and gentle handling. We lost 20% to 30% of the litter in the first few attempts; however, with repeated work, we revived the whole litter and they were all alive in 24 h. We highly doubt that the loss was caused by CO2 exposure as the maternal exposure time was short (usually 45–120 s). Moreover, it is known that newborn mice are refractory to CO2 exposure, and it has been shown that on the day of birth, pups can survive up to 50 min in 100% CO2.27
Fostering
Female CD-1 mice with a newborn litter were used for fostering purposes. Pups that were revived after a C-section were exposed to fecal/nesting material from the foster animal’s cage. The litter was removed and the premature pups were placed in the nest. The foster mother’s original litter was then humanely euthanized. Animal interactions were observed for up to 10 min to evaluate success in regard to acceptance of the pups by the foster animal. The pups were then euthanized per experimental protocol requirements.
C-section at full term
To evaluate whether the process of C-section, CO2 exposure, and physical manipulation has an effect, we performed a C-section on full-term pregnant females. We ordered 7 timed pregnant females expected to deliver on the same date. We observed the mice for spontaneous delivery. Once the 2 mice delivered by natural birth, we performed C-section in 2 more animals and used the first 2 as foster mothers; the other 3 females delivered spontaneously on the same day and the litters were used as full-term natural delivery controls. The mice were housed until they reached 30–35 days of age and then they were euthanized for glomerular counting. All were full-term animals delivered on the same day either by C-section or natural delivery.
Glomerular count
Glomeruli were counted in the kidneys extracted from mice at the age of 30–35 days using a direct maceration/counting method.28 For this method, the kidneys were minced into fine cubes, and the fragments were incubated in 5 mL of 6 mol/L HCl at 37°C for 90 min. The tissue was homogenized through repeated pipetting, and 45 mL phosphate-buffered saline was added. After overnight incubation at 4°C, glomeruli were counted in 3 × 1 mL of this solution in 3 wells of 24-well plates. The total glomerular number per kidney was extrapolated mathematically from the counting of these 3 aliquots.
Urine albumin and creatinine
Urine albumin was assayed by an enzyme-linked immunosorbent assay (ELISA) using mouse albumin-specific antibody provided in microalbuminuria ELISA kit from Exocell (Philadelphia, Pa) according to the manufacturer’s recommendations. Briefly, diluted samples and standards were added to separate wells of microtiter plates precoated with murine albumin. An aliquot of antialbumin antibody was then added. After incubation and wash, horseradish peroxidase-conjugated secondary antibody was then added to the wells. After another incubation and wash, color developer was applied to the wells and the plates were read at 450 nm. The creatinine levels were determined with the CREP2 Creatinine plus ver. 2 kit (Roche Diagnostics, Indianapolis, Ind) according to the manufacturer’s recommendations. This kit is an enzymatic reaction based on creatininase, which has been shown to be reliable in mice and correlate with high-performance liquid chromatography method.29 The method was adapted in our laboratory for use in microtiter plates. The creatinine concentration is calculated based on a standard curve using creatinine standard (Sigma Aldrich, St Louis, Mo).
Measurement of real-time glomerular filtration rate (GFR)
The real-time GFR was measured in awake mice using fluoresceinyl isothiocyanate (FITC)-inulin and the single-injection plasma disappearance method as described previously.30 Briefly, FITC-inulin was purchased from Sigma and dissolved at 5% concentration in 0.9% NaCl. Unbound FITC was removed by dialysis against NaCl. The FITC-inulin was injected at a dose of 3.74 μL/g body weight during a 20 seconds light anesthesia with isoflurane (Baxter Pharmaceutical). A total of 20–30 μL blood was collected via the saphenous vein at 3, 7, 10, 15, 35, 55, and 75 min after the injection of FITC-inulin. The red cells were removed by centrifugation and the plasma was used to measure the FITC inulin using a fluorometer. The calculation of GFR was done as described previously using GraphPad Prism graphing software (GraphPad, San Diego, Calif), which allowed for the automated generation of nonlinear regression of a 2-phase exponential decay curve.30
Catheter insertion and blood pressure measurement
The mice were anesthetized deeply with isoflurane 2% vol/vol.31 Using a sterile technique, a telemetry transmitter (model PA-C10; Data Sciences International, St Paul, Minn) was implanted between the scapulae with the catheter inserted into the carotid artery. The skin was sutured, and the mice were treated with antibiotics and analgesics. After 5–7 days of recovery, daily blood pressure measurements were recorded while the mice were housed in their home cages. The obtained readings were an average of 3 days of measurements of blood pressure recorded every 60 seconds. We noticed diurnal variation of blood pressure as well as activity related change of blood pressure, but the data were consistent from day to day.
Pathology sections and staining and morphometric analysis
The kidneys were fixed in formalin zinc and embedded in paraffin. Four-micron sections were stained with periodic acid schiff (PAS). The histology slides were examined and photographed on an Olympus light microscope equipped with color digital camera (Olympus, Center Valley, Pa). The microscope slides were also scanned at 40× magnification using a Hamamatsu NanoZoomer HT (Hamamatsu Photonics KK, Hamamatsu City, Japan) digital scanner. Image post processing and data analysis was performed using Visiomorph and Microimager software (Visiopharm, Hoersholm, Denmark). The glomeruli were picked randomly from each section by a blinded operator, and the software was used to quantitate the PAS positive material.
Statistical analysis
Mean values were determined for related experimental groups. The experimental groups were compared with controls using a 2-tailed Student t test.
RESULTS
Data from both male and female mice were combined; we did not observe statistically significant sex difference within each group. The error bars in all data represents standard error of mean.
Premature mice have lower nephron number
To determine whether premature delivery, in an otherwise normal pregnancy, can alter kidney development and nephron number, we calculated the nephron number by counting glomeruli in the premature mice kidneys and compared it with the full-term mice from the same mouse colony. The mouse kidney is expected to be fully developed at 4 weeks of age; accordingly, we performed the glomerular count between the age of 30 and 35 days. In CD1 mice spontaneously delivered at full-term, the number of nephrons averaged 12,252 ± 571 nephrons/kidney, which is in agreement with previously published data.32 The average number of nephrons per kidney was reduced in 1-day and 2-days premature mice compared with full-term mice. The mean nephron numbers were 17.4% lower for the 1-day premature mice and 23.6% for the 2-days premature mice with P values of 0.03 and 0.015, respectively (Fig 1, A). Premature mice were smaller in size, but the weight difference did not reach statistical significance. Accordingly, we normalized the nephron number in relation to body weight. The nephron number per gram body weight was still lower for the premature mice compared with full-term mice. However, it only reached statistical significance for the 2-days premature mice, P = 0.028 (Fig. 1, B). No significant difference (P = 0.365) was found in the nephron number between C-section delivered (n = 30) vs naturally delivered full-term mice (n = 18) (Fig 1, C)
Fig 1.
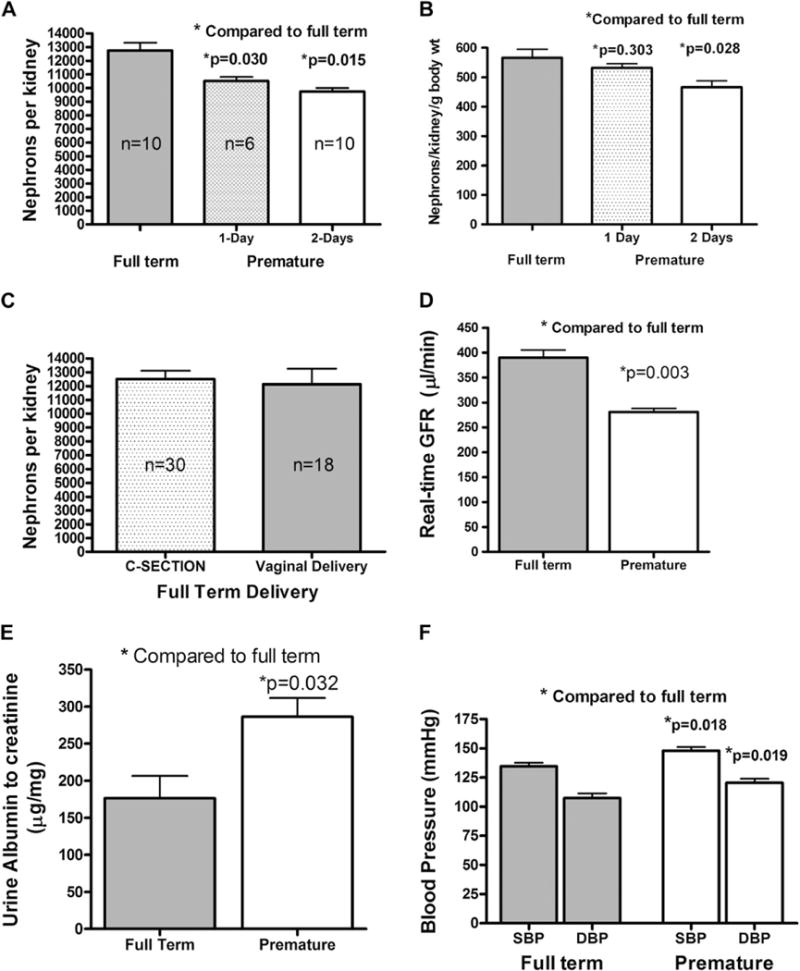
Nephron count and physiologic parameters in premature and full term mice. (A) The absolute nephron number per kidney in CD1 mice and in mice delivered 1-day or 2-days premature showing that the nephron number of the 2-days premature mice is less than the 1-day premature mice, and both are lower than the full-term mice. Nephron counting was performed when the mice were 30–35 days old. (B) The nephron number normalized to body weight. (C) The nephron number in full-term mice delivered by either C-section or spontaneous vaginal delivery. (D) The real-time GFR in full-term CD1 mice and in mice delivered 2 days premature. The GFR measurements were performed when the mice were 30–35 days old. (E) Urine albumin to creatinine ratio (μg albumin/mg creatinine) in full-term CD1 mice and in mice delivered 2 days premature. Urine measurements were made when the mice were 30–35 days old. (F) Mean systolic and diastolic arterial pressure measurements from conscious full-term CD1 mice and in mice delivered 2 days premature. Telemetry blood pressure measurements were obtained using implantable catheters when the mice were approximately 9 months old. The error bars represent the standard error of the mean.
GFR and albumin excretion rate
To examine whether the decrease in nephron number in premature mice is reflected on their kidney function, we measured real-time GFR at 30–35 days of age using FITC-inulin and the plasma disappearance method.30 The GFR was significantly lower in the premature mice compared with full-term mice. The average GFR was 283 ± 13 for the 2-day premature vs 389 ± 26 μL/min for the full-term mice (Fig 1, D).
Urinary albumin excretion
Urinary albumin excretion is one of the best predictors of renal disease.33,34 We found that the premature mice excreted significantly higher urinary albumin compared with full-term mice (286 ± 83 vs 176 ± 59 μg/mg). The urinary albumin was expressed as the albumin-to-creatinine ratio (P = 0.032) (Fig 1, E).
Blood pressure
Previous models of reduced nephron number that reported hypertension showed a minimum of 30% reduction of nephron mass compared with control. Without a priori knowledge of the expected blood pressure, we selected 9 months of age to measure blood pressure. A 9-month-old CD1 mouse is on average middle aged. The cardiovascular parameters were measured using an implantable carotid catheter connected to an implanted telemetry sensor/transmitter. The blood pressure was measured while mice were fed Purina chow (which is the standard diet fed to all mice in our animal facility unless otherwise specified). The mice were given 1 week to recover from the surgical preparation, and blood pressure was then recorded over a 3-day period. Both systolic (148 ± 19 vs 135 ± 13 mm Hg) and diastolic (120 ± 18 vs 107 ± 16 mm Hg) blood pressures were significantly higher in premature mice compared with full-term mice (Fig 1, F).
HISTOPATHOLOGY
Prior studies from human and mouse models of reduced nephron number showed that the renal lesion was in the form of sclerosis of the glomeruli.35,36 To assess whether the decrease of nephron number in our model will yield the same histopathologic picture observed in humans and other mouse models, we stained paraffin-embedded kidney sections with PAS. The glomerular surface area was measured electronically in 30 glomeruli per mouse in whole-section images captured on Hamamatsu NanoZoomer. The mesangial area was measured electronically in relation to the glomerular size using Visiomorph and Microimager software. The glomeruli from premature mice were significantly (P = 0.007, n = 6) larger in surface area and contained significantly (P = 0.001) more PAS positive material compared with full-term mice (n = 6, Fig 2).
Fig 2.
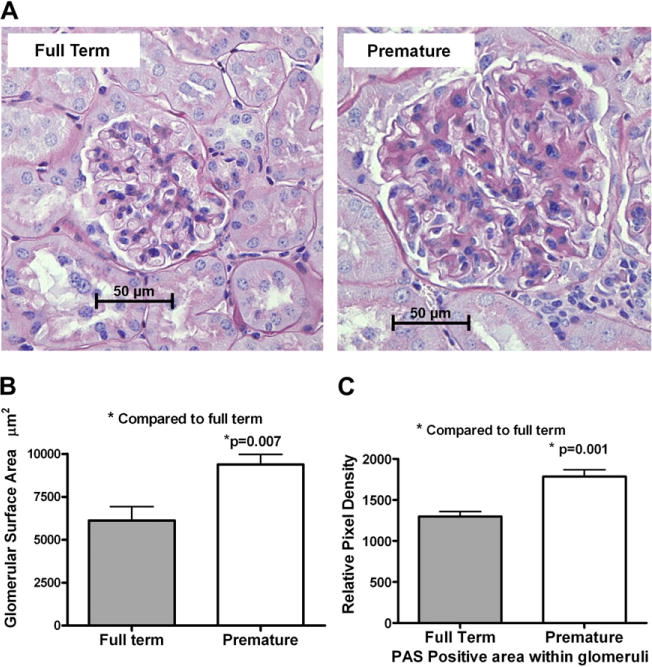
Histology of full term and premature mouse kidneys. (A) Representative histologic images of glomeruli from full-term CD1 mice and mice delivered 2 days premature stained with periodic acid schiff reagent. (B) Bar graph comparing the measured glomerular surface in premature and full-term mice. An average of 30 glomeruli per mouse was found, and 6 mice per group were used. (C) The relative pixel density of PAS positive material in the glomeruli from full-term and premature mice. (Color version of figure is available online.)
DISCUSSION
The mechanism of prematurity-induced reduction in nephron number, hypertension and chronic kidney disease (CKD) remains mostly speculative. “Organ sparing,” as a result of unfavorable intrauterine conditions, is a proposed mechanism for multiorgan underdevelopment including low nephron number,37–42 whereas mechanical stress and hyperfiltration have been proposed as a mechanism of CKD associated with the low nephron number.35 Meanwhile, placental and environmental programming of organ development is still unexplored. The animal models of reduced nephron number studied thus far are generated by targeted mutation in genes critical to renal development, inhibition of cyclooxygenase, restricting maternal protein or caloric intake, or by ligating the uterine artery leading to fetal growth restriction in utero.19,22,43,44,45 In the current studies, we describe a model of premature birth in mice, in an otherwise normal pregnancy, which displays a permanent reduction in the nephron number and a predisposition toward CKD and hypertension. The body weight of the premature mice was less than that of the full-term mice throughout the 9 months of observation, but the number of nephrons per gram body weight was still lower for the premature animals. We hypothesized that single-nephron GFR was higher in the premature mice compared with full-term mice, which is the principle of hyperfiltration theory. Consistent with this concept, the premature kidneys have enlarged glomeruli and increased matrix deposition N, which are histopathologic findings associated frequently with hyperfiltering glomeruli. With few exceptions, it is difficult to dissociate prematurity from intrauterine growth retardation in humans. However, the reduction of the nephron number in our mouse model cannot be explained by unfavorable intrauterine conditions. We hypothesize that the shift from intrauterine milieu to external environment leads to early termination of nephrogenesis by depriving the developing kidney from maternally provided factors that are essential for kidney development and maturation. The loss of such factors may lead to a switch from proliferation to final differentiation, resulting in the termination of nephrogenesis. In mice, nephrogenesis continues after birth, and an outburst of nephron formation was observed in the first 3 postnatal days.46 Also, few cortical nephrons can involute or have defective vascularization after formation. In our studies, we did not observe changes to prove or disprove such an event. One possible mechanism of low nephron number is the loss of maintenance and proliferation of progenitor cell populations. Also, the stress of premature delivery could be the inciting signal for termination of the nephrogenesis program before complete development of the kidney. More specifically, it is possible that premature delivery arrests epithelial branching morphogenesis, which is a fundamental biologic process central to the developmental theme of multiple organs including the kidney, lung, pancreas, and vascular system. Last but not least, there is a likelihood of epigenetic programming of the premature kidneys leading to increased susceptibility to damage and increased risk of hypertension.
The current data support the concept that nephron adequacy is an important predictor of the health outcome of reduced nephron number. Nephron adequacy is the functional relationship between the nephron number and the metabolic mass and/or metabolic demand of the adult individual; it was observed that very low birth weight (including premature) infants who show catch-up growth, thus creating a higher metabolic demand on the less endowed kidneys, are the individuals at risk for hypertension.7,8 Severe nephron inadequacy leads to higher degree of hyperfiltration, which leads to rapid nephron dropout and faster decline in renal function at a younger age. The longevity of the transplanted kidney survival was shown to be affected by the relationship between the functional nephron mass and the metabolic demand of the recipient.47–49
The dogma is that nephron number reduction has to occur before birth to increase the risk of hypertension. The prerequisite for the developmental (in utero) nephron number reduction to produce increased risk for hypertension and CKD could reflect the length of time needed to show an effect. Developmentally, there is a likelihood of an ancestral process common to other organ system programming including renal, vascular, and neuroendocrine. Thus, a low nephron number kidney will coexist in a host with multiorgan programming abnormalities that each cooperate with one another to produce the hypertension phenotype. Moreover, kidneys with lower nephron numbers might have less than optimum vascular tree leading to abnormalities in the renin-angiotensin system and/or alterations in pressure naturesis. Figure 3 depicts our model for prematurity-induced renal disease and hypertension. All these mechanisms need to be explored and validated in multiple models/strains of prematurity-induced reduced nephron number and hypertension. This will permit us to determine the effect of genetic background on modifying the phenotype of the animal.
Fig 3.
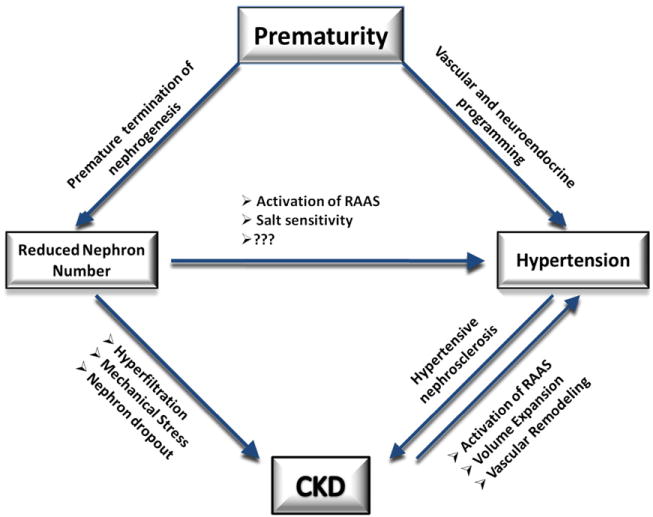
A diagram depicting the hypothetical relationship between prematurity, nephron number, hypertension, and kidney disease. (Color version of figure is available online.)
The profound phenotype we observe in the premature mice after only 2-days premature delivery could be caused by the mathematically exponential nature of nephron formation. An event, like prematurity, which may trigger termination of nephrogenesis, will lead to the loss of a significant number of nephrons. It is difficult to compare the effects of prematurity timing in relation to the biologic effects between mice and humans. Human nephrogenesis is usually concluded by 36 weeks of gestation. Children born before 33 weeks of gestation display health outcomes related to low nephron number.50,51 However, there is an undetermined percentage of later preterm infants who also suffer from intrauterine growth retardation and have a younger biologic age; they will have as high risk, similar to extreme premature individuals, for developing hypertension and CKD later in life. The hypertension and the reduced nephron number in our premature mouse model are 2 separate outcomes that might have a direct relationship. We are not sure to what extent the reduction in nephron number contributed to the hypertension phenotype in our model. The mouse model described in this report is novel because it represents a reduced nephron number and hypertension outcome from the early termination of a normal pregnancy, ie, no IGUR.
Understanding the pathobiology of prematurity-induced low nephron number, CKD and hypertension can lead to the development of pharmacologic agents to support extrauterine organ development, which will have a significant impact on the health of affected individuals later in life. Moreover, because of the commonality of the developmental controls of branching organs, these measures may be applicable to the lung, cardiovascular system, and pancreas, which all have adult disease related to prematurity and IGUR. The model developed in this study will allow future pathophysiologic studies regarding premature delivery and kidney development/function and may be used for potential treatment methodologies.
AT A GLANCE COMMENTARY.
El-Meanawy A, et al
Background
Prematurity is a worldwide, prevalent public health problem. Besides the immediate outcome in the form of increased infant mortality and morbidity, the long-term outcome resulting from organ developmental programming defect is lifelong and costly.
Translational Significance
To understand the pathophysiology of prematurity-induced adult disease and to develop specific targeted therapy to improve the outcome of premature infants, we need to understand perinatal organ programming and organ development. In this manuscript we developed a mouse model of prematurity induced nephron number reduction, hypertension, and proteinuria. We believe that this model will help us explore perinatal organ programming in-depth and might help us identify factors that are important for organ development.
Acknowledgments
Supported in part by grants HL-29587 and DK-62803 from the National Institutes of Health and by the Kidney Disease Center at the Medical College of Wisconsin.
Abbreviations
- C-section
Cesarean delivery
- CKD
chronic kidney disease
- FITC
fluorescein isothiocyanate
- ELISA
enzyme-linked immunosorbent assay
- GFR
glomerular filtration rate
- IGUR
intrauterine growth retardation
- PAS
Periodic Acid Schiff
Biography
Ashraf El-Meanawy, MD, PhD, is an Assistant Professor of Medicine in the Division of Nephrology at the Medical College of Wisconsin. His article is based on a presentation given at the Combined Annual Meeting of the Central Society for Clinical Research and Midwestern Section American Federation for Medical Research held in Chicago, Ill, April 2011.
References
- 1.Hamilton BE, Minino AM, Martin JA, et al. Annual summary of vital statistics. Pediatrics. 2005;2007(119):345–60. doi: 10.1542/peds.2006-3226. [DOI] [PubMed] [Google Scholar]
- 2.Martin JA, Hamilton BE, Sutton PD, et al. National Vital Statistics Report. 2010. [Google Scholar]
- 3.Centers for Disease Control and Prevention. National Vital Statistics Report. 58. 7-1-2008. Atlanta, Ga: Centers for Disease Control and Prevention; 2008. [Google Scholar]
- 4.Kistner A, Celsi G, Vanpee M, Jacobson SH. Increased systolic daily ambulatory blood pressure in adult women born preterm. Pediatr Nephrol. 2005;20:232–3. doi: 10.1007/s00467-004-1717-4. [DOI] [PubMed] [Google Scholar]
- 5.Kistner A, Celsi G, Vanpee M, Jacobson SH. Increased blood pressure but normal renal function in adult women born preterm. Pediatr Nephrol. 2000;15:215–20. doi: 10.1007/s004670000473. [DOI] [PubMed] [Google Scholar]
- 6.White SL, Perkovic V, Cass A, et al. Is low birth weight an antecedent of CKD in later life? A systematic review of observational studies. Amer J Kidney Dis. 2009;54:248–61. doi: 10.1053/j.ajkd.2008.12.042. [DOI] [PubMed] [Google Scholar]
- 7.Hack M, Schluchter M, Cartar L, Rahman M. Blood pressure among very low birth weight (<1.5 kg) young adults. Pediatr Res. 2005;58:677–84. doi: 10.1203/01.PDR.0000180551.93470.56. [DOI] [PubMed] [Google Scholar]
- 8.Rotteveel J, van Weissenbruch MM, Twisk JW, Delemarre-Van de Waal HA. Infant and childhood growth patterns, insulin sensitivity, and blood pressure in prematurely born young adults. Pediatrics. 2008;122:313–21. doi: 10.1542/peds.2007-2012. [DOI] [PubMed] [Google Scholar]
- 9.Barker DJP. The developmental origins of adult disease. J Am Coll Nutr. 2004;23:588S–595. doi: 10.1080/07315724.2004.10719428. [DOI] [PubMed] [Google Scholar]
- 10.Barker DJP, Eriksson JG, Forsen T, Osmond C. Fetal origins of adult disease: strength of effects and biological basis. Int J Epidemiol. 2002;31:1235–9. doi: 10.1093/ije/31.6.1235. [DOI] [PubMed] [Google Scholar]
- 11.Barker DJ. Birth weight and hypertension. Hypertension. 2006;48:357–8. doi: 10.1161/01.HYP.0000236552.04251.42. [DOI] [PubMed] [Google Scholar]
- 12.Brenner BM, Mackenzie HS. Nephron mass as a risk factor for progression of renal disease. Kidney Int Suppl. 1997;63:S124–7. [PubMed] [Google Scholar]
- 13.Mackenzie HS, Brenner BM. Fewer nephrons at birth: a missing link in the etiology of essential hypertension? Am J Kidney Dis. 1995;26:91–8. doi: 10.1016/0272-6386(95)90161-2. [DOI] [PubMed] [Google Scholar]
- 14.Mackenzie HS, Lawler EV, Brenner BM. Congenital oligonephropathy: the fetal flaw in essential hypertension? Kidney Int. 1996;49:S30–4. [PubMed] [Google Scholar]
- 15.Keller G, Zimmer G, Mall G, Ritz E, Amann K. Nephron number in patients with primary hypertension. N Engl J Med. 2003;348:101–8. doi: 10.1056/NEJMoa020549. [DOI] [PubMed] [Google Scholar]
- 16.Hughson MD, Douglas-Denton R, Bertram JF, Hoy WE. Hypertension, glomerular number, and birth weight in African Americans and white subjects in the southeastern United States. Kidney Int. 2006;69:671–8. doi: 10.1038/sj.ki.5000041. [DOI] [PubMed] [Google Scholar]
- 17.Li S, Chen SC, Shlipak M, et al. Low birth weight is associated with chronic kidney disease only in men. Kidney Int. 2008;73:637–42. doi: 10.1038/sj.ki.5002747. [DOI] [PubMed] [Google Scholar]
- 18.Brenner BM, Garcia DL, Anderson S. Glomeruli and blood pressure. Less of one, more the other? Am J Hypertens. 1988;1:335–47. doi: 10.1093/ajh/1.4.335. [DOI] [PubMed] [Google Scholar]
- 19.Cullen-McEwen LA, Kett MM, Dowling J, Anderson WP, Bertram JF. Nephron number, renal function, and arterial pressure in aged GDNF heterozygous mice. Hypertension. 2003;41:335–40. doi: 10.1161/01.hyp.0000050961.70182.56. [DOI] [PubMed] [Google Scholar]
- 20.Fogelgren B, Yang S, Sharp IC, et al. Deficiency in Six2 during prenatal development is associated with reduced nephron number, chronic renal failure, and hypertension in Br/+ adult mice. Am J Physiol Renal Physiol. 2009;296:F1166–78. doi: 10.1152/ajprenal.90550.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nishinakamura R, Matsumoto Y, Nakao K, et al. Murine homolog of SALL1 is essential for ureteric bud invasion in kidney development. Development. 2001;128:3105–15. doi: 10.1242/dev.128.16.3105. [DOI] [PubMed] [Google Scholar]
- 22.Poladia DP, Kish K, Kutay B, Bauer J, Baum M, Bates CM. Link between reduced nephron number and hypertension: studies in a mutant mouse model. Pediatr Res. 2006;59:489–93. doi: 10.1203/01.pdr.0000202764.02295.45. [DOI] [PubMed] [Google Scholar]
- 23.Sanyanusin P, McNoe LA, Sullivan MJ, Weaver RG, Eccles MR. Mutation of PAX2 in two siblings with renal-coloboma syndrome. Hum Mol Genet. 1995;4:2183–4. doi: 10.1093/hmg/4.11.2183. [DOI] [PubMed] [Google Scholar]
- 24.Kohlhase J. SALL1 mutations in Townes-Brocks syndrome and related disorders. Hum Mutat. 2000;16:460–6. doi: 10.1002/1098-1004(200012)16:6<460::AID-HUMU2>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 25.Institute for Laboratory Animal Research (ILAR) Guide for the Care and Use of Laboratory Animals. National Academies Press; 1996. Earth and Life Studies (DELS) [Google Scholar]
- 26.National Research Council. Guide for the Care and Use of Laboratory Animals. 8. Washington, DC: Academies Press; 2010. [Google Scholar]
- 27.Pritchett K, Corrow D, Stockwell J, Smith A. Euthanasia of neonatal mice with carbon dioxide. Comp Med. 2005;55:275–81. [PubMed] [Google Scholar]
- 28.Kaufman JM, Hardy R, Hayslett JP. Age-dependent characteristics of compensatory renal growth. Kidney Int. 1975;8:21–6. doi: 10.1038/ki.1975.72. [DOI] [PubMed] [Google Scholar]
- 29.Keppler A, Gretz N, Schmidt R, et al. Plasma creatinine determination in mice and rats: An enzymatic method compares favorably with a high-performance liquid chromatography assay. Kidney Int. 2007;71:74–8. doi: 10.1038/sj.ki.5001988. [DOI] [PubMed] [Google Scholar]
- 30.Qi Z, Whitt I, Mehta A, et al. Serial determination of glomerular filtration rate in conscious mice using FITC-inulin clearance. Am J Physiol. 2004;286:F590–6. doi: 10.1152/ajprenal.00324.2003. [DOI] [PubMed] [Google Scholar]
- 31.Itah R, Gitelman I, Davis C. A replacement for methoxyflurane (Metofane) in open-circuit anaesthesia. Lab Anim. 2004;38:280–5. doi: 10.1258/002367704323133664. [DOI] [PubMed] [Google Scholar]
- 32.Bonvalet JP, Champion M, Courtalon A, et al. Number of glomeruli in normal and hypertrophied kidneys of mice and guinea-pigs. J Physiol. 1977;269:627–41. doi: 10.1113/jphysiol.1977.sp011919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gansevoort RT, Nauta FL, Bakker SJ. Albuminuria: all you need to predict outcomes in chronic kidney disease? Curr Opin Nephrol Hypertens. 2010;19:513–8. doi: 10.1097/MNH.0b013e32833e4ce1. [DOI] [PubMed] [Google Scholar]
- 34.van der Velde M, Halbesma N, De Charro FT, et al. Screening for albuminuria identifies individuals at increased renal risk. J Am Soc Nephrol. 2009;20:852–62. doi: 10.1681/ASN.2008060655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hodgin JB, Rasoulpour M, Markowitz GS, D’Agati VD. Very low birth weight is a risk factor for secondary focal segmental glomerulosclerosis. Clin J Am Soc Nephrol. 2009;4:71–6. doi: 10.2215/CJN.01700408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.He C, Zalups RK, Henderson DA, Striker GE, Striker LJ. Molecular analysis of spontaneous glomerulosclerosis in Os/+ mice, a model with reduced nephron mass. Am J Physiol. 1995;269:F266–73. doi: 10.1152/ajprenal.1995.269.2.F266. [DOI] [PubMed] [Google Scholar]
- 37.Manalich R, Reyes L, Herrera M, Melendi C, Fundora I. Relationship between weight at birth and the number and size of renal glomeruli in humans: a histomorphometric study. Kidney Int. 2000;58:770–3. doi: 10.1046/j.1523-1755.2000.00225.x. [DOI] [PubMed] [Google Scholar]
- 38.Kilavuz O, Vetter K. Is the liver of the fetus the 4th preferential organ for arterial blood supply besides brain, heart, and adrenal glands? J Perinat Med. 1999;27:103–6. doi: 10.1515/JPM.1999.012. [DOI] [PubMed] [Google Scholar]
- 39.Baschat AA, Gembruch U, Reiss I, Gortner L, Diedrich K. Demonstration of fetal coronary blood flow by Doppler ultrasound in relation to arterial and venous flow velocity waveforms and perinatal outcome–the ‘heart-sparing effect’. Ultrasound Obstet Gynecol. 1997;9:162–72. doi: 10.1046/j.1469-0705.1997.09030162.x. [DOI] [PubMed] [Google Scholar]
- 40.Wladimiroff JW, vd Wijngaard JA, Degani S, et al. Cerebral and umbilical arterial blood flow velocity waveforms in normal and growth-retarded pregnancies. Obstet Gynecol. 1987;69:705–9. [PubMed] [Google Scholar]
- 41.Wladimiroff JW, Tonge HM, Stewart PA. Doppler ultrasound assessment of cerebral blood flow in the human fetus. Br J Obstet Gynaecol. 1986;93:471–5. [PubMed] [Google Scholar]
- 42.Sebire NJ, Talbert DG. The dynamic placenta: a closer look at the pathophysiology of placental hemodynamics in uteroplacental compromise. Ultrasound Obstet Gynecol. 2001;18:557–61. doi: 10.1046/j.0960-7692.2001.602.doc.x. [DOI] [PubMed] [Google Scholar]
- 43.Qiao JZ, Uzzo R, Obara-Ishihara T, et al. FGF-7 modulates ureteric bud growth and nephron number in the developing kidney. Development. 1999;126:547–54. doi: 10.1242/dev.126.3.547. [DOI] [PubMed] [Google Scholar]
- 44.Hoppe CC, Evans RG, Bertram JF, Moritz KM. Effects of dietary protein restriction on nephron number in the mouse. Am J Physiol Regul Integr Comp Physiol. 2007;292:R1768–74. doi: 10.1152/ajpregu.00442.2006. [DOI] [PubMed] [Google Scholar]
- 45.Saez F, Reverte V, Salazar F, et al. Hypertension and sex differences in the age-related renal changes when cyclooxygenase-2 activity is reduced during nephrogenesis. Hypertension. 2009;53:331–7. doi: 10.1161/HYPERTENSIONAHA.108.124354. [DOI] [PubMed] [Google Scholar]
- 46.Hartman HA, Lai HL, Patterson LT. Cessation of renal morphogenesis in mice. Dev Biol. 2007;310:379–87. doi: 10.1016/j.ydbio.2007.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brenner BM, Milford EL. Nephron underdosing: a programmed cause of chronic renal allograft failure. Am J Kidney Dis. 1993;21:66–72. doi: 10.1016/0272-6386(93)70097-i. [DOI] [PubMed] [Google Scholar]
- 48.Feldman HI, Fazio I, Roth D, et al. Recipient body size and cadaveric renal allograft survival. J Am Soc Nephrol. 1996;7:151–7. doi: 10.1681/ASN.V71151. [DOI] [PubMed] [Google Scholar]
- 49.Chertow GM, Milford EL, Mackenzie HS, Brenner BM. Antigen-independent determinants of cadaveric kidney transplant failure. JAMA. 1996;276:1732–6. [PubMed] [Google Scholar]
- 50.Keijzer-Veen MG, Schrevel M, Finken MJ, et al. Microalbuminuria and lower glomerular filtration rate at young adult age in subjects born very premature and after intrauterine growth retardation. J Am Soc Nephrol. 2005;16:2762–8. doi: 10.1681/ASN.2004090783. [DOI] [PubMed] [Google Scholar]
- 51.Keijzer-Veen MG, Dulger A, Dekker FW, Nauta J, van der Heijden BJ. Very preterm birth is a risk factor for increased systolic blood pressure at a young adult age. Pediatr Nephrol. 2010;25:509–16. doi: 10.1007/s00467-009-1373-9. [DOI] [PMC free article] [PubMed] [Google Scholar]