

Abstract
Malignant gliomas are the most common and deadly primary brain tumors, due to their infiltrative invasion of the normal neural tissue that makes them virtually impossible to completely eliminate. We have previously identified and characterized the proteoglycan BEHAB/brevican in gliomas and have demonstrated that upregulation and cleavage of this CNS-specific molecule promote glioma invasion. Here, we have further investigated if the proteolytic processing of BEHAB/brevican by metalloproteases of the ADAMTS family is a necessary step in mediating its pro-invasive effect in glioma. By generating a site-specific (396SRG398 → NVY) mutant form resistant to ADAMTS cleavage, we have shown that the predominant proteolytic processing of BEHAB/brevican by glioma cells occurs only at this site. More importantly, “uncleavable” BEHAB/brevican is unable to enhance glioma cell invasion in vitro and tumor progression in vivo. In addition, our results suggest that the full-length protein and its cleavage products may act independently because the mutant form does not exert a dominant negative effect on normal BEHAB/brevican expression or cleavage. These results illustrate how the regulated processing of major components of the neural extracellular matrix has important functional implications in glioma progression. In addition, our findings underscore the relevance of the ADAMTS family of metalloproteases as attractive targets for novel pharmacological approaches in glioma therapy.
Keywords: extracellular matrix, glioma invasion, proteoglycan, metalloprotease, aggrecanase, ADAMTS
Introduction
Malignant gliomas, the most common primary tumors in the adult CNS, are characterized by a unique ability to infiltrate and invade the surrounding neural tissue [1]. The extensive dispersion of these motile tumor cells, coupled to their high resistance to apoptosis compared to the cells at the core of the tumor, are major features that make invasive gliomas virtually impossible to eliminate by surgery followed by conventional radio- and chemotherapies [2,3].
Understanding the cellular mechanisms of glioma invasion is currently regarded a major priority for developing effective therapeutic strategies against these tumors [4]. Little is known of the molecular basis that allows glioma cells to overcome those barriers that inhibit motility in the adult nervous tissue and that prevent the infiltration of tumors that metastasize to the CNS [5,6].
A major barrier to cellular movement in the CNS is the extracellular matrix (ECM) that surrounds neurons and glial cells and occupies a considerable volume of nervous tissue. In contrast to other tissues, the ECM of the neural parenchyma lacks most fibrous proteins such as collagens, laminin-1 and fibronectin, and is instead largely composed of a hyaluronic acid scaffold with associated glycoproteins and proteoglycans [7]. Glioma cells may overcome the neural ECM by a variety of mechanisms, including the over-expression of normal and altered versions of ECM molecules and their cell-surface receptors (reviewed in [8–10]). In addition, glioma cells produce proteolytic [11,12] and hyalurolytic [13] enzymes that promote detachment from the surrounding ECM and allow the cells to move away from the core of the tumor.
From a clinical standpoint, tumor-associated proteases are very attractive targets because they promote one of the most downstream steps in the process of tumor invasion. Furthermore, the proteolytic digestion of matrix components and the subsequent invasion of the tissue are tightly linked with angiogenesis, a critical step in the progression of malignant brain tumors [14,15].
Several classes of proteases are involved in glioma invasion, including urokinase-type plasminogen activator, cathepsins and, particularly, matrix metalloproteases (MMPs) [16–20]. The upregulation in glioma cells of specific members of the MMP family, such as MMP2 and MMP9 (reviewed in [12,21,22]), and the decrease of glioma invasion through inhibition of these enzymes [23–27] have encouraged the design of adjuvant therapies based on targeting MMP activity. However, clinical trials of MMP inhibitors have proven disappointing due to lack of effect or damaging side-effects [28–30]. Because MMPs are involved in many cellular processes in addition to tumorigenesis, there has been an increased interest in identifying proteases more specifically involved in glioma invasion.
Previous work from ours and other laboratories has shown that gliomas consistently upregulate the ECM protein brevican, also known as brain enriched hyaluronan binding protein (BEHAB). BEHAB/brevican (B/b) is a neural-specific chondroitin sulfate proteoglycan of the lectican family that inhibits cell and neurite motility in the normal neural tissue [31,32]. However, experimental overexpression of B/b dramatically enhances glioma growth and invasion in vitro and in vivo [33–35]. B/b is not cleaved by MMPs in vivo, but is processed at a single site by metalloproteases of the Disintegrin and Metalloprotease with Thrombospondin Motifs (ADAMTS) family, both during normal CNS development and in malignant gliomas [36–39]. Upregulation of B/b cleavage products has been observed in human gliomas [39] and overexpression of these fragments increases glioma cell motility in vitro as well as tumor dispersion in vivo [34,35].
Our previous work has shown that overexpression and cleavage of B/b is sufficient to enhance glioma invasion but it did not provide unequivocal evidence of whether B/b processing is a necessary step for its pro-invasive effect. This, in turn, is important to establish if the dispersion of B/b-overexpressing gliomas could be reduced by specifically targeting the processing of this proteoglycan. In the present investigation we demonstrate that ADAMTS-mediated cleavage is indeed a critical step for the pro-invasive role of B/b in gliomas, suggesting that specific inhibition of lectican cleavage may reduce the progression of these tumors and thus provide an additional approach for glioma therapy.
Materials and Methods
Cells and antibodies
The rat CNS-1 glioma cell line was grown at 5% CO2 in RPMI-1640 medium supplemented with 10% fetal calf serum, 100 U/ml penicillin, and 100 μg/ml streptomycin (Invitrogen, Carlsbad CA) [40]. These cells express and cleave B/b when grown as intracranial grafts but have no production of B/b in culture, likely due to the absence of CNS-specific inducing factors [33]. Three rabbit polyclonal anti-B/b antibodies were employed in this study: B6, against the chondroitin sulfate-attachment region (aa506–529) of rat B/b; B5, against the NH2 terminus (AA 60–73) of rat B/b; and B50, against the neoepitope QEAVESE (AA 389–395) that is exposed in the NH2-terminal fragment of B/b after cleavage by ADAMTS proteases (see Figure 1A). These antibodies have been extensively characterized for the detection of rat and human B/b [38,39].
Figure 1. Mutation of the 393ES.

E/SRG398 site of BEHAB/brevican renders the sequence uncleavable by cultured CNS-1 cells.
A) Using site directed mutagenesis, the sequence 396SRG398 was mutated to NVY as indicated in the methods section (HABD, hyaluronan-binding domain; CS, chondroitin sulfate attachment region). B) CNS-1 cells were transiently or stably transfected with the normal (B/b) or mutated (BNVY) BEHAB/brevican constructs, and their culture media collected and processed for Western blotting. The antibodies B6 and B50 were used to detect, respectively, the full-length protein and its N-terminal cleavage product. The latter was completely absent in cells transfected with BNVY. C) Blots probed with the antibody B5, directed against the NH2-terminus of B/b, did not show any other major cleavage product for BNVY, indicating that the mutation rendered BNVY uncleavable in culture conditions. Blots in (B) and (C) were overexposed to maximize the detection of cleavage products in BNVY-transfected cells. D) Cells co-transfected with B/b (1 μg cDNA) and increasing amounts (0–4 μg) of BNVY expressed increased amounts of the full-length protein without detectable changes in the levels of the N-terminal cleavage product, suggesting that BNVY did not compete with B/b for ADAMTS binding. The plasmid pcDNA3.1 was used to complete the total amount of transfected DNA to 5 μg in all cases. Equal protein load in the gels was controlled by total protein measurement and by equal detection of bovine serum albumin in the blot membranes (stained with 0.1% amido-black).
Site-directed mutagenesis
Full-length rat B/b cDNA (nucleotides 60–2780 of the 3077bp full-length clone) [41] was subcloned between the BamH1-Not1 sites of the eukaryotic expression vector pCDNA3.1 (Invitrogen). The cDNA was subsequently mutated using the Quikchange site-directed mutagenesis kit (Stratagene, La Jolla, CA) following the manufacturer instructions. Mutation primer 5′-GTG GAG AGC GAG AAC GTT TAC GCG ATC TAC TCC-3′ (nucleotides 1230–1268 of the full-length clone) and its complementary primer were used to change the sequence 392VESESRGAIYS402 to VESENVYAIYS. Incorporation of the appropriate mutation was confirmed by the detection of a restriction product for the mutated sequence AACGTT (enzyme Acl-1) and by automated DNA sequencing.
Cell culture and transfection
CNS-1 cells were transfected employing LipofectAMINE 2000 (Invitrogen) according to the manufacturer protocols. Control transfections were usually performed with the original pCDNA3.1 vector. Alternatively, control cells were stably transfected with a pBOB1.5 vector carrying the cDNA for green fluorescent protein (GFP), to facilitate their detection within the neural parenchyma. Control cells transfected with either of these vectors were undistinguishable regarding proliferation rate and invasive behavior in vitro and in vivo. For biochemical experiments, transfected cells were routinely changed to serum-free OptiMEM-I culture medium (Invitrogen) 24 hours after transfection, followed by medium collection 12 to 16 hours later. For the creation of stably transfected cells, 800 μg/ml Geneticin (G418, Invitrogen) were added to the culture medium 24 hours after transfection. Stable pools of cells expressing B/b constructs or GFP were derived by maintaining cells in Geneticin-supplemented medium for 21 days. After selection, stably transfected cells were maintained in culture medium containing 200 μg/ml Geneticin.
Growth rate, cell proliferation and cell death
Cells were cultured in 96-well plates at an initial density of 2,000 cells/well in 200 μl of culture medium containing 2% fetal bovine serum. 100 μl of medium were removed daily for cell death assessment (see below) and 25 μl of 2 mg/ml MTT (3,-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide; Sigma-Aldrich, St. Louis, MO) in DPBS were added to the remaining medium and cells to measure cell growth. Cells were incubated with MTT for 3 hours at 37 °C, followed by addition of 100 μl of 40 mM hydrochloric acid in isopropanol to solubilize the resulting formazan product. Solubilization in the wells was allowed to proceed for 2 hours at 37 °C, followed by measuring of absorbance at 550 nm in a microplate reader.
To quantify the daily proportion of cell death, 100 μl of cell-free medium from each well were processed using the Cytotoxicity Detection kit (Roche Applied Science, Indianapolis, IN) according to the manufacturer’s instructions. Briefly, the amount of lactate dehydrogenase (LDH) released to the medium was quantified by measuring the absorbance at 450 nm of an LDH-dependent redox product, and the proportion of cell death was calculated as the ratio of released to total LDH. Total LDH was measured each day in additional wells by solubilization of the cells with 1% v/v Triton X-100. All absorbance measurements for cell growth and death were performed in quadruplicate for each day and cell line.
To quantify the proportion of effectively proliferating cells in vitro, cells were plated at an initial density of 3,000 cells/well in 48-well plates and 72 hours later fixed with cold acetone. Fixed and permeabilized cells were processed for fluorescence immunocytochemistry using an anti-Ki-67 antibody (Santa Cruz Biotechnologies, Santa Cruz, CA), and counterstained with 4′,6-diamino-2-phenylindole (DAPI). Ki-67 positive cells and total cell number were manually quantified in six random fields for each cell line, and the proliferative index was calculated as the averaged ratio of Ki-67 positive to total cells. Ki-67 staining in tumor-tissue sections yielded results essentially identical to those observed in vitro, with an undistinguishable and strikingly high proportion of Ki-67 positive cells in tumors formed by all cell lines (not shown).
Brain slice invasion assay
Brain slice organotypic cultures were essentially performed as described in the literature [42], with minor modifications. Briefly, postnatal day 1 CD-1 mice (Charles River Laboratories, Wilmington, MA) were decapitated on ice and their brains were removed into ice-cold HBSS containing 100 U/ml penicillin, 100 μg/ml streptomycin and 250 ng/ml ampothericin. The meninges were quickly removed and the brains were cut coronally into 300 μm slices using a McIlwain tissue chopper (Brinkmann Instruments. Westbury, NY). Brain slices were dissociated in HBSS and placed on a polytetrafluoroethylene membrane (0.4 μm-pore size Millicell-CM, Millipore), suspended inside a 35mm culture dish over 1 ml of the following slice culture medium: 60% Neurobasal-A:25% HBSS:15% DMEM (containing 25 mM HEPES), supplemented with B27 supplement (Invitrogen), G5 supplement (Invitrogen), 100 U/ml penicillin and 100 μg/ml streptomycin. Tissue slices were incubated at 37 °C in a humid incubator at 5% CO2 and the slice culture medium was changed every other day. CNS-1 cells carrying B/b constructs or pcDNA3.1 were transfected with the vector pDsRed2 (Beckton Dickinson Biosciences, Palo Alto, CA) carrying the cDNA for red fluorescent protein and plated at 1.105 cells/ml in dishes containing a 2 mm-thick layer of 1% sterile agarose, to form spherical aggregates. After 48 hours, the tumor cell aggregates were individually transferred onto the brain slices using a pulled glass capillary pipette. Fluorescent images were obtained with an inverted Nikon TE-200 microscope fitted with long working-distance objectives and a Spot II CCD camera. Images were captured at 24 hour intervals using equal exposure conditions. Total cross-sectional area occupied by the dispersed cells was quantified with ImageJ software (v1.37, National Institutes of Health), by a researcher blinded to the experimental conditions.
Animal studies
All studies involving animals were approved by the institutional Animal Care and Use Committee. Female Lewis rats (Charles River Laboratories) were anesthetized with 75 mg/kg ketamine and 5 mg/kg xylazine and positioned in a David Kopf stereotactic instrument with the incisor bar set at 3.0 mm below the interaural line. Stably transfected CNS-1 cells at 70–80% confluence were harvested by mild trypsinization, washed once in Dullbecco’s PBS and suspended at 5.104 cells/μl in sterile PBS supplemented with 1 μg/ml MgCl2, 1 μg/ml CaCl2 and 0.1 % w/v glucose. Intrathalamic injections were made with a 10 μl Hamilton syringe, fitted with a 26G beveled needle, into the right thalamus at the coordinates 2.8 mm posterior to bregma, 2.2 mm lateral to the midline, and 5.0 mm ventral to the dura. A total volume of 3 μl of the cell suspension was injected over the course of 3 minutes and the needle was left in place for an additional minute before being slowly withdrawn. For studies of survival, cells were applied in the same conditions but at a concentration of 1.105 cells/μl. Injected animals were inspected twice daily and those that were not able to right themselves within 15 seconds of being placed on their side were considered to have reached the survival endpoint and were sacrificed. The day of sacrifice was recorded as the last day of survival.
Histology and estimation of tumor volume
Tumor-bearing animals were deeply anesthetized with halothane and transcardially perfused with 100 mM sodium phosphate buffer, pH 7.4, followed by ice-cold 4% phosphate-buffered paraformaldehyde. Brains were dissected, post-fixed, cryoprotected and completely sectioned coronally at 40 μm in a cryostat. Every fifth section was stained with 0.1% w/v cresyl violet and analyzed by microscopy to identify the tumor mass and clusters of tumor cells. Using ImageJ, the perimeters of the tumor mass and tumor cell clusters in each stained section were manually drawn by a researcher blinded to the experimental conditions. The software then calculated the total tumor area (in mm2) for each section and integrated the total volume using Cavalieri’s estimator of morphometric volume: Vol= (d * ΣAi) − t * Amax, where d=distance between sections that were analyzed, ΣAi= sum of the areas of all sections, t= section thickness, and Amax= maximum value of A [43]. Individual CNS-1 cells that migrated away from the tumor clusters were not included in the quantification, but were routinely detected in the control tumors by GFP fluorescence, remarking the invasive nature of this glioma model.
Electrophoresis and Western blotting
Samples of cell-free, serum-free conditioned culture medium were immediately treated with protease inhibitors (Complete, Roche Applied Science) to prevent further B/b cleavage, followed by concentration in centrifugal concentrators (Amicon Ultra, Millipore) with a Mw cutoff of 30 kDa. Samples of CNS-1-derived tumor tissue were subjected to isotonic subcellular fractionation as previously described [39] and the final soluble fraction was prepared for protein electrophoresis. All samples, containing 10 μg of total protein, were electrophoresed on reducing 7% SDS-polyacrylamide gels and analyzed by Western blotting with anti-B/b antibodies.
Statistical Analysis
All data in the figures were expressed as (mean ± standard deviation) values. Statistical analysis of slice invasion data was performed by two-way ANOVA for repeated measures, followed by post-hoc Bonferroni’s test. Comparison of tumor volumes was performed by one-way ANOVA and post-hoc Tukey’s test. Survival curves (Kaplan-Meier plots) were compared by a logrank test with a two-tailed P value.
Results
Effect of the “NVY” mutation on B/b cleavage
To test whether the specific proteolysis of B/b by ADAMTS proteases was a necessary step for the effects of this proteoglycan in gliomas, we engineered a mutant version of B/b resistant to ADAMTS-mediated cleavage (Figure 1A). The single ADAMTS cleavage site of B/b is located at the Glu-Ser position within the sequence 393ESE/SRG398 in rat and mouse B/b, and 398ESE/SRG403 in the human protein. Previous work on rat aggrecan, which has an ADAMTS cleavage site with a similar sequence (371EGE/ARG376), has shown that mutation of the three amino acids downstream of the cleavage site from ARG to NVY completely blocks aggrecan cleavage at this site [44]. Therefore, we examined the effect of a similar mutation, ESE/SRG to ESE/NVY, on B/b cleavage.
CNS-1 rat glioma cells were transfected with the cDNAs of the full-length normal (B/b) or mutated (BNVY) B/b constructs and the resulting secreted proteins were analyzed by Western blotting (Figure 1B). Cells transfected with the normal B/b construct produced, as expected, the full-length, 150-kDa protein as well as a 50-kDa NH2-terminal fragment generated by ADAMTS activity [36]. In contrast, although a large amount of full-length protein was made by cells transfected with the BNVY construct, no ADAMTS cleavage product was detected using the antibody B50, which specifically detects the neo-epitope generated by cleavage at 395E/S396 ([36], figure 1A). To further confirm that the mutated form of B/b was completely resistant to cleavage, samples were immunoprecipitated with antibody B50 to detect trace amounts of the NH2-terminal cleavage product. Even with this higher sensitivity assay, no reactive product was detected (not shown), indicating that the BNVY mutant was not cleaved at the 395E/S396 site in culture conditions.
In addition, we probed the same samples with the antibody B5, which detects an epitope located in the NH2-terminus of B/b, to determine if BNVY was cleaved at any other site upstream or downstream of 395E/S396. The results (Figure 1C) showed that BNVY was not only resistant to cleavage at the ADAMTS site but that the mutation eliminated all major proteolytic processing of B/b in these glioma cells. Moreover, transient co-transfection of B/b with excess BNVY at DNA molar ratios up to 1:4 did not cause any reduction in the amount of N-terminal cleavage product detected with B50 (Figure 1D), indicating that BNVY was also unable to act as a dominant negative of B/b cleavage in cultured cells.
Taken together, these results indicate that a) the 395E/S396 cleavage position is the predominant site of B/b proteolytic processing in glioma cells and b) the sequence immediately downstream of the cleavage site is a major consensus element for recognition by the ADAMTS enzymes that cleave B/b. Interestingly, the ability of the SRG → NVY mutation seems to be somewhat unique in its effectiveness to produce complete inhibition of B/b cleavage at this site because other changes of this sequence were less successful to fully prevent proteolysis (RTM, unpublished observations).
Effect of uncleavable B/b on tumor cell proliferation and dispersion in vitro
Given the evident importance of cell growth and proliferation for tumor progression, we first compared the effect of the normal and mutated B/b constructs on cell growth, proliferation, and death rate prior to evaluating their effects on the invasive behavior of glioma cells. The growth rates of CNS-1 cells stably expressing B/b, BNVY, or GFP were analyzed over a 7-day period using the MTT-reduction assay, which yielded identical profiles for all the cell lines tested (Figure 2A). To verify that this result effectively reflected similar proliferation rates, we quantified the proportion of proliferating cells by Ki-67 staining and found them identical in vitro (Figure 2B). Finally, we tested the death rate in vitro by measuring the release of the enzyme LDH to the culture medium, and confirmed that all the cell lines also had similar death rates (Figure 2C). Thus, these results strongly suggested that any differences in tumor progression would likely reflect changes in the invasive rather than the proliferative ability of cells expressing B/b or BNVY. Indeed, we did not observe differences in the proliferative index of these cells against the controls in vivo (not shown).
Figure 2. Stable transfection with B/b constructs does not affect cell proliferation and death.
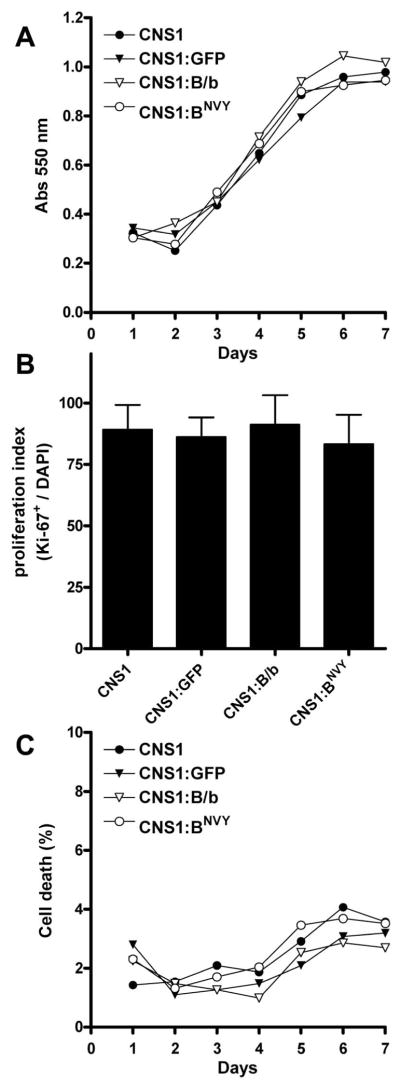
CNS-1 cells stably overexpressing normal B/b (CNS1:B/b), uncleavable B/b (CNS1:BNVY) or green fluorescent protein (CNS1:GFP) were compared to parental CNS-1 using assays to quantify growth rate (MTT assay, A), proliferative index (Ki-67 staining, B) and cell death (LDH release, C). No significant differences were observed in any of the cell lines, suggesting that transfection of B/b constructs does not affect the capacity of CNS-1 cells to grow and proliferate. Data were analyzed by two-way (A, C) and one-way (B) ANOVA, and represent the mean of four values per experimental condition. Error bars represented less than 10% variation in (A) and (C) and were removed for clarity.
To determine the effect of blocking B/b cleavage on the invasive behavior of glioma cells, we first employed the brain slice assay to closely mimic glioma dispersion in vitro. In this experimental model, glioma cells that have formed tumor “spheroids” in culture detach from those aggregates when placed onto brain slices and migrate through living neural tissue that retains most of the brain cytoarchitecture, including its natural barriers to cell movement. Importantly, cells grown as spheroids were negative for endogenous B/b expression by western blotting and qRT-PCR, but were able to cleave transfected B/b (not shown). This indicated that the change of culture conditions from monolayer to spheroid culture likely did not affect the expression of B/b or the metalloproteases involved in B/b cleavage.
Visual inspection of the tumor spheroids over 72h showed that individual cells effectively detached from the spheroids and infiltrated the surrounding parenchyma (Figure 3A–C). Analysis of the total cross-sectional area occupied by the tumor cells (Figure 3D) showed that as early as 48 hours, glioma spheroids expressing normal B/b already occupied a larger area than control spheroids (p<0.05). This area was ~50% larger than control by 72 hours post-seeding (p<0.001). In stark contrast, CNS-1 cells overexpressing the mutated BNVY showed significantly less dispersion than B/b-expressing cells, being essentially indistinguishable from control cells.
Figure 3. Overexpression of normal B/b, but not uncleavable B/b, increases CNS-1 cell dispersion on organotypic brain slices.

Spheroids of transfected CNS-1 cells were prepared and placed on slices as described in the methods section. A) Representative images obtained over 72h for cell aggregates overexpressing a control vector, B/b or BNVY; cells were visualized by co-transfection with a vector carrying red fluorescent protein. B–C) Visual inspection at higher magnification was routinely performed to confirm that cells were truly detaching from the spheroid mass and migrating throughout the brain slices (arrows). D) Migration index (Areatime=x/Areatime=0) values for CNS-1 cells transfected with B/b (n=15), BNVY (n=15) or control vector (n=12) were compared by two-way repeated measures ANOVA and post-hoc Bonferroni’s test. Results show that B/b-transfected cells dispersed more than controls or BNVY transfectants at 48h (p<0.05) and 72h (p<0.001), while BNVY-transfected cells did not differ significantly from controls (p>0.05). Bars (A, B): 20 μm; (C): 10 μm.
Effect of uncleavable B/b on tumor progression in vivo
To validate the results observed with B/b and BNVY in vitro, we further investigated the effects of these constructs in CNS-1 cells implanted intracranially. All CNS-1-derived tumors expressed B/b endogenously in vivo [33] and the expression of endogenous B/b was comparable to the levels of transfected B/b in these cells in vitro (Figure 4A). Thus, the increased expression of B/b in CNS-1:B/b and CNS-1:BNVY cells compared to CNS-1:GFP cells was likely within physiologically relevant limits, as well as comparable to the levels of B/b previously observed in human tumors [39].
Figure 4. Overexpression of normal or uncleavable B/b has a differential effect on tumor progression in vivo.

A) Endogenous soluble brevican detected in CNS-1:GFP tumors (column 2) was comparable to the level of transfected B/b in CNS-1:B/b cells in vitro (column 1), resulting in an expected overexpression of B/b and its NH2-cleavage product in CNS-1:B/b tumors (column 3). B) Whole coronal sections (left) and 4X-magnification images (right) of gliomas produced 8 days after intracranial injection of CNS-1 cells. Images on the right column show partial drawing (dotted line) of the perimeter of the tumor mass (asterisk) and cell clusters (arrows) included in the morphometric analysis. Bar= 0.5 mm. C) Tumors from B/b-overexpressing cells were significantly larger than tumors from control or BNVY-expressing cells (## p< 0.01 by one-way ANOVA).
Eight days after tumor implantation, animals were perfused and their brains processed for histology to analyze tumor infiltration in the surrounding tissue. In agreement with previous descriptions of this cell line [40,45], all CNS-1-derived gliomas were invasive and exhibited cell clusters detached from the tumor core (Figure 4B,C), as well as extensive infiltration of single cells within the brain parenchyma. CNS-1 cells stably overexpressing B/b produced larger, more diffuse and more infiltrative tumors than control CNS-1 cells (mean ± S.E.M. volumes: 9.12 ± 1.43 mm3 versus 3.85 ± 1.20 mm3; n=8, p<0.01; Figure 4D). However, tumors produced by CNS-1 cells overexpressing BNVY (mean ± S.E.M. volume: 3.79 ± 1.39 mm3; n=8) did not differ in size from control tumors.
As expected from these results on tumor burden, animals implanted with CNS-1 cells overexpressing B/b reached the survival endpoint significantly earlier than controls (median value 18.0 days versus 23.2 days; n=6, p<0.005; Figure 5). On the other hand, animals implanted with CNS-1 cells overexpressing BNVY survived as long as control animals (median value 23.6 days; n=6). While these differences in survival endpoint seem small, it is worth noting that by the time the first animal bearing a BNVY-tumor died, all the animals bearing B/b-expressing tumors were already dead, thus underscoring the increased lethality of cleavable versus non-cleavable B/b on tumor progression. More importantly, because we did not detect differences in cell proliferation, these results strongly suggest that the observed differences in tumor progression and animal survival were due to increased invasion of tumor cells overexpressing normal B/b, and they indicate that cleavage of the full-length protein was critical for its pro-invasive effect.
Figure 5. Overexpression of normal B/b, but not the uncleavable construct, decreases the survival of tumor-bearing animals.

Animals (n=6/group) were implanted as described in the methods section with a total of 3.105 stably transfected CNS-1 cells, and followed for a period of 25 days after injection. Animals with tumors overexpressing normal B/b (CNS1:B/b) survived a significantly (p<0.005) shorter time than both animals with control tumors (CNS1:GFP) or with tumors that overexpressed BNVY (CNS1:BNVY). Conversely, animals bearing CNS1:BNVY tumors were not different (p>0.05) from controls (Kaplan-Meier survival curves analyzed by two-tailed logrank test).
Discussion
B/b, one of the most abundant proteoglycans in the adult neural ECM, exhibits several features that make it a relevant target for brain tumor therapeutics, including CNS-specific expression, consistent upregulation in gliomas [46,47], enhancing effect on glioma invasion [35], correlation with decreased patient survival [48], and the presence of a glioma-specific, cell surface-associated B/b isoform [39]. The molecular mechanisms by which B/b promote glioma progression are mostly unknown, but current evidence suggests that they likely affect cell adhesion and motility on specific substrates (MSV and RTM, manuscript in preparation). It is important, therefore, to understand the key events that turn B/b, a major inhibitory component of the neural matrix, into a pro-invasive component of the glioma matrix.
Previous work from our laboratories has shown that the rat gliosarcoma cell line 9L, characterized in vivo as a poorly invasive brain tumor [40], does not express B/b endogenously [33] and, when transfected with the full-length protein, is also unable to cleave it at the ADAMTS site [34]. Moreover, both control and B/b-overexpressing 9L cells exhibit the same non-infiltrative behavior when grown as intracranial grafts and produce tumors that do not differ in size [34]. However, transfection of 9L cells with constructs corresponding to the NH2-terminal cleavage fragment of B/b allows them to disperse through neural tissue, both in vivo [34] and in organotypic brain slices (MSV and RTM, unpublished results). This phenotypic change of 9L cells after transfection with the NH2-terminal fragment of B/b makes them behave similarly to CNS-1 cells, which express and cleave B/b endogenously when grafted into the brain and exhibit the infiltrative behavior typical of high-grade astrocytomas [33]. Although these results have shown that cleavage of B/b is sufficient to increase the invasiveness of glioma cells, they have not determined whether this cleavage is necessary for the pro-invasive effect of B/b and whether this pro-invasive effect is achieved by a competition between the B/b cleavage products and the full-length protein.
Here we have analyzed these hypotheses and demonstrated that 1) cleavage of B/b at the ADAMTS site is the predominant proteolytic processing of this protein in glioma cells, 2) cleavage of B/b is a critical step for its pro-invasive role in gliomas, and 3) there is likely no competition between the B/b cleavage products and the full-length protein to promote glioma invasion.
First, while B/b has been shown to be a substrate for several types of MMPs under specific conditions in vitro [37,49], our results show that cleavage of B/b in glioma cells in culture occurs almost exclusively at the ADAMTS cleavage site. These results are in agreement with our previous observations in vivo and in several cell lines [34,36,38,39], and indicate that cleavage of B/b by proteases other than ADAMTS, while possible, may not contribute appreciably to glioma invasion.
Second, we have shown here that overexpression of normal B/b versus its uncleavable form, BNVY, does not affect the proliferation or death rate of transfected CNS-1 glioma cells, but causes marked differences in their invasive behavior. Importantly, these differences were very consistent between our in vitro and in vivo models, suggesting that the organotypic slice invasion assays accurately reproduce the exposure of glioma cells to an organized neural matrix. Our results strongly suggest that CNS-1 cells overexpressing B/b disperse more and produce more cell clusters in the brain parenchyma, resulting in more diffuse tumors that spread over a larger volume and cause a faster deadly outcome. In marked contrast, BNVY-overexpressing cells do not differ from control cells regarding tumor dispersion in vitro, tumor infiltration in vivo or total tumor volume, even though they produce significant amounts of full-length B/b. Taken together, these results indicate that only B/b cleavage products, and not the full-length protein, have a pro-invasive role in gliomas.
It is important to remark that intracranial grafts of CNS-1 cells express and cleave B/b endogenously (Figure 4A), as previously reported [33,38]. The absence of any inhibitory effects by BNVY on cell invasion indicates that this construct did not affect the expression or cleavage of endogenous B/b, a lack of effect that we verified by co-expression of B/b and BNVY in culture (Figure 1D). More importantly, these results suggest that full-length B/b and its cleavage products likely function through independent mechanisms. It is unlikely that the products generated by B/b cleavage simply out-compete the full-length protein for attachment sites in the matrix scaffold, otherwise overexpressed BNVY should have acted as a dominant negative of endogenous B/b in vivo. Because this effect was not observed, we hypothesize that the cleavage products of B/b may have unique molecular associations that are unaffected by the excess of full-length protein. This underscores the critical role of proteolytic cleavage for the pro-invasive role of B/b in gliomas.
A large body of literature has illuminated the relevance of proteases and protease inhibitors in modulating the progression of malignant brain tumors (e.g. [12,17,50]). Of the several classes of proteases, members of the MMP family of metalloproteases have received the most attention for the particularly important roles they play in gliomas. The functions of MMPs, which include activation of growth factors, suppression of apoptosis in tumor cells, and release of pro-angiogenic factors, contribute to the formation of a microenvironment that promotes the early phases of growth and division of newly transformed cells [28]. The inhibition of MMP activity has been explored as a therapeutic avenue for the treatment of gliomas, mostly with disappointing results due to the lack of knowledge of the complex physiological roles of these enzymes [28]. Our present work suggests that cleavage mediated by another family of ECM metalloproteases, the ADAMTS, may also play critical roles in enhancing the progression of glial tumors.
The identity of the specific ADAMTS proteases that cleave B/b at the ADAMTS site in vivo is still not fully defined. Current evidence strongly suggests ADAMTS-4 (aggrecanase-1) and ADAMTS-5 (aggrecanase-2) as the most likely candidates, which are also upregulated in gliomas [36,51,52]. These two proteases also cleave two other members of the lectican family, aggrecan [53] and versican [54], and have a restricted additional spectrum of ECM substrates [55–57]. Other members of the ADAMTS family have been shown or proposed to have aggrecanase activity (recently reviewed in [58]) but their expression in the CNS is low and there is currently no evidence for their role in B/b processing in gliomas [59–63].
In conclusion, the upregulation and pro-invasive effect of B/b and its processing enzymes in malignant gliomas make the ADAMTS proteases relevant therapeutic targets that exhibit certain advantages over MMPs, including a more restricted expression profile and narrower substrate specificity. Inhibition of ADAMTS activity may thus prove a useful therapeutic strategy for glioma management. Moreover, our present results show that the inhibition of cleavage of a single ADAMTS substrate such as B/b renders this ECM protein functionally inactive as a pro-invasive molecule in gliomas. Thus, inhibition of specific ADAMTS targets in gliomas could slow glioma invasion and may represent an important complementary avenue for the treatment of these tumors.
Acknowledgments
This work was supported by research grants from NIH (R01NS035228) and the Accelerate Brain Cancer Cure foundation (to RTM), and by a fellowship from the Butler Family Foundation/American Brain Tumor Association (to MSV).
References
- 1.Bolteus AJ, Berens ME, Pilkington GJ. Migration and invasion in brain neoplasms. Curr Neurol Neurosci Rep. 2001;1(3):225–232. doi: 10.1007/s11910-001-0022-x. [DOI] [PubMed] [Google Scholar]
- 2.Lefranc F, Brotchi J, Kiss R. Possible future issues in the treatment of glioblastomas: special emphasis on cell migration and the resistance of migrating glioblastoma cells to apoptosis. J Clin Oncol. 2005;23(10):2411–2422. doi: 10.1200/JCO.2005.03.089. [DOI] [PubMed] [Google Scholar]
- 3.Giese A, Bjerkvig R, Berens ME, et al. Cost of migration: invasion of malignant gliomas and implications for treatment. J Clin Oncol. 2003;21(8):1624–1636. doi: 10.1200/JCO.2003.05.063. [DOI] [PubMed] [Google Scholar]
- 4.Louis DN. Molecular Pathology of Malignant Gliomas. Annu Rev Pathol Mech Dis. 2006;1:97–117. doi: 10.1146/annurev.pathol.1.110304.100043. Ref Type: Generic. [DOI] [PubMed] [Google Scholar]
- 5.Pilkington GJ. The paradox of neoplastic glial cell invasion of the brain and apparent metastatic failure. Anticancer Res. 1997;17(6B):4103–5. [PubMed] [Google Scholar]
- 6.Subramanian A, Harris A, Piggott K, et al. Metastasis to and from the central nervous system--the ‘relatively protected site’. Lancet Oncol. 2002;3(8):498–507. doi: 10.1016/s1470-2045(02)00819-7. [DOI] [PubMed] [Google Scholar]
- 7.Novak U, Kaye AH. Extracellular matrix and the brain: components and function. J Clin Neurosci. 2000;7(4):280–90. doi: 10.1054/jocn.1999.0212. [DOI] [PubMed] [Google Scholar]
- 8.Gladson CL. The extracellular matrix of gliomas: modulation of cell function. J Neuropathol Exp Neurol. 1999;58(10):1029–40. doi: 10.1097/00005072-199910000-00001. [DOI] [PubMed] [Google Scholar]
- 9.Bellail AC, Hunter SB, Brat DJ, et al. Microregional extracellular matrix heterogeneity in brain modulates glioma cell invasion. Int J Biochem Cell Biol. 2004;36(6):1046–69. doi: 10.1016/j.biocel.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 10.Goldbrunner RH, Bernstein JJ, Tonn JC. Cell-extracellular matrix interaction in glioma invasion. Acta Neurochir (Wien) 1999;141(3):295–305. doi: 10.1007/s007010050301. [DOI] [PubMed] [Google Scholar]
- 11.Binder DK, Berger MS. Proteases and the biology of glioma invasion. J Neurooncol. 2002;56(2):149–158. doi: 10.1023/a:1014566604005. [DOI] [PubMed] [Google Scholar]
- 12.Rao JS. Molecular mechanisms of glioma invasiveness: the role of proteases. Nat Rev Cancer. 2003;3(7):489–501. doi: 10.1038/nrc1121. [DOI] [PubMed] [Google Scholar]
- 13.Novak U, Stylli SS, Kaye AH, et al. Hyaluronidase-2 overexpression accelerates intracerebral but not subcutaneous tumor formation of murine astrocytoma cells. Cancer Res. 1999;59(24):6246–50. [PubMed] [Google Scholar]
- 14.Bjerkvig R, Lund-Johansen M, Edvardsen K. Tumor cell invasion and angiogenesis in the central nervous system. Curr Opin Oncol. 1997;9(3):223–229. doi: 10.1097/00001622-199709030-00002. [DOI] [PubMed] [Google Scholar]
- 15.Bello L, Giussani C, Carrabba G, et al. Angiogenesis and invasion in gliomas. Cancer Treat Res. 2004:117263–284. doi: 10.1007/978-1-4419-8871-3_16. [DOI] [PubMed] [Google Scholar]
- 16.VanMeter TE, Rooprai HK, Kibble MM, et al. The role of matrix metalloproteinase genes in glioma invasion: co-dependent and interactive proteolysis. J Neurooncol. 2001;53(2):213–235. doi: 10.1023/a:1012280925031. [DOI] [PubMed] [Google Scholar]
- 17.Nakada M, Okada Y, Yamashita J. The role of matrix metalloproteinases in glioma invasion. Front Biosci. 2003:8e261–e269. doi: 10.2741/1016. [DOI] [PubMed] [Google Scholar]
- 18.Rooprai HK, McCormick D. Proteases and their inhibitors in human brain tumours: a review. Anticancer Res. 1997;17(6B):4151–4162. [PubMed] [Google Scholar]
- 19.Levicar N, Nuttall RK, Lah TT. Proteases in brain tumour progression. Acta Neurochir (Wien ) 2003;145(9):825–838. doi: 10.1007/s00701-003-0097-z. [DOI] [PubMed] [Google Scholar]
- 20.Tsatas D, Kaye AH. The role of the plasminogen activation cascade in glioma cell invasion: a review. J Clin Neurosci. 2003;10(2):139–145. doi: 10.1016/s0967-5868(02)00328-4. [DOI] [PubMed] [Google Scholar]
- 21.VanMeter TE, Rooprai HK, Kibble MM, et al. The role of matrix metalloproteinase genes in glioma invasion: co-dependent and interactive proteolysis. J Neurooncol. 2001;53(2):213–235. doi: 10.1023/a:1012280925031. [DOI] [PubMed] [Google Scholar]
- 22.Levicar N, Nuttall RK, Lah TT. Proteases in brain tumour progression. Acta Neurochir (Wien ) 2003;145(9):825–838. doi: 10.1007/s00701-003-0097-z. [DOI] [PubMed] [Google Scholar]
- 23.Rooprai HK, Kandanearatchi A, Maidment SL, et al. Evaluation of the effects of swainsonine, captopril, tangeretin and nobiletin on the biological behaviour of brain tumour cells in vitro. Neuropathol Appl Neurobiol. 2001;27(1):29–39. doi: 10.1046/j.0305-1846.2000.00298.x. [DOI] [PubMed] [Google Scholar]
- 24.Rooprai HK, Kandanearatachi A, Rucklidge G, et al. Influence of putative antiinvasive agents on matrix metalloproteinase secretion by human neoplastic glia in vitro. Ann N Y Acad Sci. 1999:878654–657. doi: 10.1111/j.1749-6632.1999.tb07753.x. [DOI] [PubMed] [Google Scholar]
- 25.Tonn JC, Kerkau S, Hanke A, et al. Effect of synthetic matrix-metalloproteinase inhibitors on invasive capacity and proliferation of human malignant gliomas in vitro. Int J Cancer. 1999;80(5):764–772. doi: 10.1002/(sici)1097-0215(19990301)80:5<764::aid-ijc22>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 26.Price A, Shi Q, Morris D, et al. Marked inhibition of tumor growth in a malignant glioma tumor model by a novel synthetic matrix metalloproteinase inhibitor AG3340. Clin Cancer Res. 1999;5(4):845–854. [PubMed] [Google Scholar]
- 27.Watanabe K, Yoshida D, Noha M, et al. Suppression of matrix metalloproteinase-2 and -9 mediated invasiveness by a novel matrix metalloproteinase inhibitor, BE16627B. J Neurooncol. 2001;52(1):1–9. doi: 10.1023/a:1010639313832. [DOI] [PubMed] [Google Scholar]
- 28.Folgueras AR, Pendas AM, Sanchez LM, et al. Matrix metalloproteinases in cancer: from new functions to improved inhibition strategies. Int J Dev Biol. 2004;48(5–6):411–424. doi: 10.1387/ijdb.041811af. [DOI] [PubMed] [Google Scholar]
- 29.Heath EI, Grochow LB. Clinical potential of matrix metalloprotease inhibitors in cancer therapy. Drugs. 2000;59(5):1043–1055. doi: 10.2165/00003495-200059050-00002. [DOI] [PubMed] [Google Scholar]
- 30.Steward WP, Thomas AL. Marimastat: the clinical development of a matrix metalloproteinase inhibitor. Expert Opin Investig Drugs. 2000;9(12):2913–2922. doi: 10.1517/13543784.9.12.2913. [DOI] [PubMed] [Google Scholar]
- 31.Yamaguchi Y. Lecticans: organizers of the brain extracellular matrix. Cell Mol Life Sci. 2000;57(2):276–89. doi: 10.1007/PL00000690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Viapiano MS, Matthews RT. From barriers to bridges: chondroitin sulfate proteoglycans in neuropathology. Trends Mol Med. 2006;12(10):488–496. doi: 10.1016/j.molmed.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 33.Jaworski DM, Kelly GM, Piepmeier JM, et al. BEHAB (brain enriched hyaluronan binding) is expressed in surgical samples of glioma and in intracranial grafts of invasive glioma cell lines. Cancer Res. 1996;56(10):2293–8. [PubMed] [Google Scholar]
- 34.Zhang H, Kelly G, Zerillo C, et al. Expression of a cleaved brain-specific extracellular matrix protein mediates glioma cell invasion In vivo. J Neurosci. 1998;18(7):2370–6. doi: 10.1523/JNEUROSCI.18-07-02370.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nutt CL, Zerillo CA, Kelly GM, et al. Brain enriched hyaluronan binding (BEHAB)/brevican increases aggressiveness of CNS-1 gliomas in Lewis rats. Cancer Res. 2001;61(19):7056–9. [PubMed] [Google Scholar]
- 36.Matthews RT, Gary SC, Zerillo C, et al. Brain-enriched hyaluronan binding (BEHAB)/brevican cleavage in a glioma cell line is mediated by a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS) family member. J Biol Chem. 2000;275(30):22695–703. doi: 10.1074/jbc.M909764199. [DOI] [PubMed] [Google Scholar]
- 37.Nakamura H, Fujii Y, Inoki I, et al. Brevican is degraded by matrix metalloproteinases and aggrecanase-1 (ADAMTS4) at different sites. J Biol Chem. 2000;275(49):38885–90. doi: 10.1074/jbc.M003875200. [DOI] [PubMed] [Google Scholar]
- 38.Viapiano MS, Matthews RT, Hockfield S. A Novel Membrane-associated Glycovariant of BEHAB/Brevican Is Up-regulated during Rat Brain Development and in a Rat Model of Invasive Glioma. J Biol Chem. 2003;278(35):33239–47. doi: 10.1074/jbc.M303480200. [DOI] [PubMed] [Google Scholar]
- 39.Viapiano MS, Bi WL, Piepmeier J, et al. Novel tumor-specific isoforms of BEHAB/brevican identified in human malignant gliomas. Cancer Res. 2005;65(15):6726–6733. doi: 10.1158/0008-5472.CAN-05-0585. [DOI] [PubMed] [Google Scholar]
- 40.Kruse CA, Molleston MC, Parks EP, et al. A rat glioma model, CNS-1, with invasive characteristics similar to those of human gliomas: a comparison to 9L gliosarcoma. J Neurooncol. 1994;22(3):191–200. doi: 10.1007/BF01052919. [DOI] [PubMed] [Google Scholar]
- 41.Yamada H, Watanabe K, Shimonaka M, et al. Molecular cloning of brevican, a novel brain proteoglycan of the aggrecan/versican family. J Biol Chem. 1994;269(13):10119–26. [PubMed] [Google Scholar]
- 42.Ohnishi T, Matsumura H, Izumoto S, et al. A novel model of glioma cell invasion using organotypic brain slice culture. Cancer Res. 1998;58(14):2935–2940. [PubMed] [Google Scholar]
- 43.Rosen GD, Harry JD. Brain volume estimation from serial section measurements: a comparison of methodologies. J Neurosci Methods. 1990;35(2):115–124. doi: 10.1016/0165-0270(90)90101-k. [DOI] [PubMed] [Google Scholar]
- 44.Mercuri FA, Maciewicz RA, Tart J, et al. Mutations in the interglobular domain of aggrecan alter matrix metalloproteinase and aggrecanase cleavage patterns. Evidence that matrix metalloproteinase cleavage interferes with aggrecanase activity. J Biol Chem. 2000;275(42):33038–33045. doi: 10.1074/jbc.275.42.33038. [DOI] [PubMed] [Google Scholar]
- 45.Candolfi M, Curtin JF, Nichols WS, et al. Intracranial glioblastoma models in preclinical neuro-oncology: neuropathological characterization and tumor progression. J Neurooncol. 2007 doi: 10.1007/s11060-007-9400-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boon K, Edwards JB, Eberhart CG, et al. Identification of astrocytoma associated genes including cell surface markers. BMC Cancer. 2004;4(1):39. doi: 10.1186/1471-2407-4-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Phillips HS, Kharbanda S, Chen R, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9(3):157–173. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 48.Liang Y, Diehn M, Watson N, et al. Gene expression profiling reveals molecularly and clinically distinct subtypes of glioblastoma multiforme. Proc Natl Acad Sci U S A. 2005;102(16):5814–5819. doi: 10.1073/pnas.0402870102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Muir EM, Adcock KH, Morgenstern DA, et al. Matrix metalloproteases and their inhibitors are produced by overlapping populations of activated astrocytes. Brain Res Mol Brain Res. 2002;100(1–2):103–117. doi: 10.1016/s0169-328x(02)00132-8. [DOI] [PubMed] [Google Scholar]
- 50.Vihinen P, la-aho R, Kahari VM. Matrix metalloproteinases as therapeutic targets in cancer. Curr Cancer Drug Targets. 2005;5(3):203–220. doi: 10.2174/1568009053765799. [DOI] [PubMed] [Google Scholar]
- 51.Held-Feindt J, Paredes EB, Blomer U, et al. Matrix-degrading proteases ADAMTS4 and ADAMTS5 (disintegrins and metalloproteinases with thrombospondin motifs 4 and 5) are expressed in human glioblastomas. Int J Cancer. 2006;118(1):55–61. doi: 10.1002/ijc.21258. [DOI] [PubMed] [Google Scholar]
- 52.Nakada M, Miyamori H, Kita D, et al. Human glioblastomas overexpress ADAMTS-5 that degrades brevican. Acta Neuropathol (Berl) 2005;110(3):239–246. doi: 10.1007/s00401-005-1032-6. [DOI] [PubMed] [Google Scholar]
- 53.Arner EC. Aggrecanase-mediated cartilage degradation. Curr Opin Pharmacol. 2002;2(3):322–329. doi: 10.1016/s1471-4892(02)00148-0. [DOI] [PubMed] [Google Scholar]
- 54.Westling J, Gottschall PE, Thompson VP, et al. ADAMTS4 (aggrecanase-1) cleaves human brain versican V2 at Glu405-Gln406 to generate glial hyaluronate binding protein. Biochem J. 2004;377(Pt 3):787–795. doi: 10.1042/BJ20030896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kashiwagi M, Enghild JJ, Gendron C, et al. Altered proteolytic activities of ADAMTS-4 expressed by C-terminal processing. J Biol Chem. 2004;279(11):10109–10119. doi: 10.1074/jbc.M312123200. [DOI] [PubMed] [Google Scholar]
- 56.Tortorella MD, Arner EC, Hills R, et al. Alpha2-macroglobulin is a novel substrate for ADAMTS-4 and ADAMTS-5 and represents an endogenous inhibitor of these enzymes. J Biol Chem. 2004;279(17):17554–17561. doi: 10.1074/jbc.M313041200. [DOI] [PubMed] [Google Scholar]
- 57.Melching LI, Fisher WD, Lee ER, et al. The cleavage of biglycan by aggrecanases. Osteoarthritis Cartilage. 2006 doi: 10.1016/j.joca.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 58.Porter S, Clark IM, Kevorkian L, et al. The ADAMTS metalloproteinases. Biochem J. 2005;386(Pt 1):15–27. doi: 10.1042/BJ20040424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cal S, Obaya AJ, Llamazares M, et al. Cloning, expression analysis, and structural characterization of seven novel human ADAMTSs, a family of metalloproteinases with disintegrin and thrombospondin-1 domains. Gene. 2002;283(1–2):49–62. doi: 10.1016/s0378-1119(01)00861-7. [DOI] [PubMed] [Google Scholar]
- 60.Georgiadis KE, Hirohata S, Seldin MF, et al. ADAM-TS8, a novel metalloprotease of the ADAM-TS family located on mouse chromosome 9 and human chromosome 11. Genomics. 1999;62(2):312–315. doi: 10.1006/geno.1999.6014. [DOI] [PubMed] [Google Scholar]
- 61.Somerville RP, Longpre JM, Jungers KA, et al. Characterization of ADAMTS-9 and ADAMTS-20 as a distinct ADAMTS subfamily related to Caenorhabditis elegans GON-1. J Biol Chem. 2003;278(11):9503–9513. doi: 10.1074/jbc.M211009200. [DOI] [PubMed] [Google Scholar]
- 62.Jungers KA, Le GC, Somerville RP, et al. Adamts9 is widely expressed during mouse embryo development. Gene Expr Patterns. 2005;5(5):609–617. doi: 10.1016/j.modgep.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 63.Dunn JR, Reed JE, du Plessis DG, et al. Expression of ADAMTS-8, a secreted protease with antiangiogenic properties, is downregulated in brain tumours. Br J Cancer. 2006;94(8):1186–1193. doi: 10.1038/sj.bjc.6603006. [DOI] [PMC free article] [PubMed] [Google Scholar]