

Abstract
The Tsuji-Trost allylic substitution reaction provides a useful and efficient approach to construct C-C bonds between sp3-hybridized carbons. The widely accepted paradigm for classifying the mode of attack of nucleophiles on palladium π-allyl intermediates in the Tsuji-Trost reaction is based on the pKa of the pronucleophile: (1) stabilized or “soft” carbon nucleophiles and heteroatom nucleophiles (e.g., pronucleophiles with pKa’s < 25), and (2) unstabilized or “hard” nucleophiles (those from pronucleophiles with pKa’s > 25). One of the keys to the continuing development of allylic substitution processes remains broadening the scope of “soft” nucleophiles. Herein we report a general method for the room temperature Pd-catalyzed allylic substitution with diarylmethane derivatives (pKa’s up to 32). The synthetic significance of the method is that it provides a rapid access to products containing allylated diarylmethyl motifs. The method is general for a wide range of nucleophiles derived from diarylmethanes and heterocyclic derivatives. A procedure for the Pd-catalyzed allylic substitutions to afford diallylation products with quaternary centers is also described. With triarylmethanes and, alkylated diarylmethanes the corresponding allylated products are isolated. We anticipate that the described method will be a valuable complement to the existing arsenal of nucleophiles in Pd-catalyzed allylic substitutions. Mechanistic studies show that the nucleophile derived from diphenylmethane undergoes external attack on π-allyl palladium species under our reaction conditions. This unexpected observation indicates that diarylmethane derivatives behave as “soft” or stabilized nucleophiles. The results of this study indicate that the cutoff between “soft” and “hard” nucleophiles should be raised from a pronucleophile pKa of 25 to at least 32.
1. INTRODUCTION
Palladium-catalyzed C-C bond-forming reactions are among the most important and well-developed processes in modern organic chemistry.1–10 Of these, the Tsuji-Trost allylic substitution reaction provides a useful and efficient approach to construct C-C bonds between sp3-hybridized carbons. As a result, it has been widely used to synthesize natural products and bioactive molecules. 11–17 The widely accepted paradigm for classifying the mode of attack of nucleophiles on transition metal η3-π-allyl intermediates in the Tsuji-Trost reaction is based on the pKa of the pronucleophile.18 Nucleophiles are divided into two classes: 1) stabilized or “soft” carbon nucleophiles and heteroatom nucleophiles (e.g., enolates and those from pronucleophiles with pKa’s < 25), and (2) unstabilized or “hard” nucleophiles (those from pronucleophiles with pKa’s > 25). The distinction between these two classes is “soft” nucleophiles directly attack the π-allyl moiety while “hard” nucleophiles first attack the metal center (via transmetallation) before bond formation with the allyl group. Importantly, these two pathways lead to distinct stereochemical outcomes (soft nucleophiles result in net retention in the Tsuji-Trost allylic substitution whereas hard nucleophiles react by single inversion). The scope of “soft” nucleophiles has received significant attention in asymmetric catalysis,11,13,14,19 although nonenantioselective Pd-catalyzed allylic substitution with “hard” nucleophiles are also known.20–22
One of the keys to the continuing development of allylic substitution processes remains broadening the scope of “soft” nucleophiles. With this in mind, Trost and co-workers increased the reach of “soft” nucleophiles in the allylic substitution with use of 2-picoline-derived nucleophiles (pKa = 34)23. Essential to their success was to increase the acidity of the 2-picoline CH3. This was accomplished by BF3 coordination to the pyridine nitrogen to facilitate deprotonation. The resulting “softened” nucleophile was successfully employed in Pd-catalyzed asymmetric allylic alkylation (AAA, Scheme 1A).18,24 The same group later reported no such activation was necessary with more acidic heterocycles, including pyrazine, pyrimidine, pyridazine, quinoxaline, and benzimidazole derivatives. (Scheme 1B)25
Scheme 1.

Palladium-Catalyzed Benzylic Allylations
Our group recently introduced a strategy to employ toluene derivatives (RC6H4–CH2R’, pKa ∼ 44)26 as “soft” pronucleophiles in Pd-catalyzed allylic substitution reactions using Cr(CO)3 to increase the acidity of the benzylic C–H’s (Scheme 1C).27 The drawback of this approach is the stoichiometric use of chromium. We, therefore, set out to develop benzylic nucleophiles in the absence of chromium activating groups.
Application of unactivated diarylmethane derivatives as pronucleophiles in Pd-catalyzed allylic substitution reactions are unknown, but would have significant potential in medicinal chemistry. Hundreds of bioactive drug-like molecules contain allylated diarylmethyl motifs (Figure 1), with applications in treatment of breast cancer28, inhibitor of HIV protease29, blocker of human T-cells30 and antagonist of the thyroid hormone receptor,31 among others. Given these important applications, we set out to introduce diarylmethane-derived nucleophiles for the Pd-catalyzed allylic substitution. To achieve this objective, it is essential to find conditions to deprotonate diarylmethane derivatives that are compatible with the catalyst and substrates. Based on our previous studies on deprotonative cross-coupling processes (DCCP) using diarylmethane derivatives,32–35 we hypothesized that diarylmethane derivatives could be reversibly deprotonated in situ by MN(SiMe3)2 (M = Li, Na, K) under mild conditions. These conditions would be more amenable to catalysis than deprotonation under traditional conditions with n-BuLi at low temperature.36 Herein we report an approach to the room temperature Pd-catalyzed allylic substitution with diarylmethane derivatives (Scheme 1D). This method enables rapid access to a variety of allylated products, including heteroaryl-containing derivatives as well as molecules bearing quaternary centers. The mild reaction conditions that have been identified employ MN(SiMe3)2 at room temperature and a Pd catalyst based on van Leeuwen’s Xantphos ligand.37 Surprisingly, stereochemical studies indicate that nucleophiles derived from diarylmethane derivatives (pKa = 25–33)26,38 behave as “soft” nucleophiles, significantly extending the range of nucleophiles undergoing the double inversion mechanism in the Tsuji–Trost allylic substitution.
Figure 1.

Selected bioactive compounds containing the allylated diarylmethyl motif.
2. RESULTS AND DISCUSSION
As mentioned above, we recently disclosed a Pd-Xantphos catalyst system to promote the allylic substitution with Cr(CO)3-stabilized toluene-derived nucleophiles (eq 1).27 We hypothesized the reaction conditions in eq 1 would be a good starting point for allylic substitution with diarylmethanes and related pronucleophiles.
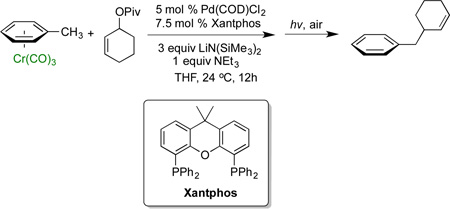 |
(1) |
2.1. Development and Optimization of Palladium-Catalyzed Allylic Substitution with Diarylmethanes
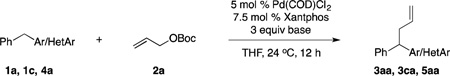
Given the perceived challenge to the application of diphenylmethane (pKa = 32.2)39 in allylic substitutions, we initiated our studies with the more acidic 2-benzylpyridine (1a, pKa = 28.2)38 under the reaction conditions in eq 1. The desired product 3aa was formed in 80% assay yield (Table 1, entry 1). Switching the base to NaN(SiMe3)2 led to 3aa in 99% assay yield (entry 2). The additive NEt3, which proved very useful in eq 1, was not necessary for the Pd-catalyzed allylic substitution with 2-benzylpyridine (entry 3 vs 2). The allylic substitution product formed with 2-benzylpyridine was isolated in 99% yield.
Table 1.
Preliminary Results of Allylic Substitution Reactionsa
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Pronuelephiles | Base | Product | Yieldb (%) | ||
| 1d | 1a | LiN(SiMe3)2 |  |
3aa | 80 | |
| 2d | 1a | NaN(SiMe3)2 |  |
3aa | (99c) | |
| 3 | 1a | NaN(SiMe3)2 | 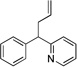 |
3aa | (99c) | |
| 4 | 1c | NaN(SiMe3)2 | 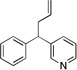 |
3ca | 27 | |
| 5 | 1c | KN(SiMe3)2 | 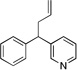 |
3ca | 70 | |
| 6 | 4a | KN(SiMe3)2 | 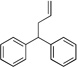 |
5aa | 10 | |
Reaction conducted on a 0.1 mmol scale with 1 equiv of pronucleophile and 2 equiv of 2a at 0.1 M.
Yield determined by 1H NMR spectroscopy of the crude reaction mixture.
Isolated yield after chromatographic purification.
1 equiv of NEt3.
We next sought to increase the pKa of the pronucleophile. The less acidic 3-benzylpyridine (1c) (pKa = 30.1),38 however, led to only 27% assay yield (entry 4). We increased the assay yield of 3ca to 70% by using the more reactive base, KN(SiMe3)2 (entry 5). Unfortunately, further decreasing the acidity of the pronucleophile was challenging: using diphenylmethane (4aa, pKa = 32.3)39 gave desired product 5aa in only 10% assay yield (entry 6). We next set out to optimize the reaction conditions for Pd-catalyzed allylic substitution with pronucleophiles with pKa’s >30, such as diphenylmethane.
From the results in Table 1, we hypothesized that the generation of the deprotonated diphenylmethane was problematic, and that choice of base would significantly impact the conversion. Since the traditional protocols preparing allylated diarylmethanes require strong bases (such as n-BuLi) at low temperature36 or under other harsh reaction conditions40, we limited ourselves to use of KN(SiMe3)2 as the strongest base in this study. We then screened different ethereal solvents (THF, 2-methyl THF, 1,4-dioxane, DME and CPME) and found that DME was the leading solvent of those examined (Table 2, entry 2, see the Supporting Information for details). Another important variable in optimizing Pd-catalyzed allylic substitution with diphenylmethane is the ratio of reagents. We observed decomposition of allyl tert-butyl carbonate 2a during the reaction. Increasing 2a to 3 equiv led to significant improvement in yield (entry 3). In addition, increasing the concentration of base led to higher concentrations of the nucleophile and improved yields (entries 4–5). Under these conditions, the allylic substitution product 5aa was obtained in 95% isolated yield in DME with diphenylmethane 4a as the limiting reagent, 5 equiv of KN(SiMe3)2 and 3 equiv of the allyl electrophile 2a. The yield dropped if the equivalence of 2a (entry 6) or the catalyst loading (entry 7) were decreased. With the optimized conditions in Tables 1 and 2, we examined various benzylic heterocycles and diarylmethanes as pronucleophiles in the allylic substitution.
Table 2.
Optimization of Allylic Substitution with Diphenyl-methane 4aa
 | |||
|---|---|---|---|
| Entry | Ratio (4a:base:2a) |
Solvent | yieldb(%) |
| 1 | 1:3:2 | THF | 10 |
| 2 | 1:3:2 | DME | 55 |
| 3 | 1:3:3 | DME | 79 |
| 4 | 1:4:3 | DME | 88 |
| 5 | 1:5:3 | DME | (95c) |
| 6 | 1:5:2 | DME | 62 |
| 7d | 1:5:3 | DME | 68 |
Reaction conducted on a 0.1 mmol scale at 0.1 M.
Yield determined by 1H NMR spectroscopy of the crude reaction mixture.
Isolated yield after chromatographic purification.
2.5 mol % Pd(COD)Cl2/3.75 mol % Xantphos.
2.2. Scope of Heterocyclic Diarylmethanes in Palladium-Catalyzed Allylic Substitution
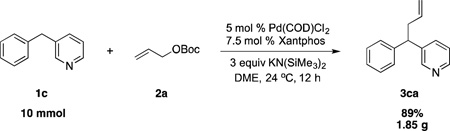
Based on the optimization process above, we anticipated that the choice of base would be critical to expand the scope of pronucleophiles and that each substrate class might require reexamination of bases. We first evaluated the scope of heterocyclic diarylmethanes as pronucleophiles in Pd-catalyzed allylic substitution (Table 3). The diarylmethane derivatives containing heterocycles are interesting targets in medicinal chemistry.41–43 Our method provides a rapid access to the heteroaryl-containing allylated products in good to excellent yields using 1–5 mol % catalyst loading (80–99% yield). Silyl-amide bases, MN(SiMe3)2 (M = Li, Na, K), were used to accommodate the wide range of pronucleophile pKa’s. In general, LiN(SiMe3)2 was used for the most acidic pronucleophiles (pKa < 28), NaN(SiMe3)2 for moderately acidic (pKa = 28–31) and KN(SiMe3)2 for the least acidic (pKa > 31). The 2-, 4- and 3-benzylpyridines underwent allylation in 91–99% yield (Table 3, entries 1–3). Xanthene (1d) derivatives are important components of dyes.44 Xanthene underwent monoallylation in 82% yield (entry 4). Furan45 and thiophene46 derivatives are valuable building blocks in agrochemicals and pharmaceuticals. Under the reaction conditions with KN(SiMe3)2 as the base, we observed decomposition of 2-benzylfuran (1e). Under the same reaction conditions with NaN(SiMe3)2 as the base, the desired product 3ea was obtained in only 32% yield. Using NaN(SiMe3)2, in combination with 15-crown-5 (2.5 equiv), the allylic substitution reaction afforded the desired product 3ea in 80% yield (entry 5). This result demonstrates that the use of additives, such as crown ethers, are useful in transition metal catalyzed processes other than deprotonative cross-coupling processes.32 With 2-benzylthiophene (1f) the desired substitution product 3fa was isolated in 93% with NaN(SiMe3)2 (entry 6). Di(3-pyridyl)methane (1g) was applied to give 3ga in 85% yield (entry 7). To establish the scalability of this method, the allylation of 3-benzylpyridine (1c) was examined on a 10 mmol scale, affording the allylated product 3ca in 89% yield (eq 2).
Table 3.
Scope of Heterocyclic Diarylmethanes in the Allylic Substitutiona
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Pronucleophlles | Base | Products | Yieldb (%) | ||
| 1c | 1a | NaN(SiMe3)2 |  |
3aa | 99 | |
| 2c | 1b | LiN(SiMe3)2 |  |
3ba | 99 | |
| 3 | 1c | KN(SiMe3)2 | 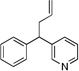 |
3ca | 91 | |
| 4 | 1d | NaN(SiMe3)2 | 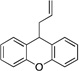 |
3da | 82 | |
| 5d | 1e | NaN(SiMe3)2 |  |
3ea | 80 | |
| 6 | 1f | NaN(SiMe3)2 | 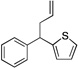 |
3fa | 93 | |
| 7 | 1g | LiN(SiMe3)2 | 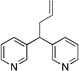 |
3ga | 85 | |
Reaction conducted on a 0.1 mmol scale with 1 equiv of 1 and 2 equiv of 2a at 0.1 M .
Isolated yield after chromatographic purification.
1 mol % Pd(COD)Cl2, 1.5 mol % Xantphos.
2.5 equiv of NaN(SiMe3)2 and 2.5 equiv of 15-crown-5.
 |
(2) |
2.3. Scope of Diphenylmethane Derivatives in the Allylic Substitution
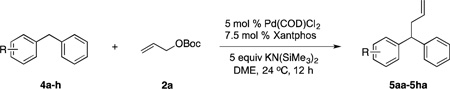
Diphenylmethane derivatives are more challenging pronucleophiles due to their higher pKa’s.39 To compensate for the less favorable equilibrium for deprotonation of these pronucleophiles, the amount of base was increased to 5 equiv. With the optimized reaction conditions for 4a (Table 4, entry 1), we investigated 4-halogenated diphenylmethanes (entries 2–4). Compounds containing fluorine are important because of their bioactivities and uses in material science.47 The reaction with 4-fluoro diphenylmethane (4b) as pronucleophile afforded the desired product 5ba in 84% yield (entry 2). We then applied our reaction conditions to 4-bromo and 4-chloro diphenylmethanes (4c and 4d). A potential problem with these substrates lies in the competing oxidative addition of C–X (X = Cl, Br) bonds to the active Pd(0) species. We were pleased to find that Pd-catalyzed allylic substitution afforded the allylated products 5ca and 5da in 95% and 73% yield, respectively (entries 3 and 4). These results suggest that generation of the π-allyl palladium intermediate is significantly faster than the oxidative addition of C–X bonds to the Pd(0) species under our reaction conditions. Fluorene (4e) derivatives have interesting characteristics and can be used in organic lightemitting diodes.44 By using 1.5 equiv LiN(SiMe3)2 and 1.1 equiv of 2a, 5ea was generated in 87% yield (entry 5). 4-Cyano diphenylmethane (4fa) is potentially problematic because benzonitriles are known to undergo additions with strong bases and organometallic reagents.48 Nonetheless, this substrate provided the product 5fa in 90% yield (entry 6). As might be anticipated, pronucleophiles with electron donating groups are less acidic and, therefore, more difficult to deprotonate. We found that 4-methyl diphenylmethane 4g underwent substitution to give 5ga in 68% yield at 50 °C (entry 7). Notably, 2-methyl diphenylmethane (4h) reacted to provide 5ha in 70% yield at 50 °C (entry 8). Unfortunately, 4-methoxy diphenylmethane did not react under these, or a variety of other conditions.
Table 4.
Scope of Diphenylmethane Derivatives in Allylic Substitution Reactionsa
 | |||||
|---|---|---|---|---|---|
| Entry | Pronucleophlles | Products | Yieldb(%) | ||
| 1 | 4a | 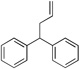 |
5aa | 95 | |
| 2 | 4b | 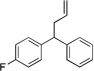 |
5ba | 84 | |
| 3 | 4c | 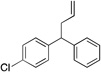 |
5ca | 95 | |
| 4 | 4d | 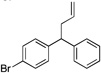 |
5da | 73 | |
| 5c | 4e | 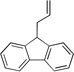 |
5ea | 87 | |
| 6d | 4f | 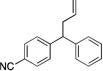 |
5fa | 90 | |
| 7e, g | 4g | 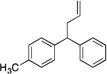 |
5ga | 68 | |
| 8f, g | 4h | 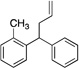 |
5ha | 70 | |
Reaction conducted on a 0.1 mmol scale with 1 equiv of 4 and 3 equiv of 2a at 0.1 M.
Isolated yield after chromatographic purification.
1.5 equiv of LiN(SiMe3)2 and 1.1 equiv of 2a.
1.5 equiv of KN(SiMe3)2 and 2 equiv of 2a.
8 equiv of KN(SiMe3)2.
10 equiv of KN(SiMe3)2.
Reaction conducted at 50 °C.
2.4. Diallylation with Heteroaryl Diarylmethanes
Having demonstrated the monoallylation of diarylmethanes and heteroaryl-containing derivatives, we next explored the possibility of adding a second allyl group to the allylated products prepared above. We were encouraged by the formation of small amounts of diallylation products with more acidic pronucleophiles (pKa < 30) in the monoallylation optimization process. We, therefore, reoptimized the conditions to enable allylation of the purified monoallylated products. By employing 5 equiv of KN(SiMe3)2 and 3 equiv of 2a with the monoallylated substrates (3aa–3ga, Table 3), we obtained the corresponding diallylation products (6aa–6ga, 70–95% yield). These results indicate that the allylation chemistry outlined here can be used to establish quaternary centers.
A more efficient method to prepare the diallylated products would be from the diarylmethane derivatives. We, therefore, set out to develop a one-pot diallylation protocol. We rationalized that the conditions mentioned above for the allylation of the mono-allylated diarylmethanes would be suitable for the one-pot diallylation. This approach proved fruitful: the yields for the double allylation ranged from 65 to 85% (Table 5, entries 1–5). The one-pot syntheses were not efficient for 2-benzylpyridine (1a) and 3-benzylpyridine (1c) as pronucleophiles, because the deprotonation events are more difficult for these substrates. Nonetheless, starting from monoallylation products 3aa and 3ca we obtained the bis-allylated products 6aa and 6ca in 84 and 80% yield (entries 6–7). We were also able to use 1,1-diarylethane (e.g., 8a) as pronucleophile to install an allyl group to afford a quaternary stereocenter (entry 8). Notably, triphenylmethane (9a) was used as pronucleophile to give the sterically congested 9aa in 90% yield (entry 9). To summarize, our method afforded diaryl-or triarylmethane products containing at least one allyl group on the quaternary carbon. Some of the products in Table 5 could presumably be employed in ring-closing metathesis reactions49 to prepare 1,1-diarylcyclopent-3-ene or Pauson–Khand type [2+2+1] reactions50 to afford bicyclic scaffolds.
Table 5.
Diallylation with Diarylmethanesa
 | |||||
|---|---|---|---|---|---|
| Entry | Pronucleophllu | Products | Yieldb (%) | ||
| 1c, d | 1b | 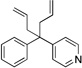 |
6ba | 85 | |
| 2 | 1d | 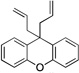 |
6da | 65 | |
| 3e | 4e | 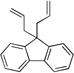 |
7ea | 85 | |
| 4f | 1f | 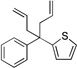 |
6fa | 70 | |
| 5e | 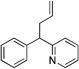 |
3aa | 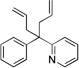 |
6aa | 84 |
| 6 | 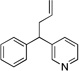 |
3ca | 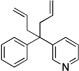 |
6ca | 80 |
| 7 | 8a | 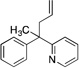 |
8aa | 90 | |
| 8 | 9a | 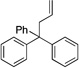 |
9aa | 90 | |
Reaction conducted on a 0.1 mmol scale with 1 equiv of pro-nucleophile and 3 equiv of 2a at 0.1 M.
Isolated yield after chromatographic purification.
Reaction conducted at 0.066 M.
Reaction conducted at 50 °C.
3 equiv of KN(SiMe3)2 and 2 equiv of 2a.
8 equiv of KN(SiMe3)2.
2.5. Scope of Electrophiles in Palladium-Catalyzed Allylic Substitutions
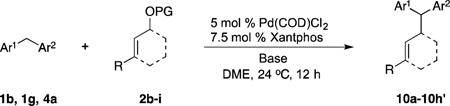
Having demonstrated that the simplest allyl electrophile can be used with a variety of pronucleophiles, we turned our attention to the nature of the electrophilic partner. We chose the more acidic pronucleophile di(3-pyridyl)methane (1g, Table 3, entry 7) and examined the influence of different leaving groups on Pd-catalyzed allylic substitution with cyclohexenyl electrophiles (2b, 2c). Both the Boc (2b) and benzoate ester (2c) derivatives gave similar yields of 10a (89 and 90% yield, entries 1–2). Changing the ring size of the electrophile from six to five (2d) resulted in 90% yield (entry 3) when the Boc analogue was used. The classic 1,3-diphenyl allyl precursor 2e afforded the allylic substitution product 10c in 87% yield with a 10:1 trans:cis ratio (entry 4).
Next, the less acidic diphenylmethane (4a) was employed as pronucleophile (entries 5–7). The Boc derived cyclohexenyl substrate 2b underwent reaction leading to the product in 85% yield (entry 5). In contrast, the benzoate derivative 2c resulted in only 20% yield (entry 6). The low yield is likely due to attack of nucleophile on the ester carbonyl. Changing the benzoate ester 2c to the pivalate ester 2f resulted in an increase in the yield to 70% (entry 7). Cyclopentenyl OBoc electrophile 2d underwent substitution in the presence of diphenylmethane in 94% yield (entry 8).
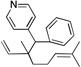
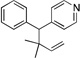
Many electrophilic partners used in allylic substitution reactions lead to unsymmetrical η3-allyl groups. We, therefore, examined the regioselectivity with unsymmetrical linear Bocprotected electrophiles such as cinnamyl alcohol (2g), geranyl alcohol (2h) and prenyl alcohol (2i) (Table 6, entries 9–11). It is well known that π-allyl palladium complexes are prone to react with carbon nucleophiles at the less substituted terminus of the π-allyl.51 For the Pd-catalyzed allylic substitution with Boc-protected cinnamyl alcohol (2g), the linear product 10f was the major product, albeit with moderate regioselectivity (2.6:1, entry 9). The reduced regioselectivity in this case, relative to cases with less basic nucleophiles, is likely a manifestation of the high reactivity of the 4-benzylpyridine-derived nucleophile.27 Interestingly, the prenylation and geranylation exhibited opposite regioselectivities (entries 10–11). The Boc activated geranyl underwent reaction with 4-benzylpyridine slightly favoring the terminal substitution product (1.9:1.0, linear:branched). In contrast, the prenylation afforded the branched product 10h’ with a linear:branched ratio of 1.0:4.5 (entry 11). We hypothesize that the origin of the regioselectivity in the prenylation is a result of the non-bonded interaction between the bulky, wide-bite angle Xantphos ligand and the more substituted carbon of the η3-allyl. This interaction places a larger δ+ partial charge on the more substituted terminus and, therefore, nucleophilic attack at this position prevails. The additional substituent (=R) on the η3-allyl in the geranylation causes this group to adopt a conformation positioning it anti to the bulky (Xantphos)Pd center (Figure 2). The substituent partially obstructs the nucleophilic attack at the more substituted terminus, resulting in a shift of the regioselectivity toward the less substituted carbon of the allyl.
Table 6.
Scope of Electrophiles in Allylic Substitution Reactionsa
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Electrophiles | Pronueleophlies | Products | Yieldb (%) | ||
| 1c | PG = Boc | 2b | 1g | 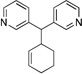 |
10a | 89 |
| 2c | PG = Bz | 2c | 1g | 90 | ||
| 3d | 2d | 1g | 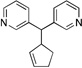 |
10b | 90 | |
| 4e | 2e | 1g | 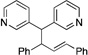 |
10c | 87 | |
| (tranc: cis = 10:1)i | ||||||
| 5f | PG = Boc | 2b | 4a | 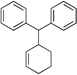 |
10d | 85 |
| 6f | PG = Bz | 2c | 4a | 20 | ||
| 7f | PG = Piv | 2f | 4a | 70 | ||
| 8f, g | 2d | 4a | 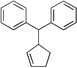 |
10e | 94 | |
| 9e, h | 2g | 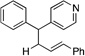 |
10f | 91 (L:B=2.6:1)i |
||
| 1b | ||||||
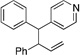 |
10f’ | |||||
| 10e, h | 2h | 1b | 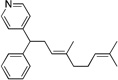 |
10g | 88 (L:B=1.9:1)i |
|
 |
10g’ | |||||
| 11e, h | 2i | 1b | 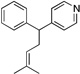 |
10h | 93 (L:B=1:4.5)i |
|
 |
10h’ | |||||
Reaction conducted on a 0.1 mmol scale at 0.1 M.
Isolated yield after chromatographic purification.
3 equiv of NaN(SiMe3)2 and 2 equiv of 2.
3 equiv of KN(SiMe3)2 and 2 equiv of 2.
3 equiv of LiN(SiMe3)2 and 2 equiv of 2.
5 equiv of KN(SiMe3)2 and 3 equiv of 2.
Reaction conducted at 50 °C.
Reaction conducted at 0 °C.
Ratio of trans:cis or line-ar:branched (L:B) determined by 1H NMR.
Figure 2.

Proposed conformational model to explain the reversal in regioselectivity between the prenylation and geranylation (Table 6, entries 10–11). When R=H nucleophilic attack is favored at the more substituted terminus (solid arrow). When R=alkyl, attack follows the dashed arrow leading to the linear product.
2.6. Internal vs. External Attack of Diarylmethane on π-Allyl Palladium Intermediate
As outlined in the Introduction, nucleophiles in allylic substitutions can directly add to the π-allyl or to the metal in their reactions with [(η3-allyl)MLn]+ intermediates. It is generally accepted that nucleophiles with conjugate acids of pKa < 25, classified as “soft” nucleophiles, undergo external attack on π-allyl palladium complexes. Soft nucleophiles, therefore, result in stereoretentive allylic substitutions reactions (via double inversion).52 On the other hand, “hard” nucleophiles, those with pKa > 25 are proposed undergo attack on the metal center of the π-allyl palladium complexes (i.e., transmetallation) followed by reductive elimination to afford net inversion for the allylic substitution.53
To examine the mechanistic pathway of the reaction, di(3-pyridyl)methane (1g) was reacted with cis-disubstituted stereoprobe 2j (Scheme 2A) and afforded cis-product 11a in 90% isolated yield as a single diastereomer (determined by 1H NMR spectroscopy). Reaction of the less acidic pronucleophile diphenylmethane (4a) (pKa = 32.3) with 2j (Scheme 2B) similarly furnished the cis-product 11b as a single diastereomer (determined by 1H NMR spectroscopy and single crystal X-ray diffraction) in 88% yield. The results indicated both nucleophiles derived from di(3-pyridyl)methane (1g) and diphenylmethane (4a) behave as “soft” nucleophiles and that the pKa limit for “soft” nucleophiles should be raised from 25 to 32, a change of 7 orders of magnitude.
Scheme 2.

Allylic Substitution with Retention of Configuration
3. SUMMARY AND OUTLOOK
Herein we have developed a general method for Pd-catalyzed allylic substitution with diarylmethane derivatives at room temperature. The synthetic significance of the method is that it provides a rapid access to products containing allylated diarylmethanyl motifs. The method is general for a wide range of nucleophiles derived diarylmethanes and heteroaryl derivatives. A tandem procedure for the Pd-catalyzed allylic substitutions to afford diallylation products with quaternary centers is also described. With alkylated diarylmethanes and triarylmethanes, the method is also efficient to afford the corresponding allylated products. We anticipate that the described method will be a valuable complement to the existing arsenal of nucleophiles in Pd-catalyzed allylic substitutions. Mechanistic studies show that diarylmethane derivatives behave as “soft” or stabilized nucleophiles. The nucleophile derived from diphenylmethane undergoes external attack on π-allyl palladium species under our reaction conditions. The results of this study indicate that the cutoff between “soft” and “hard” nucleophiles should be raised from a pKa of 2 5 to at least 32.
4. EXPERIMENTAL SECTION
Representative procedures are described herein. Full experimental details and characterization of all compounds are provided in the Supporting Information.
4.1. General Methods
All reactions were performed under nitrogen using oven-dried glassware and standard Schlenk or vacuum line techniques. Air- and moisture-sensitive solutions were handled under nitrogen and transferred via syringe. The solvents (DME and THF) were sparged for 20 min with dry N2 and dried using a commercial two-column solvent purification system comprising columns packed with neutral alumina. Unless otherwise stated, reagents were commercially available and used as purchased without further purification. Chemicals were obtained from Sigma-Aldrich, Acros, TCI America, Strem Chemicals or Matrix Scientific, and solvents were purchased from Fisher Scientific. The progress of the reactions was monitored by thin-layer chromatography using Whatman Partisil K6F 250 µm precoated 60 Å silica gel plates and visualized by short-wavelength ultraviolet light as well as by treatment potassium permanganate (KMnO4) stain or iodine. Silica gel (230–400 mesh, Silicycle) was used for flash chromatography. The 1H NMR and 13C{1H} NMR spectra were obtained using a Brüker AM-500 Fourier transform NMR spectrometer at 500 and 126 MHz, respectively. Chemical shifts are reported in units of parts per million (ppm) downfield from tetramethylsilane (TMS), and all coupling constants are reported in hertz. The infrared spectra were obtained with KBr plates using a Perkin-Elmer Spectrum 100 Series FTIR spectrometer. High-resolution mass spectrometry (HRMS) data were obtained on a Waters LC–TOF mass spectrometer (model LCT-XE Premier) using chemical ionization (CI) or electrospray ionization (ESI) in positive or negative mode, depending on the analyte. Melting points were determined on a Unimelt Thomas-Hoover melting point apparatus and are uncorrected.
4.2. General Procedure A: Palladium-Catalyzed Allylic Substitution with Heterocyclic Diarylmethanes Under Room Temperature
An oven-dried 10 mL reaction vial equipped with a stir bar was charged with NaN(SiMe3)2 (55 mg, 0.30 mmol, 3 equiv) under a nitrogen atmosphere. A solution (from a stock solution) of Pd(COD)Cl2 (1.43 mg, 0.0050 mmol) and Xantphos (4.34 mg, 0.0075 mmol) in 1 mL of dry DME was taken up by syringe and added to the reaction vial. After stirring for 5 min at 24 °C, 2-benzylpyridine 1a (16 µL, 0.1 mmol, 1 equiv) was added to the reaction mixture followed by 2a (34 µL, 0.2 mmol, 2 equiv). Note that the diarylmethanes or allyl-OBoc in a solid form was added to the reaction vial prior to NaN(SiMe3)2. The reaction mixture was stirred for 12 h at 24 °C, quenched with two drops of H2O, diluted with 3 mL of ethyl acetate and filtered over a pad of MgSO4 and silica. The pad was rinsed with additional ethyl acetate, and the solution was concentrated in vacuo. The crude material was loaded onto a silica gel column and purified by flash chromatography eluting with EtOAc/hexanes.
4.3. General Procedure B: Palladium-Catalyzed Allylic Substitution with Heterocyclic Diarylmethanes Under 50°C
An oven-dried 10 mL reaction vial equipped with a stir bar was charged with KN(SiMe3)2 (160 mg, 0.80 mmol, 8 equiv) under a nitrogen atmosphere. A solution (from a stock solution) of Pd(COD)Cl2 (1.43 mg, 0.0050 mmol) and Xantphos (4.34 mg, 0.0075 mmol) in 1 mL of dry DME was taken up by syringe and added to the reaction vial. After stirring for 5 min at 24 °C, 4-methyl diphenylmethane 4g (18.5 µL, 0.1 mmol, 1 equiv) was added to the reaction mixture followed by 2a (51 µL, 0.3 mmol, 3 equiv). The reaction mixture was stirred for 12 h at 50 °C, quenched with two drops of H2O, diluted with 3 mL of ethyl acetate, and filtered over a pad of MgSO4 and silica. The pad was rinsed with additional ethyl acetate and the solution was concentrated in vacuo. The crude material was loaded onto a silica gel column and purified by flash chromatography eluting with EtOAc/hexanes.
4.4 General Procedure C: Palladium-Catalyzed Allylic Substitution with Heterocyclic Diarylmethanes Under 0 °C
An oven-dried 10 mL reaction vial equipped with a stir bar was charged with LiN(SiMe3)2 (51 mg, 0.30 mmol, 3 equiv) under a nitrogen atmosphere. A solution (from a stock solution) of Pd(COD)Cl2 (1.43 mg, 0.0050 mmol) and Xantphos (4.34 mg, 0.0075 mmol) in 1 mL of dry DME was taken up by syringe and added to the reaction vial. After stirring for 5 min at 0 °C, 4-benzylpyridine 1b (16 µL, 0.1 mmol, 1 equiv) was added to the reaction mixture followed by Boc-protected cinnamyl alcohol 2g (46 µL, 0.2 mmol, 2 equiv). The reaction mixture was stirred for 12 h at 0 °C, quenched with two drops of H2O, diluted with 3 mL of ethyl acetate, and filtered over a pad of MgSO4 and silica. The pad was rinsed with additional ethyl acetate and the solution was concentrated in vacuo. The crude material was loaded onto a silica gel column and purified by flash chromatography eluting with EtOAc/hexanes.
Supplementary Material
ACKNOWLEDGMENT
We thank the National Science Foundation [CHE-1152488] and the National Institutes of Health (NIGMS GM104349) for financial support. We are grateful to Prof. Per Ola Norrby for helpful discussions.
Footnotes
ASSOCIATED CONTENT
Supporting Information
Procedures and full characterization of new compounds. This material is available free of charge via the Internet at http://pubs.acs.org
The authors declare no competing financial interest.
REFERENCES
- 1.Cárdenas DJ. Angew. Chem., Int. Ed. 2003;42:384. doi: 10.1002/anie.200390123. [DOI] [PubMed] [Google Scholar]
- 2.Netherton MR, Fu GC. Adv. Synth. Catal. 2004;346:1525. [Google Scholar]
- 3.Frisch AC, Beller M. Angew. Chem., Int. Ed. 2005;44:674. doi: 10.1002/anie.200461432. [DOI] [PubMed] [Google Scholar]
- 4.Rudolph A, Lautens M. Angew. Chem., Int. Ed. 2009;48:2656. doi: 10.1002/anie.200803611. [DOI] [PubMed] [Google Scholar]
- 5.Jana R, Pathak TP, Sigman MS. Chem. Rev. 2011;111:1417. doi: 10.1021/cr100327p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burger EC, Tunge JA. Org. Lett. 2004;6:4113. doi: 10.1021/ol048149t. [DOI] [PubMed] [Google Scholar]
- 7.Waetzig SR, Tunge JA. J. Am. Chem. Soc. 2007;129:14860. doi: 10.1021/ja077070r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Behenna DC, Stoltz BM. J. Am. Chem. Soc. 2004;126:15044. doi: 10.1021/ja044812x. [DOI] [PubMed] [Google Scholar]
- 9.Behenna DC, Liu Y, Yurino T, Kim J, White DE, Virgil SC, Stoltz BM. Nat. Chem. 2012;4:130. doi: 10.1038/nchem.1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trost BM, Xu J, Schmidt T. J. Am. Chem. Soc. 2009;131:18343. doi: 10.1021/ja9053948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trost BM, Van Vranken DL. Chem. Rev. 1996;96:395. doi: 10.1021/cr9409804. [DOI] [PubMed] [Google Scholar]
- 12.Trost BM, Crawley ML. Chem. Rev. 2003;103:2921. doi: 10.1021/cr020027w. [DOI] [PubMed] [Google Scholar]
- 13.Trost BM. J. Org. Chem. 2004;69:5813. doi: 10.1021/jo0491004. [DOI] [PubMed] [Google Scholar]
- 14.Trost BM, Machacek MR, Aponick A. Acc. Chem. Res. 2006;39:747. doi: 10.1021/ar040063c. [DOI] [PubMed] [Google Scholar]
- 15.Lu Z, Ma S. Angew. Chem., Int. Ed. 2008;47:258. doi: 10.1002/anie.200605113. [DOI] [PubMed] [Google Scholar]
- 16.Watson IDG, Styler SA, Yudin AK. J. Am. Chem. Soc. 2004;126:5086. doi: 10.1021/ja049242f. [DOI] [PubMed] [Google Scholar]
- 17.Watson IDG, Yudin AK. J. Am. Chem. Soc. 2005;127:17516. doi: 10.1021/ja055288c. [DOI] [PubMed] [Google Scholar]
- 18.Trost BM, Thaisrivongs DA. J. Am. Chem. Soc. 2008;130:14092. doi: 10.1021/ja806781u. [DOI] [PubMed] [Google Scholar]
- 19.Trost BM, Toste FD. J. Am. Chem. Soc. 1999;121:4545. [Google Scholar]
- 20.Castanet Y, Petit F. Tetrahedron Lett. 1979;20:3221. [Google Scholar]
- 21.Keinan E, Roth Z. J. Org. Chem. 1983;48:1769. and references therein. [Google Scholar]
- 22.Shukla KH, DeShong P. J. Org. Chem. 2008;73:6283. doi: 10.1021/jo8010254. [DOI] [PubMed] [Google Scholar]
- 23.Dewick PM. Essentials of Organic Chemistry:For Students of Pharmacy, Medicinal Chemistry and Biological Chemistry. West Sussex, UK: John Wiley & Sons, Ltd; 2006. [Google Scholar]
- 24.Trost BM, Thaisrivongs DA. J. Am. Chem. Soc. 2009;131:12056. doi: 10.1021/ja904441a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Trost BM, Thaisrivongs DA, Hartwig J. J. Am. Chem. Soc. 2011;133:12439. doi: 10.1021/ja205523e. [DOI] [PubMed] [Google Scholar]
- 26.Bordwell FG, Bares JE, Bartmess JE, Drucker GE, Gerhold J, McCollum GJ, Van Der Puy M, Vanier NR, Matthews WS. J. Org. Chem. 1977;42:326. [Google Scholar]
- 27.Zhang J, Stanciu C, Wang B, Hussain MM, Da C-S, Carroll PJ, Dreher SD, Walsh PJ. J. Am. Chem. Soc. 2011;133:20552. doi: 10.1021/ja208935u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Das B, Reddy CR, Kashanna J, Mamidyala SK, Kumar CG. Med. Chem. Res. 2011;21:3321. [Google Scholar]
- 29.Craig C. 2-Aminothiazole Compounds Useful as Aspartyl Protease Inhibitors. 2005 WO2005US10224. [Google Scholar]
- 30.Burgess LE, Koch K, Cooper K, Biggers MS, Ramchandani M, Smitrovich JH, Gilbert EJ, Bruns MJ, Mather RJ, Donovan CB. Bioorg. Med. Chem. Lett. 1997;7:1047. [Google Scholar]
- 31.Yoshihara HA, Apriletti JW, Baxter JD, Scanlan TS. Bioorg. Med. Chem. Lett. 2001;11:2821. doi: 10.1016/s0960-894x(01)00521-2. [DOI] [PubMed] [Google Scholar]
- 32.Bellomo A, Zhang J, Trongsiriwat N, Walsh PJ. Chem. Sci. 2013;4:849. [Google Scholar]
- 33.Zhang J, Bellomo A, Creamer AD, Dreher SD, Walsh PJ. J. Am. Chem. Soc. 2012;134:13765. doi: 10.1021/ja3047816. [DOI] [PubMed] [Google Scholar]
- 34.Jia T, Bellomo A, Baina KE, Dreher SD, Walsh PJ. J. Am. Chem. Soc. 2013;135:3740. doi: 10.1021/ja4009776. [DOI] [PubMed] [Google Scholar]
- 35.Zheng B, Jia T, Walsh PJ. Org. Lett. 2013;15:1690. doi: 10.1021/ol400472v. [DOI] [PubMed] [Google Scholar]
- 36.Hajri M, Blondelle C, Martinez A, Vasse J-L, Szymoniak J. Tetrahedron Lett. 2013;54:1029. [Google Scholar]
- 37.Kranenburg M, van der Burgt YEM, Kamer PCJ, van Leeuwen PWNM, Goubitz K, Fraanje J. Organometallics. 1995;14:3081. [Google Scholar]
- 38.Bordwell FG. Acc. Chem. Res. 1988;21:456. [Google Scholar]
- 39.Bordwell FG, Matthews WS, Vanier NR. J. Am. Chem. Soc. 1975;97:442. [Google Scholar]
- 40.Cook JW, Moffatt JS. J. Chem. Soc. 1951:2487. [Google Scholar]
- 41.Hsin L-W, Dersch CM, Baumann MH, Stafford D, Glowa JR, Rothman RB, Jacobson AE, Rice KC. J. Med. Chem. 2002;45:1321. doi: 10.1021/jm010430f. [DOI] [PubMed] [Google Scholar]
- 42.Wai JS, Egbertson MS, Payne LS, Fisher TE, Embrey MW, Tran LO, Melamed JY, Langford HM, Guare JP, Zhuang L, Grey VE, Vacca JP, Holloway MK, Naylor-Olsen AM, Hazuda DJ, Felock PJ, Wolfe AL, Stillmock KA, Schleif WA, Gabryelski LJ, Young SD. J. Med. Chem. 2000;43:4923. doi: 10.1021/jm000176b. [DOI] [PubMed] [Google Scholar]
- 43.Boyd RE, Rasmussen CR, Press JB, Raffa RB, Codd EE, Connelly CD, Li QS, Martinez RP, Lewis MA, Almond HR, Reitz AB. J. Med. Chem. 2001;44:863. doi: 10.1021/jm0003891. [DOI] [PubMed] [Google Scholar]
- 44.Griesbaum K, Behr A, Biedenkapp D, Voges H-W, Garbe D, Paetz C, Collin G, Mayer D, Höke H. Hydrocarbons. Weinheim, Germany: Wiley-VCH Verlag GmbH & Co. KGaA; 2000. [Google Scholar]
- 45.Hoydonckx HE, Van Rhijn WM, Van Rhijn W, De Vos DE, Jacobs PA. Furfural and Derivatives. Weinheim, Germany: Wiley-VCH Verlag GmbH & Co. KGaA; 2000. [Google Scholar]
- 46.Swanston J. Thiophene. Weinheim, Germany: Wiley-VCH Verlag GmbH & Co. KGaA; 2000. [Google Scholar]
- 47.Liu P, Sharon A, Chu CK. J. Fluorine Chem. 2008;129:743. doi: 10.1016/j.jfluchem.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hou G, Gosselin F, Li W, McWilliams JC, Sun Y, Weisel M, O’Shea PD, Chen C-Y, Davies IW, Zhang X. J. Am. Chem. Soc. 2009;131:9882. doi: 10.1021/ja903319r. [DOI] [PubMed] [Google Scholar]
- 49.Kotha S, Manivannan E, Ganesh T, Sreenivasachary N, Deb A. Synlett. 1999;10:1618. [Google Scholar]
- 50.Wender PA, Croatt MP, Deschamps NM. J. Am. Chem. Soc. 2004;126:5948. doi: 10.1021/ja0489487. [DOI] [PubMed] [Google Scholar]
- 51.For linear regioselectivity: Trost BM, Crawley ML. Chem. Rev. 2003;103:2921–2944. doi: 10.1021/cr020027w. For branched regioselectivity in Pd-catalyzed AAA reactions: Trost BM, Crawley ML. Chem.—Eur. J. 2004;10:2237. doi: 10.1002/chem.200305634. Trost BM, Bunt RC, Lemoine RC, Calkins TL. J. Am. Chem. Soc. 2000;122:5968. Trost BM, Toste FD. J. Am. Chem. Soc. 1999;121:4545. Helmchen G, Pfaltz A. Acc. Chem. Res. 2000;33:336. doi: 10.1021/ar9900865. Pretot R, Pfaltz A. Angew. Chem., Int. Ed. 1998;37:323. doi: 10.1002/(SICI)1521-3773(19980216)37:3<323::AID-ANIE323>3.0.CO;2-T. You SL, Zhu XZ, Luo YM, Hou XL, Dai LX. J. Am. Chem. Soc. 2001;123:7471. doi: 10.1021/ja016121w. Zheng WH, Sun N, Hou XL. Org. Lett. 2005;7:5151. doi: 10.1021/ol051882f. Trost BM, Malhotra S, Chan WH. J. Am. Chem. Soc. 2011;133:7328. doi: 10.1021/ja2020873.
- 52.Trost BM, Verhoeven TR. J. Org. Chem. 1976;41:3215. [Google Scholar]
- 53.Matsushita H, Negishi E-I. J. Chem. Soc., Chem. Commun. 1982:160. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.