

Significance
All plants carry distinctive bacterial communities on and inside organs such as roots and leaves, collectively called the plant microbiota. How this microbiota diversifies in related plant species is unknown. We investigated the diversity of the bacterial root microbiota in the Brassicaceae family, including three Arabidopsis thaliana ecotypes, its sister species Arabidopsis halleri and Arabidopsis lyrata, and Cardamine hirsuta. We show that differences in root microbiota profiles between these hosts are largely quantitative and that host phylogenetic distance alone cannot explain the observed microbiota diversification. Our work also reveals a largely conserved and taxonomically narrow root microbiota, which comprises stable community members belonging to the Actinomycetales, Burkholderiales, and Flavobacteriales.
Keywords: Brassicaceae species, bacterial communities, 16S amplicon ribotyping
Abstract
Plants host at the contact zone with soil a distinctive root-associated bacterial microbiota believed to function in plant nutrition and health. We investigated the diversity of the root microbiota within a phylogenetic framework of hosts: three Arabidopsis thaliana ecotypes along with its sister species Arabidopsis halleri and Arabidopsis lyrata, as well as Cardamine hirsuta, which diverged from the former ∼35 Mya. We surveyed their microbiota under controlled environmental conditions and of A. thaliana and C. hirsuta in two natural habitats. Deep 16S rRNA gene profiling of root and corresponding soil samples identified a total of 237 quantifiable bacterial ribotypes, of which an average of 73 community members were enriched in roots. The composition of this root microbiota depends more on interactions with the environment than with host species. Interhost species microbiota diversity is largely quantitative and is greater between the three Arabidopsis species than the three A. thaliana ecotypes. Host species-specific microbiota were identified at the levels of individual community members, taxonomic groups, and whole root communities. Most of these signatures were observed in the phylogenetically distant C. hirsuta. However, the branching order of host phylogeny is incongruent with interspecies root microbiota diversity, indicating that host phylogenetic distance alone cannot explain root microbiota diversification. Our work reveals within 35 My of host divergence a largely conserved and taxonomically narrow root microbiota, which comprises stable community members belonging to the Actinomycetales, Burkholderiales, and Flavobacteriales.
Plants host distinct bacterial communities associated with roots and leaves (1, 2). Both the leaf and root microbiota contain bacteria that provide indirect pathogen protection, but root microbiota members appear to serve additional host functions through the acquisition of nutrients from soil supporting plant growth (2). The plant-root microbiota emerges as a fundamental trait that includes mutualism enabled through diverse biochemical mechanisms, as exemplified by previous studies on numerous plant growth and plant health-promoting bacteria (2).
Recent deep profiling of the root microbiota of Arabidopsis thaliana ecotypes, grown under controlled environments, confirmed soil type as major source of variation in root microbiota membership and provided evidence for limited host genotype-dependent variation (3, 4). Using four soil types on two continents and based on two 16S rRNA gene PCR primer sets, these replicated experiments revealed a similar phylogenetic structure of the root-associated microbiota at high taxonomic rank, including the phyla Actinobacteria, Bacteroidetes, and Proteobacteria. In addition, these studies revealed a minor “rhizosphere effect” in A. thaliana, i.e., a weak differentiation of the bacterial communities in the rhizosphere (soil that is firmly attached to roots) compared with the corresponding unplanted bulk soil.
The genus Arabidopsis consists of the four major lineages Arabidopsis thaliana, Arabidopsis lyrata, Arabidopsis halleri and Arabidopsis arenosa. The former is the sole self-fertile species and diverged from the rest of the genus ∼13 Mya whereas the other three species radiated approximately ∼8 Mya (5; Fig. 1). Cardamine hirsuta diverged from the Arabidopsis species ∼35 Mya and often shares the same habitat with A. thaliana. A. thaliana has a cosmopolitan distribution whereas the other species occur in spatially restricted populations or developed even endemic subspecies, indicative of their adaptation to specific ecological niches (6). The two diploid species, A. halleri and A. lyrata, co-occur in Eurasia, but, in contrast to A. lyrata (Northern rock-cress), the geographical distribution of A. halleri rarely extends into northern latitudes. A. lyrata primarily colonizes, similar to A. thaliana, low-competition habitats as, for example, tundra, stream banks, lakeshores, or rocky slopes, whereas A. halleri (Meadow rock-cress) is tolerant of shading and competition, growing in habitats such as mesic meadow sites (7). In contrast to its sister species, A. halleri can grow on heavy metal-contaminated soils and serves as a model species for metal hyperaccumulation and associated metal hypertolerance and for extremophile adaptation (8).
Fig. 1.

Phylogenetic placement of the Arabidopsis species A. halleri, A. lyrata, and A. thaliana and relative species Cardamine hirsuta. The relationships and divergence time estimates are based on molecular systematics using combined data of NADH dehydrogenase subunit F and phytochrome A sequences anchored by four fossil age constraints (5).
The bacterial root microbiota of plants—“plants wear their guts on the outside” (9)—is conceptually analogous to the gut microbiota of animals owing to a shared primary physiological function of root and gut organs for nutrient uptake. The idea of a core microbiota within a species has been initially explored in humans by revealing an extensive array of shared microbial genes among sampled individuals, comprising an identifiable gut core microbiome at the gene, rather than at the level of organismal lineages (10, 11). However, using a phylogroup- and tree-independent approach, two prevalent core phylogroups belonging to the clostridial family Lachnospiraceae were identified in the human colon among a total of 210 human beings with widespread geographic origin, ethnic background, and diet (12). These phylogroups were also detected in a wide range of other mammals and are thought to play a conserved role in gut homeostasis and health. The findings of a core set of species in the human gut microbiota remain contentious as a wider set of samples including developing countries and a broader age range becomes available (13). However, spatial stratification of the gut microbiota, which is normally missing in fecal samples, led to the definition of a crypt-specific core microbiota in the mouse colon, dominated by aerobic Acinetobacter, regardless of the mouse line used or breeding origin of these mice (14). Finally, evidence for a shared core gut microbiota was found in domesticated and recently caught zebrafish, dominated by Proteobacteria, some Fusobacteria, and Firmicutes (15). This shared core is believed to reflect common selective pressures governing microbial community assembly within this intestinal habitat. Although root microbiota profiles of numerous plant species, including crops, have been examined (16–19), different sampling protocols and low-resolution profiling methods make it difficult to reexamine and compare these for the existence of a conserved core microbiota between plant species.
Here, we present a systematic investigation of host–microbiota diversification within a phylogenetically defined plant species framework, combined with replicated experiments under controlled conditions and sampling in natural habitats. Using deep 16S rRNA gene profiling of root and corresponding soil samples of four host species of the Brassicaceae family, together with rigorous statistical analysis, we show that interhost species microbiota diversity is largely quantitative, and we discuss a possible microbiota coevolution with these hosts. We also compared bacterial community structure variation within and between the tested host species. We provide evidence for the existence of a largely conserved and taxonomically narrow root microbiota between the tested host species, which remains stable in natural and controlled environments. This identified core comprises Actinomycetales, Burkholderiales, and Flavobacteriales. Members of each of these bacterial families are known to promote plant growth and plant health. It is possible that the conserved microbiota represents a standing reservoir of retrievable host services independent of environmental parameters and host species-specific niche adaptations.
Results
We collected side-by-side growing A. thaliana and C. hirsuta plants at two natural sites, designated “Cologne” and “Eifel,” and prepared quadruplicate root and rhizosphere samples for bacterial 16S rRNA gene community profiling (Table 1, Dataset S1, and SI Appendix). In parallel, we conducted two replicate greenhouse experiments using two seasonal batches of natural experimental Cologne soil (SI Appendix, Table S1) on which we cultivated A. halleri (Auby), A. lyrata (Mn47), and C. hirsuta (Oxford), together with the three A. thaliana accessions Shakdara (Sha), Landsberg (Ler) and Columbia (Col), and prepared triplicate root samples for microbiota analysis (Table 1, Dataset S1, and SI Appendix). Rhizosphere samples are defined as firmly root-adhering soil particles removed by a washing step and collected by centrifugation. Root samples were washed a second time and treated with ultrasound to deplete root surface-associated bacteria and to enrich for endophytic bacteria (3) (SI Appendix). To quantify the start inoculum for the root-associated bacterial communities, we prepared triplicate samples from unplanted pots of each greenhouse experiment, as well as four samples from bulk soil collected at each natural site (Table 1 and Dataset S1). Barcoded pyrosequencing of bacterial 16S rRNA gene amplicon libraries generated with the PCR primers 799F (20) and 1193R (21) was used to display bacterial communities (SI Appendix).
Table 1.
Experimental design
| Sample | Species | Natural sites |
Greenhouse |
||
| Cologne | Eifel | Exp. 1 | Exp. 2 | ||
| Soil | 4 | 4 | 3 | 3 | |
| Rhizosphere | A. thaliana | 4 | 4 | — | — |
| Root | A. thaliana | 4 | 4 | 8* | 9† |
| Rhizosphere | C. hirsuta | 4 | 3 | — | — |
| Root | C. hirsuta | 4 | 3 | 3 | 3 |
| Root | A. halleri | — | — | 3 | 2 |
| Root | A. lyrata | — | — | 2 | 3 |
Numerical overview of biological replicate samples per sample type, plant species, and experiments. —, sample types which were not prepared. See Dataset S1 for the detailed experimental design, including the sequencing effort.
Three samples of genotype Col, 3x Ler and 2x Sha.
Three samples of genotype Col, 3x Ler and 3x Sha.
We generated 2,110,506 raw reads from 77 samples of the replicated natural-site and greenhouse experiments (Dataset S1). For subsequent analysis, we included 1,567,657 quality sequences (SI Appendix), resulting in a median of 15,603 quality sequences per sample (range 6,339–58,150 sequences per sample). Quality sequences were binned at >97% sequence identity using QIIME (22) to define operational taxonomic units (OTUs), corrected for differences in sequencing depth between samples by rarefaction to 6,000 sequences per sample. OTU representative sequences were taxonomically classified based on the Greengenes database (23) (SI Appendix) and we identified a total of 88,731 unique bacterial OTUs and a single archea OTU across all samples.
Defining Abundant Community Members.
Technical reproducibility of community profiles was determined by repeated library sequencing, and we defined a minimum of 20 sequences per OTU for reproducible quantification of OTU abundance (SI Appendix, Fig. S1, and Dataset S2). This reproducibility threshold is similar to previous studies (3, 14, 15). We noted a low reproducibility for soil microbiota profiles and found that OTU richness does not reach a plateau even at a sequencing depth of 50,000 quality sequences per sample (SI Appendix, Fig. S2A). These observations, together with the exclusion of low-abundant (<20 sequences) and nonreproducible OTUs for rarefaction analysis (SI Appendix, Fig. S2B), suggested that OTU richness in soil is the result of a vast number of low-count OTUs. In the root samples, the richness of the abundant community members (OTUs with >20 sequences) was sufficiently captured at a sequencing depth of 6,000 sequences per sample. Consequently, we focus our analyses on the abundant community members (ACMs) of the dataset, which we define to comprise OTUs reaching the threshold of 20 quality sequences in at least one sample (SI Appendix and Dataset S3). Without application of this abundance threshold, we refer to community profiles rarefied to 6,000 sequences as threshold-independent communities (TICs) (Dataset S4).
The ACM, including soil, rhizosphere, and root samples, was represented by 237 bacterial OTUs comprising 55.3% of rarefied quality sequences (Datasets S1 and S3). Soil and rhizosphere samples contained fewer sequences after thresholding compared with root samples, likely due to increased richness by low-count OTUs in the former two compartments (SI Appendix, Figs. S2 and S3). We normalized the counts of the ACM OTUs per sample, expressed their relative abundance as per mille, and used log2-transformed values for statistical comparisons (SI Appendix).
Community Composition Is Defined More Strongly by Environmental Parameters than by Host Species.
We first examined taxonomic composition and ecological diversity parameters in the whole dataset consisting of samples from two natural sites and two greenhouse experiments. All OTUs of the ACM belonged to the domain of bacteria. In root samples, the majority of OTUs belonged to Proteobacteria (4.2%, 33.6%, 6.4%, and 1.8% in the Alpha-, Beta-, Gamma-, and Deltaproteobacteria subphyla, respectively), Bacteroidetes (27.5%), Actinobacteria (22.1%), and Chloroflexi (2.2%) (SI Appendix, Fig. S4A). Soil samples also contain Proteobacteria (52.2%) and Actinobacteria (26.8%), but few Bacteroidetes (3.5%) and, characteristic for this compartment, Firmicutes (10.1%) and Nitrospirae (2.4%). Similar taxonomic characteristics of soil and root samples were also found for TICs (SI Appendix, Fig. S4B). We noted the dominance of a single Flavobacterium (OTU162362) in root communities of natural-site and greenhouse experiments, representing, in some of the samples, more than half of the total community. A high OTU diversity in family-rich phyla, such as the Proteobacteria (128 OTUs in 24 families) or Actinobacteria (67 OTUs in 17 families), contrasts with a low taxonomic diversity within the root-specific Bacteroidetes (20 OTUs), all belonging to the family of the Flavobacteriaceae (Dataset S3).
To compare community diversity between samples, we used the weighted UniFrac metric (24) (SI Appendix). Consistent with previous studies (3, 4, 16), the hierarchical clustering of UniFrac distances revealed that compartments and environmental conditions (soil types/soil batches, controlled/noncontrolled climates) are the major sources of variation both in the ACMs (Fig. 2) and TICs (SI Appendix, Fig. S5). Due to independent library preparation and sequencing, we validated that the variation in the replicate greenhouse experiments reflects biological rather than technical variation (SI Appendix, Fig. S6, Dataset S5, and SI Appendix). For both natural-site and controlled environment samples, we did not detect a consistent clustering by host species, evidencing that the present sample-to-sample variation obscures a possible host species effect on beta diversity.
Fig. 2.
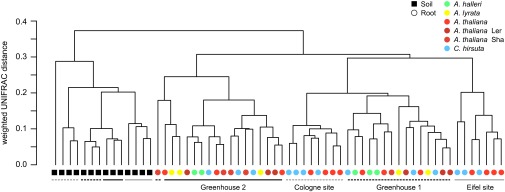
Beta diversity of the ACM. Between-sample similarities were estimated on 1,400 sequences per sample using the phylogeny-based UniFrac distance metric. Weighted UniFrac is sensitive to the sequence abundances. The A. thaliana ecotype Col (nonshaded red) was used in the greenhouse experiments.
We estimated OTU diversity within samples based on the number of OTUs detected (richness) and Faith’s Phylogenetic Diversity (PD) metric (25). Root TICs are of lower richness and diversity compared with the soil and rhizosphere microbiota (SI Appendix, Fig. S7). Of note, roots of plants grown under natural conditions host bacterial communities of increased richness and Faith’s PD compared with greenhouse-grown plants. Root TICs and root ACMs did not differ in richness and Faith’s PD among the tested host species in natural and greenhouse experiments (SI Appendix, Fig. S7). This finding further supports the existence of qualitatively similar root-associated bacterial assemblies among A. thaliana relatives.
Naturally Grown A. thaliana and C. hirsuta Host a Taxonomically Narrow Root Microbiota.
In a second step, we investigated the variation in root microbiota composition between the plant species A. thaliana and C. hirsuta from both natural-site experiments. We compared the root bacterial communities using ANOVA-based statistics to detect taxonomic groups of OTUs (“community modules”) and individual OTUs (“community members”) that differ quantitatively between sites and/or host species (SI Appendix). The community member analysis was performed on the ACM, and, for the community module analyses, we prepared abundance matrices at phylum and family rank of all ACM OTUs representing 9 and 51 taxa, respectively.
We searched the abundance matrices at phylum and family rank for modules that differ between the root communities as a function of the variables site and host species (SI Appendix). The taxonomic structure of the root communities varies mainly by site (six phyla, 35 families) and less by host species (two phyla, two families; ANOVA, P < 0.1 [false discovery rate (FDR) corrected]) (Dataset S6, worksheet A). At both sites, A. thaliana and C. hirsuta root communities displayed similar relative distributions of bacterial phyla, except for an increased abundance of Bacteroidetes in C. hirsuta at the Eifel site [SI Appendix, Fig. S8, Tukey, P < 0.1 (FDR) and Dataset S6B]. The single dominant Flavobacterium OTU mentioned earlier (OTU162362) was more abundant in C. hirsuta compared with A. thaliana root communities. We conclude that A. thaliana and C. hirsuta root microbiota consist of similar community modules and that the root communities differ quantitatively by their biogeography.
Next, we identified individual community members that differ quantitatively between the two host species. For this analysis, we initially defined for both natural sites the “RootOTUs” (SI Appendix), which represent 70 OTUs that are enriched in A. thaliana or C. hirsuta roots compared with the corresponding soil communities [Tukey, P < 0.1 (FDR); Fig. 3 A and B, SI Appendix, Fig. S9, and Dataset S6, worksheets C and D]. Spearman rank correlation coefficients of the RootOTU communities between these two hosts are 0.89 and 0.74 for the Cologne and Eifel sites, respectively, indicating an overall similar RootOTU composition. The RootOTUs in root communities vary mainly by the variable site (50 of the 70 RootOTUs) followed by host species [18 RootOTUs; ANOVA, P < 0.1 (FDR)] (Dataset S6, worksheet E). The comparison of RootOTUs between sites revealed a taxonomically narrow and shared set of 14 RootOTUs, consisting of seven Actinomycetales, three Burkholderiales, three Flavobacteriales, and a Myxococcales OTU (SI Appendix, Fig. S10). These shared RootOTUs were validated by parametric Tukey and nonparametric Mann–Whitney and Bayesian statistics (SI Appendix) and represent in their abundance half of the community. At the Eifel site, quantitative differences between the two plant species were found in 9 of the 70 RootOTU members, where 7 RootOTUs were more abundant in A. thaliana compared with C. hirsuta and 2 RootOTUs were less abundant [Tukey, P < 0.1 (FDR)] (SI Appendix, Fig. S11 and Dataset S6, worksheet F). This finding and the few aforementioned host species-differentiating community modules point to the existence of largely shared bacterial root communities with similar relative abundances in A. thaliana and C. hirsuta.
Fig. 3.

Root microbiota comparisons of A. thaliana and C. hirsuta at the natural sites Cologne and Eifel. The ternary plots depict the relative occurrence of individual OTUs (circles) in root samples of A. thaliana and C. hirsuta compared with the respective soil samples for the Cologne (A) and the Eifel site (B). RootOTUs [OTUs enriched in roots compared with soil; Tukey, P < 0.1 (FDR)] are colored by taxonomy, and OTUs, which are not enriched in root communities, are plotted in gray. The size of the circles is proportional to the mean abundance in the community. (C) Variation in mean relative abundance (RA) of individual OTUs (circles) across species and sites, where axes depict logtwofold variation [x axis is log2(A. thaliana/C. hirsuta) and y axis is log2(Eifel/Cologne)]. Color coding as in A and B.
Previous studies revealed a weak rhizosphere effect for A. thaliana (3, 4). To quantify the rhizosphere effect, we determined for both sites the OTUs that are enriched in the rhizosphere of A. thaliana or C. hirsuta compared with the corresponding soil (termed “RhizoOTUs”) (SI Appendix). Similar to these studies, we detected only few RhizoOTUs at the Cologne site [Tukey, P < 0.1 (FDR)] (SI Appendix, Fig. S12 and Dataset S6, worksheets C and D). The occurrence of a rhizosphere effect was found to be site-dependent: 6 (A. thaliana) and 11 RhizoOTUs (C. hirsuta) discriminated the rhizosphere from soil communities at the Cologne site whereas no RhizoOTUs (both host species) were found at the Eifel site. We conclude that the magnitude of the rhizosphere effect is site-dependent but independent of the tested host species.
Phylogenetic Distance of Host Species Contributes to Microbiota Diversification.
Next, we examined the bacterial root communities retrieved from the A. thaliana and the relative species A. halleri, A. lyrata, and C. hirsuta grown under controlled environmental conditions in replicated greenhouse experiments. A similar overall rank abundance profile of the ACMs in root communities between these four hosts reveals qualitatively similar community structures, indicating that variation in root microbiota is largely quantitative (Fig. 4A). We used ANOVA-based statistics to detect community modules and members that vary in abundance between the tested host species (SI Appendix). A few taxonomic modules differed in relative abundance between the root microbiota of the tested plant species (SI Appendix, Fig. S13), exemplified by significantly lower Bacteroidetes levels in A. halleri [Tukey, P < 0.1 (FDR)] (Dataset S6, worksheet G). This phenotype was again due to the differential abundance of the dominant Flavobacterium mentioned above (OTU162362). At family rank, A. halleri and A. lyrata display a species-specific quantitative reduction of Flavobacteriaceae and Oxalobacteriaceae, respectively (SI Appendix, Fig. S13B and Dataset S6, worksheet G).
Fig. 4.
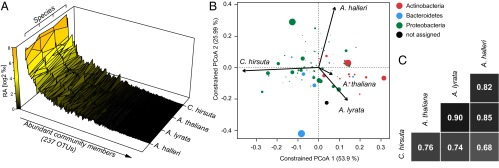
Root microbiota comparisons of A. halleri, A. lyrata, A. thaliana, and C. hirsuta. (A) The mean abundance of individual OTUs (both replicate experiments) was calculated for the indicated species and plotted ranked by average OTU abundance across all species. (B) OTU scores of principal coordinate analysis of the RootOTU community, constrained by host species and based on Bray–Curtis distances among root samples (see corresponding SI Appendix, Fig. S15). The arrows point to the centroid of the constrained factor. Circle sizes correspond to relative abundances of RootOTUs, and colors are assigned to different phyla. The percentage of variation explained by each axis refers to the fraction of the total variance of the data explained by host species. (C) Pairwise Spearman rank correlation analysis of the RootOTU communities between the indicated species.
Analogous to the community member analysis of the natural-site experiments, we identified 76 RootOTUs enriched in roots of at least one plant species compared with soil [Tukey, P < 0.1 (FDR)] (SI Appendix, Fig. S14 and Dataset S6, worksheets H and I). We then examined the between-sample (beta diversity) variation in the composition of RootOTUs among the host species using canonical analysis of principal coordinates (CAP) (26). CAP analysis constrained for the variable species revealed that 17% of the variation in beta diversity, as measured by Bray–Curtis distance metric, was explained by the host species (SI Appendix, Fig. S15) (P < 0.005; 95% confidence interval = 12%, 25%). The samples clustered by host species and distances between host species revealed that the root communities of A. thaliana were more similar to A. lyrata than to A. halleri and that the root microbiota of the three Arabidopsis species are more similar to each other than to the root microbiota of C. hirsuta. Thus, within the genus Arabidopsis (A. thaliana, A. halleri, and A. lyrata), microbiota diversification is incongruent with the phylogenetic distance of these hosts (compare Fig. 1 and SI Appendix, Fig. S15). Further exploration of the CAP analysis revealed a correspondence between the taxonomy of the RootOTUs and their contribution to the microbial diversity between host species: RootOTUs of the phylum Actinobacteria showed the strongest influence on the variation between the Arabidopsis species and C. hirsuta root communities (Fig. 4B). Similarly, the abundance of Bacteroidetes largely explains the differentiation between A. halleri and the other host species.
Community similarities were confirmed by pairwise correlation analysis of the RootOTU communities between the four host species, revealing Spearman rank coefficients ranging from 0.68 to 0.90 (Fig. 4C). The RootOTU composition of A. thaliana correlated best with each of its sister species A. halleri and A. lyrata, and all three pair-wise comparisons of C. hirsuta with the Arabidopsis species showed low correlation coefficients, suggesting that the evolutionarily most ancient plant species hosts a RootOTU community, which is quantitatively most diversified. Thus, inclusion of the more distant C. hirsuta suggests that phylogenetic distance of host species contributes to microbiota diversification across all four tested hosts. These observations were supported by the highest number of species-specific RootOTU accumulation patterns for C. hirsuta [Tukey, P < 0.1 (FDR)] (SI Appendix, Fig. S16 and Dataset S6, worksheet J). In total, we identified 14 species-specific RootOTUs that consisted of 1, 2, 4, and 7 RootOTUs for A. thaliana, A. halleri, A. lyrata, and C. hirsuta, respectively (SI Appendix, Fig. S16). The lower accumulation of the A. halleri-specific Flavobacterium (OTU162362) and the Oxalobacteriaceae member (OTU91279) in A. lyrata (SI Appendix, Fig. S16) contributed to the species-specific accumulation of the corresponding community modules (SI Appendix, Fig. S13). Similarly, the Actinocorallia RootOTU (OTU97580) contributed to the trend of lower accumulation of the Thermomonosporaceae, a distinctive feature of C. hirsuta (SI Appendix, Fig. S13). We independently validated the lower accumulation of Thermononosporaceae in C. hirsuta using quantitative PCR with taxon-specific PCR primers (SI Appendix, Fig. S17 and SI Appendix).
We assessed variation in microbiota composition between and within host species by a direct comparison of the three A. thaliana ecotypes with the three Arabidopsis sister species (SI Appendix). Ternary plots revealed a larger spread of abundant OTUs between the sister species than between the A. thaliana ecotypes (SI Appendix, Fig. S18). This observation is supported by the identification of 13 host species-dependent OTUs and one host genotype-dependent OTU [ANOVA, P < 0.1 (FDR)] (Dataset S6, worksheet K). This direct comparison demonstrates a greater inter- compared with intraspecies variation in microbiota composition.
Members of the Actinomycetales, Burkholderiales, and Flavobacteriales Are Stable Across Host Species and Environments.
We identified in the controlled environment experiments 26 RootOTUs shared among the four tested host species (SI Appendix, Fig. S19B). These shared RootOTUs belonged to the orders Burkholderiales (11 RootOTUs), Actinomycetales (7), Rhizobiales (3), Flavobacteriales (2), Myxococcales (1), Xanthomonadales (1), and Herpetosiphonales (1). These OTUs were validated by parametric Tukey and nonparametric Mann–Whitney and Bayesian statistics and constituted by their relative abundance the bulk of the root community (∼75%). Remarkably, the most abundant orders were also recovered in the shared RootOTUs in the natural-site experiments (SI Appendix, Fig. S10). The intersection of RootOTUs shared between plant species found at natural sites and in greenhouse experiments determined the core microbiota (Fig. 5A and SI Appendix, Fig. S20). This core consisted of nine RootOTUs assigned to the orders Actinomycetales (four RootOTUs, genus Actinocorallia), Burkholderiales (three, family Comamonadaceae), and Flavobacteriales (two, genus Flavobacterium) (Fig. 5A). This core represented a taxonomically extremely reduced subcommunity of the microbiota in all tested host species, and together these RootOTUs constituted by their abundance up to half of the root microbiota in all samples tested (SI Appendix, Fig. S20A). The enrichment of these core microbiota members relative to soil across plant species and sites was identified by three statistical methods (SI Appendix, Figs. S10B and S19B) and confirmed by a subsampling technique (i.e., bootstrapping) (SI Appendix, Fig. S20B, and SI Appendix). In addition, bootstrapping predicted OTUs of the orders Rhizobiales and Myxococcales to be part of the core microbiota (SI Appendix, Fig. S20B). However, the abundance of the latter two orders was less stable between environments, and, therefore, they did not pass the stringent identification of significant RootOTUs in the original data set using three different statistical methods (Fig. 5). Taken together, the core RootOTUs found across all host species and sites belonged to only three bacterial orders: the Actinomycetales, Burkholderiales, and Flavobacteriales. We compared the composition of this core microbiota to A. thaliana root endophyte communities from previous studies (3, 4, 21), which were based on different soil types, environments, and PCR primer combinations (SI Appendix, Fig. S21). The raw 16S rRNA gene sequences of these studies were coclustered with the sequences of this study, and the common OTU table was examined for the core microbiota in each data subset using the statistical procedure of this study (SI Appendix, Fig. S21 A and B, Dataset S7, and SI Appendix). A common core at OTU level cannot be confirmed in other A. thaliana root microbiome studies (SI Appendix, Fig. S21C) whereas, at order rank, the presence of Actinomycetales presents the common denominator across all studies. Burkholderiales and Flavobacteriales were detected in three and two of the four studies, respectively. Additionally, Rhizobiales and Sphingomonadales were each detected once as core members.
Fig. 5.

Identification of the core microbiota. (A) Core members result from the intersection of the shared RootOTUs found at the natural sites (SI Appendix, Fig. S10) and the shared RootOTUs detected in the greenhouse experiments (SI Appendix, Fig. S19). The pie chart segments are colored by the bacterial phyla of the corresponding taxa. The taxonomic assignments of the core RootOTUs are reported at order and family rank in the center and the first ring of the pie chart, respectively. The genera of the core members are noted at the periphery of the pie chart. (B) OTU scores of principal coordinate analysis of the RootOTU community, constrained by sample groups and based on Bray–Curtis distances among soil and root samples (see corresponding SI Appendix, Fig. S22). Sample groups include root samples by species and soil samples. The arrows point to the centroid of the constrained factor. Circle sizes correspond to relative abundances, colors are assigned to different phyla, and core members are marked as solid circles. The percentage of variation explained by each axis refers to the fraction of the total variance of the data explained by host species.
Using CAP analysis, we finally investigated the contribution of the core RootOTUs to the overall variation in root—compared with soil samples in all experimental conditions. Therefore, we constrained the analysis for all sample groups, i.e., root samples by species and the soil samples as additional group (SI Appendix, Fig. S22 and Fig. 5B). Consistent with the unconstrained beta diversity analysis (Fig. 2), the compartment constituted the major source of variation (SI Appendix, Fig. S22). We observed a clear differentiation between soil and root samples along the first principal coordinate, which explained the largest fraction of the variation (82.87%). We noted that the core RootOTUs (filled circles in Fig. 5B)—having the largest species descriptors—contributed most to the formation of the ordination space. This observation was consistent with their definition (enriched in root samples) and identification in all experimental conditions. Importantly, we confirmed the correspondence between the taxonomy of the RootOTUs and root microbiota diversity across host species over all experimental conditions: the root microbiota of Arabidopsis species were distinguished by RootOTUs of the phylum Actinobacteria (open and closed red circles in Fig. 5B) whereas root bacterial communities of C. hirsuta were differentiated by Bacteroidetes (open and closed blue circles in Fig. 5B). We interpreted these correspondences as evidence of a host impact on the root microbiota at a high taxonomic rank.
Discussion
A Conserved Core Root Microbiota?
Here, we have examined the bacterial root microbiota of A. thaliana along with its sister species A. lyrata and A. halleri and of C. hirsuta. This study revealed the existence of a core root microbiota comprising members from the three bacterial orders Actinomycetales, Burkholderiales, and Flavobacteriales (Fig. 5A). Previous studies (3, 4, 21), using different 16S rRNA PCR primer combinations, reported that A. thaliana roots host mainly Actinobacteria, Bacteroidetes, and Proteobacteria. This taxonomic structure at phylum level is congruent with the core composition described here because the order Actinomycetales belongs to the phylum Actinobacteria, the Burkholderiales belongs to the subphylum Betaproteobacteria, and the Flavobacteriales to the phylum Bacteroidetes. Bootstrapping also identified the core members and revealed additional RootOTUs, expanding the core composition (SI Appendix, Fig. S20B). These Rhizobiales and Myxococcales members became apparent in approximately half of random subsets of the original data. Enhanced variation in their abundance between replicate samples and tested environments could explain their absence from the core microbiota.
The significance of our definition of the core microbiota is potentially constrained by the PCR primer used and a low number of tested environments (Cologne and Eifel natural sites and controlled environment). It remains to be seen whether additional samples, also from extreme environments, modify the composition of the core. Corroborating evidence for its stability in additional environments comes from a recent A. thaliana field study comprising four disturbed sites in the United States using the same PCR primer combination (21). In these root samples, Actinomycetales, Burkholderiales, and Flavobacteriales were found by 16S rRNA gene pyrosequencing among other prevalent taxa, and the taxonomic composition of the core at order level is similar to our study (SI Appendix, Fig. S21C). Differences in the selectivity of different 16S PCR primers and variation in 16S rRNA gene copy number likely distort the composition of the core root microbiota. The comparison across A. thaliana root microbiome studies (3, 4, 21) did not reveal a common core at the OTU level (SI Appendix, Fig S21C). However, we cannot discriminate whether PCR primer bias, the soil type/start inoculum, or combinations thereof account for the lack of a common OTU core. Despite this lack of clarity, we noted that, at higher taxonomic rank, the enrichment of members of the Actinomycetales in roots was a common feature of all A. thaliana root microbiome studies (SI Appendix, Fig. S21B). Actinobacteria, including the Actinomycetales, appear to be enriched from soil by cues from living A. thaliana roots (3). Our study suggests that such host-derived assembly signal(s) are evolutionarily conserved, at least in the Brassicaceae. Future examination of root microbiomes from additional plant species in the same environments and using the same PCR primer combination will test whether this core is a lineage-specific innovation of the Brassicaceae family.
The core root microbiota members accounted for up to half of the total community size (SI Appendix, Fig. S20A). The core consisted of both abundant and low-abundant community members, suggesting that assembly and physiological function(s) depend on selective membership and regulation of their relative abundance. In addition, we found a correspondence between the taxonomy of the bacterial communities and the diversity pattern of the root microbiota across host species and three different environments (Fig. 5B). The core members largely supported this correspondence. These observations, together with the reduced taxonomic complexity of the core community, point to the existence of a common organizing principle for their establishment. We speculate about two potential and mutually not exclusive mechanisms that take part in the establishment process: (i) each bacterial lineage autonomously responds to host-derived cues and (ii) microbe–microbe interactions enable a selective advantage for cocolonization by core members. For example, the commensal relationship between Bacillus cereus and bacteria of the Cytophaga-Flavobacterium (CF) group in the soybean rhizosphere is mediated by peptidoglycan, which is produced by B. cereus and stimulates the growth of CF bacteria (27). From future root-microbiota metagenome and -transcriptome analyses, we expect insights into the connectivity among microbes (28). Such approaches will define the core microbiota at the level of genes rather than taxonomic lineages and will provide a deeper understanding of host services of the core as pioneered by human gut microbiota research (10, 11).
Members of each taxon of the core are potentially beneficial for their hosts. Grassland rhizosphere-derived cultured strains belonging to the Burkholderiales were shown to have antagonistic activities toward multiple soilborne oomycete and fungal pathogens (29). Wheat rhizosphere-derived Comamonadaceae strains appear to be functional specialists in soil sulfonate transformation as part of the biogeochemical sulfur cycle, likely enabling mineralization of organic sulfur for sulfate acquisition by high-affinity sulfate transporters at the root surface (30, 31). Flavobacterium, a common soil and water bacterium, was positively correlated with potato biomass and frequently isolated from barley and bell pepper rhizospheres (32–34). A reference Flavobacterium isolated from the rhizosphere of the latter plant tested positive for several biochemical assays associated with plant growth promotion and pathogen protection, and accessible genomes of soil/rhizosphere-derived Flavobacteria indicate that these bacteria define a distinct clade compared with Flavobacteria from aquatic environments (34). Finally, root-derived isolates of the Thermomonosporaceae and other core members such as Polaromonas isolates are capable of fixing atmospheric nitrogen (35, 36), potentially increasing the amount of bioavailable nitrogen for plant growth.
Root Microbiota Interactions with the Environment.
If the aforementioned physiological function(s) of the core hold true for isolates present in Brassicaceae roots, then these functions could present a standing reservoir of retrievable host services independently of environmental parameters and host species-specific niche adaptations (e.g., metal tolerance of A. halleri) or life history traits (perennialism of A. lyrata and A. halleri). Although the conserved core is established in both controlled and natural environments, the composition of the entire root microbiota depends most on interactions with the environment (Fig. 2). This dependence is consistent with prior findings that soil type is the key determinant of root community structure (3, 4, 16). Thus, the whole root microbiota consists of two parts, the conserved core and an environment-responsive subcommunity. For example, members of the order Rhizobiales represent a root community module found only as shared RootOTUs in the controlled environment using experimental Cologne soil (SI Appendix, Fig. S19). In addition, the relative abundance of the family Oxalobacteriaceae is environment-responsive (SI Appendix, Figs. S8 and S13), resulting in a change in rank abundance from rank 2 in the greenhouse experiments to rank 3 at the natural sites. Likewise, dominant Streptomycetaceae in the controlled environment (rank 3) are a low-abundant taxon at the natural sites (rank 14). Thus, the relative abundance of these taxa is tunable by the environment. We speculate that these environment-responsive community modules provide services to all tested host species in an environment-dependent manner.
The strong responsiveness of the root microbiota to the environment might be explained by the fact that soil does not only define the start inoculum but also the “diet” for plants, e.g., bioavailability of macro- and micronutrients. Diet as a major driver for community structure was previously reported in microbiota hosted by other eukaryotes. For example, the mammalian gut microbiota follows primarily the dietary habits of the animals, where communities from herbivores, carnivores, and omnivores clustered clearly apart from each other (37). Dietary patterns in humans appear to determine gut microbiota enterotypes (38, 39). However, it remains to be examined whether dietary effects independent of the soil start inoculum are sufficiently strong to provoke consistent shifts in root microbiota community composition. A further exploration of this question would require the modulation of individual nutrients or their composition in the same soil type/start inoculum and subsequent determination of possible effects on community structure.
Host Microbiota Coevolution.
A systematic investigation of host-microbiota diversification within a phylogenetically defined plant species framework, combined with replicated experiments under controlled conditions, has not been reported before. A major finding of our work is that the diversification of the root microbiota of the tested host species is largely quantitative. This conclusion is based on abundant community members (ACM, >20 sequences per OTU in at least one sample of the dataset), and, therefore, qualitative differences might exist in the rare biosphere, which is currently not quantifiable. Despite an overall interhost species microbiota similarity, we found host species-specific community modules (SI Appendix, Figs. S8 and S13) and members (SI Appendix, Figs. S11 and S16). The host species-specific community modules are clearly part of the environment-responsive subcommunity, as illustrated by the observation that they are site-dependent (Fig. 3). Both the most divergent root microbiota (Figs. 4 B and C and 5B) and the highest number of species-specific community members (SI Appendix, Fig. S16) were found in the phylogenetically most distant C. hirsuta (Fig. 1), suggesting that phylogenetic distance of the hosts could contribute to microbiota diversification. Future studies using additional plant lineages are required to conclude whether phylogenetic distance time correlates with microbiota diversification. For example, in primates, the branching order of host-species phylogeny was found to be congruent with gut community composition (40). The greatest similarities in root microbiota were found between A. thaliana and A. lyrata whereas the root microbiota of A. halleri was more dissimilar (Fig. 4 B and C), demonstrating that, within the genus Arabidopsis (A. thaliana, A. halleri, and A. lyrata), microbiota diversification is incongruent with the phylogenetic distances of these hosts (Fig. 1). The two species A. thaliana and A. lyrata occur in similar habitats whereas A. halleri has evolved a distinctive lifestyle enabling growth in mesic sites and tolerance to high-competition habitats. This particularity could imply that the recent speciation event of A. halleri, coupled to an adaptation to a distinctive habitat, resulted in the selection of a distinctive microbiota with habitat-specific services. Taken together, both host species-specific ecological adaptation and phylogenetic distance might have driven microbiota diversification among the tested hosts. Whether the proportion of species-specific community members/modules increases when the host species are exposed to stressful conditions where a plant species has an adaptive advantage (e.g., tolerance of A. halleri to metalliferous soils) (8) remains to be tested. Similarly, it will be interesting to examine whether the proportion of species-specific community members/modules increases when the perennials A. lyrata and A. halleri are grown according to their lifestyle for longer than 1 y. Finally, future experimentation using synthetic bacterial communities with isolates of the core microbiota members and gnotobiotic Arabidopsis plants will directly test whether their presumed beneficial roles in plant growth and health can be reproduced under laboratory conditions and are retrievable by the host under normal and stressful conditions.
Materials and Methods
We collected roots of naturally occurring A. thaliana and C. hirsuta growing side by side at the two replicate sites Cologne and Eifel. Additionally, we sampled in two replicate experiments roots of A. thaliana and the relative species A. lyrata, A. halleri, and C. hirsuta, which were grown under controlled conditions in the greenhouse in pots containing natural microbe-rich soil. We used a root-sampling protocol similar to Bulgarelli et al. (3) to examine the root-inhabiting bacterial microbiota. For comparison, we also sampled bulk soil and rhizosphere compartments. Bacterial communities were characterized by pyrosequencing 16S rRNA gene amplicons derived from the PCR primers 799F (20) and 1193R (21). The pyrosequencing reads were processed and analyzed with the software QIIME (22), and custom R scripts were used for statistical analyses. For details, see SI Appendix.
Supplementary Material
Acknowledgments
We thank Dr. Bruno Hüttel and Dr. Kurt Stüber (Max Planck Genome Centre Cologne) for excellent support in amplicon library preparation and pyrosequencing. We thank Dr. Yang Bai and Dr. Davide Bulgarelli (Department of Plant Microbe Interactions, Max Planck Institute for Plant Breeding Research) for critical comments on the manuscript. Dr. Pierre Saumitou-Laprade (Laboratoire de Génétique et Évolution des Populations Végétales, Formations de Recherche en Évolution, Centre National de la Recherche Scientifique 3268, Université de Lille) kindly provided seeds of A. halleri. K.S. was supported by Swiss National Science Foundation Grants PBFRP3-133544 and PA00P3-136473. This work was supported by funds to P.S.-L. from the Max Planck Society (M.IF.A. ZUCH8048), a European Research Council advanced grant (ROOTMICROBIOTA), and the “Cluster of Excellence on Plant Sciences” program funded by the Deutsche Forschungsgemeinschaft.
Footnotes
The authors declare no conflict of interest.
Data deposition: The data reported in this paper have been deposited in the European Nucleotide Archive database (accession no. PRJEB5058).
See Profile on page 570.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1321597111/-/DCSupplemental.
References
- 1.Vorholt JA. Microbial life in the phyllosphere. Nat Rev Microbiol. 2012;10(12):828–840. doi: 10.1038/nrmicro2910. [DOI] [PubMed] [Google Scholar]
- 2.Bulgarelli D, Schlaeppi K, Spaepen S, Ver Loren van Themaat E, Schulze-Lefert P. Structure and functions of the bacterial microbiota of plants. Annu Rev Plant Biol. 2013;64:807–838. doi: 10.1146/annurev-arplant-050312-120106. [DOI] [PubMed] [Google Scholar]
- 3.Bulgarelli D, et al. Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature. 2012;488(7409):91–95. doi: 10.1038/nature11336. [DOI] [PubMed] [Google Scholar]
- 4.Lundberg DS, et al. Defining the core Arabidopsis thaliana root microbiome. Nature. 2012;488(7409):86–90. doi: 10.1038/nature11237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beilstein MA, Nagalingum NS, Clements MD, Manchester SR, Mathews S. Dated molecular phylogenies indicate a Miocene origin for Arabidopsis thaliana. Proc Natl Acad Sci USA. 2010;107(43):18724–18728. doi: 10.1073/pnas.0909766107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoffmann MH. Evolution of the realized climatic niche in the genus Arabidopsis (Brassicaceae) Evolution. 2005;59(7):1425–1436. [PubMed] [Google Scholar]
- 7.Clauss MJ, Koch MA. Poorly known relatives of Arabidopsis thaliana. Trends Plant Sci. 2006;11(9):449–459. doi: 10.1016/j.tplants.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 8.Krämer U. Metal hyperaccumulation in plants. Annu Rev Plant Biol. 2010;61:517–534. doi: 10.1146/annurev-arplant-042809-112156. [DOI] [PubMed] [Google Scholar]
- 9.Janzen DH. The natural history of mutualisms. In: Boucher DH, editor. The Biology of Mutualism: Ecology and Evolution. New York: Oxford Univ Press; 1985. [Google Scholar]
- 10.Turnbaugh PJ, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457(7228):480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qin JJ, et al. MetaHIT Consortium A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464(7285):59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sekelja M, Berget I, Næs T, Rudi K. Unveiling an abundant core microbiota in the human adult colon by a phylogroup-independent searching approach. ISME J. 2011;5(3):519–531. doi: 10.1038/ismej.2010.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;489(7415):220–230. doi: 10.1038/nature11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pedron T, et al. (2012) A crypt-specific core microbiota resides in the mouse colon. Mbio 3(3):e00116-12. [DOI] [PMC free article] [PubMed]
- 15.Roeselers G, et al. Evidence for a core gut microbiota in the zebrafish. ISME J. 2011;5(10):1595–1608. doi: 10.1038/ismej.2011.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peiffer JA, et al. Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc Natl Acad Sci USA. 2013;110(16):6548–6553. doi: 10.1073/pnas.1302837110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Inceoğlu O, Al-Soud WA, Salles JF, Semenov AV, van Elsas JD. Comparative analysis of bacterial communities in a potato field as determined by pyrosequencing. PLoS ONE. 2011;6(8):e23321. doi: 10.1371/journal.pone.0023321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hardoim PR, et al. Rice root-associated bacteria: Insights into community structures across 10 cultivars. FEMS Microbiol Ecol. 2011;77(1):154–164. doi: 10.1111/j.1574-6941.2011.01092.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sharma S, Aneja MK, Mayer J, Munch JC, Schloter M. Characterization of bacterial community structure in rhizosphere soil of grain legumes. Microb Ecol. 2005;49(3):407–415. doi: 10.1007/s00248-004-0041-7. [DOI] [PubMed] [Google Scholar]
- 20.Chelius MK, Triplett EW. The diversity of Archaea and Bacteria in association with the roots of Zea mays L. Microb Ecol. 2001;41(3):252–263. doi: 10.1007/s002480000087. [DOI] [PubMed] [Google Scholar]
- 21.Bodenhausen N, Horton MW, Bergelson J. Bacterial communities associated with the leaves and the roots of Arabidopsis thaliana. PLoS ONE. 2013;8(2):e56329. doi: 10.1371/journal.pone.0056329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caporaso JG, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McDonald D, et al. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012;6(3):610–618. doi: 10.1038/ismej.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lozupone C, Knight R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 2005;71(12):8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Faith DP. Conservation evaluation and phylogenetic diversity. Biol Conserv. 1992;61(1):1–10. [Google Scholar]
- 26.Anderson MJ, Willis TJ. Canonical Analysis of Principal Coordinates: A useful method of constrained ordination for ecology. Ecology. 2003;84:511–525. [Google Scholar]
- 27.Peterson SB, Dunn AK, Klimowicz AK, Handelsman J. Peptidoglycan from Bacillus cereus mediates commensalism with rhizosphere bacteria from the Cytophaga-Flavobacterium group. Appl Environ Microbiol. 2006;72(8):5421–5427. doi: 10.1128/AEM.02928-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shade A, Handelsman J. Beyond the Venn diagram: The hunt for a core microbiome. Environ Microbiol. 2012;14(1):4–12. doi: 10.1111/j.1462-2920.2011.02585.x. [DOI] [PubMed] [Google Scholar]
- 29.Benítez MS, Gardener BBM. Linking sequence to function in soil bacteria: Sequence-directed isolation of novel bacteria contributing to soilborne plant disease suppression. Appl Environ Microbiol. 2009;75(4):915–924. doi: 10.1128/AEM.01296-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schmalenberger A, et al. The role of Variovorax and other Comamonadaceae in sulfur transformations by microbial wheat rhizosphere communities exposed to different sulfur fertilization regimes. Environ Microbiol. 2008;10(6):1486–1500. doi: 10.1111/j.1462-2920.2007.01564.x. [DOI] [PubMed] [Google Scholar]
- 31.Yoshimoto N, Takahashi H, Smith FW, Yamaya T, Saito K. Two distinct high-affinity sulfate transporters with different inducibilities mediate uptake of sulfate in Arabidopsis roots. Plant J. 2002;29(4):465–473. doi: 10.1046/j.0960-7412.2001.01231.x. [DOI] [PubMed] [Google Scholar]
- 32.Manter DK, Delgado JA, Holm DG, Stong RA. Pyrosequencing reveals a highly diverse and cultivar-specific bacterial endophyte community in potato roots. Microb Ecol. 2010;60(1):157–166. doi: 10.1007/s00248-010-9658-x. [DOI] [PubMed] [Google Scholar]
- 33.Johansen JE, Nielsen P, Binnerup SJ. Identification and potential enzyme capacity of flavobacteria isolated from the rhizosphere of barley (Hordeum vulgare L.) Can J Microbiol. 2009;55(3):234–241. doi: 10.1139/w08-116. [DOI] [PubMed] [Google Scholar]
- 34.Kolton M, et al. Draft genome sequence of Flavobacterium sp. strain F52, isolated from the rhizosphere of bell pepper (Capsicum annuum L. cv. Maccabi) J Bacteriol. 2012;194(19):5462–5463. doi: 10.1128/JB.01249-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Valdés M, et al. Non-Frankia actinomycetes isolated from surface-sterilized roots of Casuarina equisetifolia fix nitrogen. Appl Environ Microbiol. 2005;71(1):460–466. doi: 10.1128/AEM.71.1.460-466.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hanson BT, Yagi JM, Jeon CO, Madsen EM. Role of nitrogen fixation in the autecology of Polaromonas naphthalenivorans in contaminated sediments. Environ Microbiol. 2012;14(6):1544–1557. doi: 10.1111/j.1462-2920.2012.02743.x. [DOI] [PubMed] [Google Scholar]
- 37.Ley RE, et al. Evolution of mammals and their gut microbes. Science. 2008;320(5883):1647–1651. doi: 10.1126/science.1155725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Arumugam M, et al. MetaHIT Consortium Enterotypes of the human gut microbiome. Nature. 2011;473(7346):174–180. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu GD, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334(6052):105–108. doi: 10.1126/science.1208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ochman H, et al. Evolutionary relationships of wild hominids recapitulated by gut microbial communities. PLoS Biol. 2010;8(11):e1000546. doi: 10.1371/journal.pbio.1000546. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.