

Significance
Drug resistance is a major problem in treating malaria infections. Resistance pathways are limited by evolutionary fitness constraints, and these pathways can be anticipated and preemptively blocked. We identified compounds selective for mutant malaria parasites that are resistant to either clinically used (chloroquine) or in-development dihydroorotate dehydrogenase (DHODH) inhibitor therapies. In both cases, resistance selections of mutant parasites with the matching mutant-selective drug led to a reversion to the wild-type phenotype. Additionally, combination selection of wild-type parasites with paired wild-type and mutant-type DHODH inhibitors failed to produce resistant parasites. These findings indicate that pairing inhibitors that target the bulk wild-type population and the small, emerging resistant population can suppress the rise and spread of drug resistance.
Keywords: evolution, target identification, combination therapy
Abstract
Drug resistance emerges in an ecological context where fitness costs restrict the diversity of escape pathways. These pathways are targets for drug discovery, and here we demonstrate that we can identify small-molecule inhibitors that differentially target resistant parasites. Combining wild-type and mutant-type inhibitors may prevent the emergence of competitively viable resistance. We tested this hypothesis with a clinically derived chloroquine-resistant (CQr) malaria parasite and with parasites derived by in vitro selection with Plasmodium falciparum dihydroorotate dehydrogenase (PfDHODH) inhibitors. We screened a chemical library against CQs and CQr lines and discovered a drug-like compound (IDI-3783) that was potent only in the CQr line. Surprisingly, in vitro selection of Plasmodium falciparum resistant to IDI-3783 restored CQ sensitivity, thereby indicating that CQ might once again be useful as a malaria therapy. In parallel experiments, we selected P. falciparum lines resistant to structurally unrelated PfDHODH inhibitors (Genz-666136 and DSM74). Both selections yielded resistant lines with the same point mutation in PfDHODH:E182D. We discovered a compound (IDI-6273) more potent against E182D than wild-type parasites. Selection of the E182D mutant with IDI-6273 yielded a reversion to the wild-type protein sequence and phenotype although the nucleotide sequence was different. Importantly, selection with a combination of Genz-669178, a wild-type PfDHODH inhibitor, and IDI-6273, a mutant-selective PfDHODH inhibitor, did not yield resistant parasites. These two examples demonstrate that the compromise between resistance and evolutionary fitness can be exploited to design therapies that prevent the emergence and spread of resistant organisms.
Malaria is a vector-borne infectious disease transmitted by female Anopheles mosquitoes and caused by protozoan parasites of the genus Plasmodium. Malaria is widespread in tropical and subtropical regions, including parts of the Americas, Asia, and Africa. Malaria afflicts 350–500 million people each year, resulting in ∼800,000 deaths per year (1). The majority of malaria-related deaths occur in young children below the age of five in Sub-Saharan Africa (1). Resistance has emerged to nearly all antimalarial drugs, including the front-line artemisinin-based combination therapies (2). Widespread resistance necessitates both the discovery and development of new antimalarial therapies as well as strategies to protect current and future therapies from the threat of resistance. We explore here the possibility of blocking the emergence of resistance with a population biology trap: by identifying situations where resistance to one compound confers hypersensitivity to another, we can design combination therapies that not only kill the parasite, but also guide its evolution away from resistance.
Drug resistance pathways in Plasmodium falciparum are sharply limited by tradeoffs among growth, transmissibility, and resistance. For example, there is a single dominant resistance-fitness maximum for the dihydrofolate reductase inhibitor pyrimethamine, and the resistance phenotype is accessible by a very small number of mutational pathways (3). Resistance pathways can be identified through human or animal treatment failures or through in vitro resistance selections. These strategies expose parasites to different selective pressures, and so do not necessarily yield identical results. However, in vitro resistance selections have been key in ethically predicting resistance pathways, including identifying pfmdr1 as a resistance locus for mefloquine (4).
Resistance has fitness costs and can leave organisms vulnerable to other stresses. For example, M184V/I mutations in HIV-1 reverse transcriptase confer resistance to nucleoside reverse transcriptase inhibitors such as lamivudine. However, these mutants are hypersensitive to zidovudine, stavudine, and tenofovir-DF and have a compromised ability to incorporate natural nucleotide substrates (5). Point mutations in the BCR-ABL kinase that drives chronic myeloid leukemia can give resistance to imatinib (6), but these differ in their transforming ability. In addition, second-generation BCR-ABL inhibitors such as nilotinib and dasatinib successfully target many of the imatinib-resistant kinases (7). From these observations, we propose that drug resistance creates a tractable target for drug discovery.
We set out to identify inhibitors that selectively targeted drug-resistant P. falciparum mutant parasites. We then used these mutant-selective inhibitors to drive parasite evolution toward a wild-type phenotype. To do so, we performed two sets of experiments: one with an existing, clinically-relevant chloroquine-resistant (CQr) mutant, and the other with de novo-derived resistance to in-development dihydroorotate dehydrogenase (PfDHODH) inhibitors.
CQ resistance is widespread in the field and is proposed to have occurred through at least four independent founder events: one in Asia that spread to Africa (K76T in the chloroquine resistance transporter PfCRT), one in Papua New Guinea, and two in South America. These mutations spread across the globe in only 20–80 sexual generations due to strong directional selective sweeps (8). Despite selective sweeps, CQ resistance has a fitness cost and is untenable in the absence of drug. For example, CQ sensitivity returned to Malawi after a 12-y CQ absence (9). As CQ is widely-used, economical, and one of very few antimalarials safe for use during pregnancy, a method for restoring CQ’s effectiveness and protecting against future resistance would be tremendously valuable.
Verapamil and related “sensitizer” compounds have been shown to reverse chloroquine resistance. When used alone, verapamil has only modest effects against P. falciparum, with IC50 values in the range of 6–8 µg/mL in culture. However, subtherapeutic doses of verapamil restored CQ sensitivity to the CQ-resistant line W2 and had no effect on the CQ-sensitive line D6 (10). This sensitization was later shown to extend across multiple CQ-resistant lines (11), suggesting a general mechanism for resistance. Later studies demonstrated that verapamil interferes with CQ transport through the Plasmodium falciparum chloroquine resistance transporter (PfCRT), which effluxes CQ from the food vacuole (12). Verapamil has a relatively modest fivefold effect (13), and it has not been used clinically in its 25-y history as an antimalarial sensitizer. A more potent, more drug-like sensitizer to restore the efficacy of chloroquine, malaria’s “spent magic bullet” (14), would be useful.
DHODH is an enzyme that catalyzes the rate-limiting step of pyrimidine biosynthesis. Malaria parasites lack pyrimidine salvage pathways and are thus reliant upon de novo biosynthesis. Although humans and Plasmodium both have class II DHODH enzymes, crystal structures have shown substantial differences in the pocket where all known inhibitors bind (15). Multiple groups are developing PfDHODH inhibitors (16), and DHODH is considered a promising target for the future of antimalarial chemotherapy (17). We sought to catalog DHODH resistance mechanisms and then develop strategies to contain and suppress resistance before clinical deployment.
Results and Discussion
We performed a high-throughput small-molecule screen against both CQs and CQr parasite lines to identify compounds differentially active in the CQr parasites. The screening effort provided a cluster of fourteen 1,4-dibenzothiazepene amides. Of these screening hits, compound IDI-3783 was resynthesized and shown to have an EC50 of 3 nM in the CQr Dd2 strain compared with >10 μM EC50 against wild-type 3D7 parasites (Fig. 1 A and B). Resistance selections were carried out in Dd2 parasites using IDI-3783 at 10-fold the EC50 concentration. Drug-resistant parasites emerged after 35 d that showed a 1,000-fold increase in the EC50 for IDI-3783, shifting from 3 nM to 4,000 nM. Surprisingly, IDI-3783 resistance was coupled with restored sensitivity to CQ, with the CQ EC50 dropping from 100 nM to 14 nM, a 10-fold shift (Fig. 1C). IDI-3783 acts synergistically with CQ when administered together, implying that IDI-3783 blocks the efflux of CQ through PfCRT K76T in addition to its own toxic effect (Fig. S1). The EC50 of atovaquone and mefloquine, both unrelated antimalarial drugs, was unchanged between the sensitive and resistant parasites (Fig. 1D and Table S1).
Fig. 1.

Chloroquine and IDI-3783 resistance are mutually exclusive. (A) The thiazepine amide IDI-3783 is highly potent against CQr parasites (Dd2, black bars) while demonstrating µM activity against CQs parasites (3D7, white bars). Selection of the Dd2 parental line with IDI-3783 led to a highly significant increase in EC50 (***P ≤ 0.0001) rendering the parasites resistant to the compound (Dd2: Q352R, gray bars). (B) Selection for IDI-3783–resistant parasites results in a significant decrease in chloroquine EC50 (***P ≤ 0.0001) rendering them sensitive to the drug at levels comparable to 3D7. (C and D) The control compounds atovaquone and mefloquine do not demonstrate significant changes in EC50 between the parasite lines tested (n.s., not significant). (E) Topology prediction of PfCRT showing the location of the Q352R amino acid change in the IDI-3783–resistant parasites (red dot). The remainder of the sequence was unchanged relative to the Dd2 parent, including the important K76T mutation found in CQr parasites, highlighted in yellow. EC50 values were determined using a whole-cell SYBR Green assay (28). Error bars indicate the SD of three biological replicates, each with triplicate measurements. Significance relative to Dd2 EC50 was determined by one-way analysis of variance (ANOVA) with Dunnett’s multiple comparison post test; n = 3.
Whole-genome sequencing of the IDI-3783-resistant Dd2 parasite revealed a Q352R mutation in PfCRT, in the ninth transmembrane segment of the protein (Fig. 1E). The preexisting K76T mutation reduces the positive character of the PfCRT channel, and this more neutral milieu is believed to allow protonated, positively charged CQ to leave the food vacuole (18), thereby preventing accumulation of toxic concentrations of drug. The Q352R mutation reintroduces positive charge into the PfCRT channel, thereby restoring sensitivity to CQ by preventing efflux. The behavior of the Q352R mutation is consistent with a model where IDI-3783 inhibits the channel function of PfCRT in the presence of the K76T CQr mutation, and that introduction of the Q352R mutation to PfCRT simultaneously disrupts IDI-3783 activity and prevents the efflux of CQ from the food vacuole. From these results, a new type of combination therapy might be proposed even in the face of widespread CQ resistance in the field: namely, the combination of CQ and a molecule that kills CQr parasites.
To extend these findings, we hypothesized that we could anticipate the emergence of resistance for molecules in the drug-development pipeline. To test this hypothesis, we focused on dihydroorotate dehydrogenase (DHODH). We developed an enzyme assay for P. falciparum dihydroorotate dehydrogenase (PfDHODH) and initiated an HTS campaign that identified a unique chemotype, 5-benzimidazolyl-N-alkylthiophene-2-carboxamide, as an inhibitor of PfDHODH (Fig. 2A) (Genz-669178) (16, 19). The alkylthiophene inhibitors have low nanomolar potency against purified enzyme and excellent selectivity against human DHODH. Further investigations of these compounds showed potency at double-digit nanomolar concentrations against P. falciparum in vitro and were also found to be efficacious against the rodent malaria parasite Plasmodium berghei in vivo (16). Phillips and co-workers (20, 21) have also published a novel series of triazolopyrimidine-based PfDHODH inhibitors with comparable activity.
Fig. 2.

Genz-669178 and IDI-6273 resistance are mutually exclusive. (A) The wild-type PfDHODH inhibitor Genz-669178 is highly potent against wild-type 3D7 parasites (black bars) and weakly potent against parasites raised in a resistance selection (3D7 E182D, white bars) (***P < 0.0001). Sequential selection of the 3D7 E182D line with IDI-6273, a PfDHODH-E182D selective inhibitor, led to increased sensitivity to Genz-669178 (3D7 E182D: D182E, gray bars) (***P < 0.0001). However, the revertant was significantly different from the 3D7 wild-type (**P < 0.001). (B) DSM74 is highly potent against wild-type 3D7 parasites (black bars), and weakly potent against parasites raised in a resistance selection (3D7 E182D, white bars) (***P < 0.0001). The 3D7 E182D: D182E revertant regained sensitivity to DSM74 (***P < 0.0002). The revertant was not significantly different from the wild-type 3D7 (n.s., not significant). (C) IDI-6253 shows no significant difference between 3D7 and 3D7 E182D, but it is less potent against the 3D7 E182D: D182E wild-type revertant (**P = 0.0043). (D) The mutant PfDHODH inhibitor IDI-6273 is highly potent against 3D7 E182D, but much less so against wild-type 3D7 or the wild-type revertant 3D7 E182D: D182E (***P < 0.0001). The 3D7 E182D: D182E wild-type revertant is significantly different from the 3D7 wild-type (***P < 0.0001). (E) GSK-3 is highly potent against 3D7 and is markedly less potent against 3D7 E182D (***P < 0.0001). The 3D7 E182D: D182E wild-type revertant shows no significant difference with wild-type 3D7. (F) The control compound dihydroartemisinin does not demonstrate significant changes in EC50 between the parasite lines tested (n.s., not significant). (G) Sequence alignment showing nucleotide changes in the pfdhodh gene. The remainder of the sequence was unchanged relative to the 3D7 parent. EC50 values were calculated using a whole-cell SYBR Green (28) assay. Error bars indicate the SD of three biological replicates, each with triplicate measurements. Significance relative to 3D7: E182D EC50 was determined by one-way ANOVA with Tukey’s multiple comparison post test; n =3. (H) Residue E182 lines the drug-binding site of PfDHODH and makes a salt bridge with R262. This interaction may be important for holding the helix-turn-helix lid of the binding pocket against the body of the protein. The shortened side chain in an E182D mutant may weaken this interaction, affecting drug binding to this pocket. The image was prepared using PDB code 3o8a with CCP4mg software (30).
We conducted resistance selections using either a representative alkylthiophene inhibitor (Genz-666136) or a triazolopyrimidine inhibitor (DSM74) in wild-type 3D7 parasites. Resistant parasites from these selections were subjected to targeted sequencing of the pfdhodh gene and whole-genome sequencing. Both compounds independently selected an E182D mutation in PfDHODH, indicating that the E182D mutation may be an optimal fitness–resistance compromise. E182 resides in the lid of the PfDHODH species-selective inhibitor site (Fig. 2H) (22).
To further characterize the 3D7 E182D PfDHODH inhibitor-resistant parasites, we rescreened 59 compounds from our earlier HTS and two compounds from a similar program at GlaxoSmithKline against the resistant line. In this small set, we found examples of compounds more potent against 3D7 E182D than 3D7. Compound IDI-6273 (Fig. 2D) was 10.95-fold more active against 3D7 E182D than 3D7, with EC50 values of 2,300 nM for the 3D7 parent improving to 210 nM for 3D7 E182D. Further resistance selections were carried out in the 3D7 E182D parasites with IDI-6273. Drug-resistant parasites emerged after 41 d. These parasites showed a 19-fold increase in the EC50 for IDI-6273, shifting from 210 nM in the 3D7 E182D parent to 4,000 nM in the newly resistant line. Both selections had no effect on response to dihydroartemisinin, a control compound (Fig. 2F and Table S2).
Sequencing the pfdhodh gene in the 3D7 E182D selected line uncovered another mutation in codon 182: GAT became GAG. This nucleotide mutation resulted in a reversion to the wild-type 3D7 protein sequence (Fig. 2G). Thus, the sequentially selected line is 3D7 E182D: D182E. Although it is promising that sequential selection of a mutant parasite with a mutant-selective inhibitor led to a wild-type reversion, the relatively long timeline under single-drug pressure increases the chances of compensatory fitness mutations that could bolster a less-fit resistance pathway and allow it to emerge and spread.
To limit the risk of minor variant resistance pathways, we tested simultaneous selection of a wild-type population with both a wild-type and mutant-type inhibitor. When dosed together, these compounds are highly synergistic (Fig. S1). Simultaneous selection with IDI-6273 and Genz-669178, a later derivative of the Genz-666136 used to produce the 3D7 E182D line, did not yield any drug-resistant parasites in forty generations of exposure (80 d) (Fig. 3 and Table S3). This result supports the idea of combining compounds that target wild-type and resistant parasites for treating malaria and reducing the risk of developing resistance. All compounds used in this study were synthesized as described or purchased (Table S4).
Fig. 3.
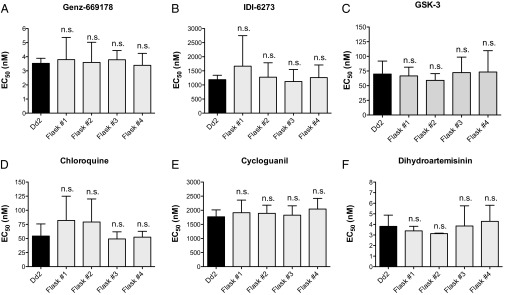
Combination selection with Genz-669178 and IDI-6273 fails to give resistance. (A–C) Sensitivity to the PfDHODH inhibitors Genz-669178, IDI-6273, and GSK-3 are unchanged after 40 generations of dual selection (n.s., not significant). (D–F) Sensitivity to the unrelated control compounds chloroquine, cycloguanil, is unchanged (n.s., not significant). EC50 values were calculated using a whole-cell SYBR Green (28) assay. Error bars indicate the SD of three biological replicates, each with triplicate measurements. Significance relative to Dd2 EC50 was determined by one-way ANOVA with Tukey’s multiple comparison post test; n =3.
The two examples described have shown that resistant parasites are hypersensitive to other agents. Sequential treatment with these potentiated agents resulted in an evolutionary loop back to wild-type sensitivity to the initial treatment. In the example of CQ, this phenotypic reversion occurred through a unique mutation in the PfCRT channel. In the example of PfDHODH inhibitors, the phenotypic reversion occurred through a mutation restoring the wild-type protein sequence of PfDHODH. Simultaneous rather than sequential treatment with both the wild-type and mutant-selective PfDHODH inhibitors failed to give parasites resistant to either compound. We believe that evolutionary fitness constraints allow few pathways to resistance, and these pathways can be anticipated and preemptively blocked. It would be interesting to extend this work to suppressive combinations, as they have been demonstrated to prevent the emergence and spread of resistant bacteria (23) and could do the same in malaria. Another possible direction is to combine wild-type and mutant-selective inhibitors into a single bifunctional molecule, as in the natural product DNA gyrase inhibitor simocyclinone D8 (24). The combination of well-chosen anti-malarial agents active against sensitive and resistant parasites effectively kills parasites in the short-term and, in the long-term, can help shape evolution away from the development of drug resistance.
Materials and Methods
Resistance Selection.
Approximately 2 × 108 mixed stage parasites were treated at 30 nM IDI-3783 or 480 nM Genz-666136 for 6–8 d in each of three independent flasks to eliminate all parasites visible by microscopy. After this treatment, compound pressure was removed, and the cultures were fed on alternate days with complete compound-free RPMI media. Once healthy parasites reappeared in the culture flasks and parasitemia reached 2–4%, compound exposure was repeated. These steps were executed for 30–60 d until the parasites were growing in the presence of compound at a typical multiplication rate. To compensate for the lysis of red blood cells, 30–40% of parasite culture was replaced with freshly washed cells once a week during the entire selection period. Selected parasites were cloned by limiting dilution in a 96-well plate in the presence of 30 nM IDI-3783 or 500 nM Genz-666136, with an inoculum of 0.2 infected RBCs per well. Parasite clones were detected after 3 wk of growth by thin smear microscopy. Frozen stocks of resistant parasites were prepared using Glycerolyte 57 (Baxter Healthcare).
Genomic DNA Extraction and PCR Resequencing.
Genomic DNA extractions from parasites were performed using Qiagen DNeasy kit (Qiagen) for whole-genome sequencing and mutation analysis of the pfdhodh (PlasmoDB ID: PFF0160c) gene. A 2.2-kb fragment encompassing the complete pfdhodh ORF was PCR amplified from Genz-666136-resistant clones and parental lines. PCR-amplified fragments were subcloned and fully sequenced from both strands using T7, M13-reverse and pfdhodh- specific primers.
Whole-Genome Sequencing and Analysis.
Genomic DNA was sheared and made into a 200-bp fragment Illumina sequencing library and sequenced with paired-end reads on an Illumina GAIIx machine. The sequenced reads were aligned against the P. falciparum 3D7 reference from PlasmoDB (version 7.1*) (25) using BWA version 0.5.7 (20). Duplicate reads were marked using the Picard MarkDuplicates tool (http://picard.sourceforge.net/). The consensus bases were called using the Genome Analysis Toolkit's (GATK) Unified Genotyper (version 1.0.5974) (26) and the SAMtools (version 0.1.16) (27) mpileup command. Only bases that were called as homozygous for the reference or the alternate allele with a genotype quality of at least 30 were considered. The GATK Unified Genotyper called 3,685 SNPs in the Genz-666136-resistant strain relative to stock (2,108 intergenic, 248 intronic, 423 synonymous coding, and 906 nonsynonymous coding). By contrast, only 47 SNPs passed all of the quality filters when using the SAMtools genotyper (25 intergenic, 2 intronic, 3 synonymous, 17 nonsynonymous). Of these, only eight overlapped with the GATK set (2 intergenic and 6 nonsynonymous). The nonsynonymous mutations fell in four genes: (i) F227I in PFF0160c (the dhodh gene), (ii) I352T and E355D in PF07_0111, a highly polymorphic gene of unknown function, (iii) adjacent mutations in the PfEMP PF08_0106, and (iv) I1548M in PFI1280c, a putative protein kinase that is also highly polymorphic.
In Vitro Drug Sensitivity and EC50 Determinations.
Drug susceptibility was measured using the SYBR Green method (28). Twelve-point curves based on twofold dilutions of the test compound were carried out in triplicate each day and replicated on three different days. EC50 values were calculated using a nonlinear regression curve fit in Prism 5.0 for Mac (GraphPad Software, Inc.).
Isobologram experiments were performed in similar fashion using the modified fixed-ratio methodology (29). Briefly, IDI-3783 and CQ were mixed at multiple fixed volumetric ratios (10:0, 8:2, 6:4, 5:5, 4:6, 2:8, and 0:10) and then serially diluted in 12-point twofold dilutions and dispensed in triplicate to 384-well assay plates and replicated on two different days. Genz-669178 was tested in combination with IDI-6273 at multiple fixed volumetric ratios (15:0, 12:3, 9:6, 6:9, 3:12, and 0:15) and then serially diluted in 12-point twofold dilutions and dispensed in triplicate to 384-well assay plates and replicated on three different days. EC50 values were calculated as above, and fractional inhibitory concentrations (FICs) were calculated for each drug combination as described (29). Synergy was defined as an FIC < 1.0, additivity as FIC = 1.0, and antagonism as FIC > 1.0.
Synthesis of DSM74.
DSM74 was prepared following the literature procedure (21) and was recrystallized from ethanol. 1H NMR spectra matched that reported (20), and HPLC analysis indicated > 95% purity.
Synthesis of Genz-669178.
Genz-669178 was prepared following the literature procedure with 1H NMR spectra that matched those reported (19) and HPLC analysis indicated >95% purity.
Statistical Analyses.
EC50 data are shown as mean ± SD and were analyzed by ANOVA with multiple comparison post hoc tests (Prism 5; GraphPad Software, Inc.) as noted in the figure legends. Differences were considered significant for P < 0.05.
Supplementary Material
Acknowledgments
We thank Tim Lewis of the Broad Novel Therapeutics Platform for his synthesis of DSM74. We thank Amar bir Singh Sidhu for the selection of the PfDHODH E182D mutant P. falciparum. We thank Margaret Phillips of the University of Texas at Southwestern for valuable discussions on PfDHODH as an antimalarial drug target. Funding for this work was provided by grants from the Medicines for Malaria Venture, the Bill and Melinda Gates Foundation, the Broad Institute Scientific Planning and Allocation of Resources Committee program, the Genzyme Humanitarian Assistance for Neglected Disease Program, and National Institutes of Health Grant R01 AI093716-01A1 (to R.C.W.).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1320886110/-/DCSupplemental.
References
- 1.World Health Organization . World Malaria Report. Geneva: World Health Organization; 2011. [Google Scholar]
- 2.Dondorp AM, et al. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med. 2009;361(5):455–467. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Costanzo MS, Hartl DL. The evolutionary landscape of antifolate resistance in Plasmodium falciparum. J Genet. 2011;90(2):187–190. doi: 10.1007/s12041-011-0072-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nzila A, Mwai L. In vitro selection of Plasmodium falciparum drug-resistant parasite lines. J Antimicrob Chemother. 2010;65(3):390–398. doi: 10.1093/jac/dkp449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sarafianos SG, Das K, Hughes SH, Arnold E. Taking aim at a moving target: Designing drugs to inhibit drug-resistant HIV-1 reverse transcriptases. Curr Opin Struct Biol. 2004;14(6):716–730. doi: 10.1016/j.sbi.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 6.Shah NP, et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002;2(2):117–125. doi: 10.1016/s1535-6108(02)00096-x. [DOI] [PubMed] [Google Scholar]
- 7.Quintás-Cardama A, Kantarjian H, Cortes J. Flying under the radar: The new wave of BCR-ABL inhibitors. Nat Rev Drug Discov. 2007;6(10):834–848. doi: 10.1038/nrd2324. [DOI] [PubMed] [Google Scholar]
- 8.Wootton JC, et al. Genetic diversity and chloroquine selective sweeps in Plasmodium falciparum. Nature. 2002;418(6895):320–323. doi: 10.1038/nature00813. [DOI] [PubMed] [Google Scholar]
- 9.Laufer MK, et al. Return of chloroquine antimalarial efficacy in Malawi. N Engl J Med. 2006;355(19):1959–1966. doi: 10.1056/NEJMoa062032. [DOI] [PubMed] [Google Scholar]
- 10.Martin SK, Oduola AM, Milhous WK. Reversal of chloroquine resistance in Plasmodium falciparum by verapamil. Science. 1987;235(4791):899–901. doi: 10.1126/science.3544220. [DOI] [PubMed] [Google Scholar]
- 11.Martiney JA, Cerami A, Slater AF. Verapamil reversal of chloroquine resistance in the malaria parasite Plasmodium falciparum is specific for resistant parasites and independent of the weak base effect. J Biol Chem. 1995;270(38):22393–22398. doi: 10.1074/jbc.270.38.22393. [DOI] [PubMed] [Google Scholar]
- 12.Martin RE, et al. Chloroquine transport via the malaria parasite’s chloroquine resistance transporter. Science. 2009;325(5948):1680–1682. doi: 10.1126/science.1175667. [DOI] [PubMed] [Google Scholar]
- 13.Ye ZG, Van Dyke K. Reversal of chloroquine resistance in falciparum malaria independent of calcium channels. Biochem Biophys Res Commun. 1988;155(1):476–481. doi: 10.1016/s0006-291x(88)81111-2. [DOI] [PubMed] [Google Scholar]
- 14.Bruce-Chwatt LJ. Chemoprophylaxis of malaria in Africa: The spent “magic bullet”. Br Med J (Clin Res Ed) 1982;285(6343):674–676. doi: 10.1136/bmj.285.6343.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hurt DE, Widom J, Clardy J. Structure of Plasmodium falciparum dihydroorotate dehydrogenase with a bound inhibitor. Acta Crystallogr D Biol Crystallogr. 2006;62(Pt 3):312–323. doi: 10.1107/S0907444905042642. [DOI] [PubMed] [Google Scholar]
- 16.Booker ML, et al. Novel inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase with anti-malarial activity in the mouse model. J Biol Chem. 2010;285(43):33054–33064. doi: 10.1074/jbc.M110.162081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Phillips MA, Rathod PK. Plasmodium dihydroorotate dehydrogenase: A promising target for novel anti-malarial chemotherapy. Infect Disord Drug Targets. 2010;10(3):226–239. doi: 10.2174/187152610791163336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fidock DA, et al. Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol Cell. 2000;6(4):861–871. doi: 10.1016/s1097-2765(05)00077-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Skerlj RT, et al. Optimization of potent inhibitors of P. falciparum dihydroorotate dehydrogenase for the treatment of malaria. ACS Medicinal Chem Lett. 2011;2(9):708–713. doi: 10.1021/ml200143c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gujjar R, et al. Identification of a metabolically stable triazolopyrimidine-based dihydroorotate dehydrogenase inhibitor with antimalarial activity in mice. J Med Chem. 2009;52(7):1864–1872. doi: 10.1021/jm801343r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Phillips MA, et al. Triazolopyrimidine-based dihydroorotate dehydrogenase inhibitors with potent and selective activity against the malaria parasite Plasmodium falciparum. J Med Chem. 2008;51(12):3649–3653. doi: 10.1021/jm8001026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Malmquist NA, Gujjar R, Rathod PK, Phillips MA. Analysis of flavin oxidation and electron-transfer inhibition in Plasmodium falciparum dihydroorotate dehydrogenase. Biochemistry. 2008;47(8):2466–2475. doi: 10.1021/bi702218c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bollenbach T, Quan S, Chait R, Kishony R. Nonoptimal microbial response to antibiotics underlies suppressive drug interactions. Cell. 2009;139(4):707–718. doi: 10.1016/j.cell.2009.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Edwards MJ, et al. A crystal structure of the bifunctional antibiotic simocyclinone D8, bound to DNA gyrase. Science. 2009;326(5958):1415–1418. doi: 10.1126/science.1179123. [DOI] [PubMed] [Google Scholar]
- 25.Aurrecoechea C, et al. PlasmoDB: A functional genomic database for malaria parasites. Nucleic Acids Res. 2009;37(Database issue):D539–D543. doi: 10.1093/nar/gkn814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.DePristo MA, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H, et al. 1000 Genome Project Data Processing Subgroup The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson JD, et al. Assessment and continued validation of the malaria SYBR green I-based fluorescence assay for use in malaria drug screening. Antimicrob Agents Chemother. 2007;51(6):1926–1933. doi: 10.1128/AAC.01607-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fivelman QL, Adagu IS, Warhurst DC. Modified fixed-ratio isobologram method for studying in vitro interactions between atovaquone and proguanil or dihydroartemisinin against drug-resistant strains of Plasmodium falciparum. Antimicrob Agents Chemother. 2004;48(11):4097–4102. doi: 10.1128/AAC.48.11.4097-4102.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McNicholas S, Potterton E, Wilson KS, Noble ME. Presenting your structures: The CCP4mg molecular-graphics software. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 4):386–394. doi: 10.1107/S0907444911007281. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.