

Abstract
Nearly all colorectal cancers (CRCs) and varied subsets of other cancers have somatic mutations leading to β-catenin stabilization and increased β-catenin/TCF transcriptional activity. Inhibition of stabilized β-catenin in CRC cell lines arrests their growth and highlights the potential of this mechanism for novel cancer therapeutics. We have pursued efforts to develop small molecules that inhibit β-catenin/TCF transcriptional activity. We used xanthothricin, a known β-catenin/TCF antagonist of microbial origin, as a lead compound to synthesize related analogues with drug-like features such as low molecular weight and good metabolic stability. We studied a panel of six candidate Wnt/β-catenin/Tcf-regulated genes and found that two of them (Axin2, Lgr5) were reproducibly activated (9–10 fold) in rat intestinal epithelial cells (IEC-6) following β-catenin stabilization by Wnt-3a ligand treatment. Two previously reported β-catenin/TCF antagonists (calphostin C, xanthothricin) and XAV939 (tankyrase antagonist) inhibited Wnt-activated genes in a dose-dependent fashion. We found that four of our compounds also potently inhibited Wnt-mediated activation in the panel of target genes. We investigated the mechanism of action for one of these (8c) and demonstrated these novel small molecules inhibit β-catenin transcriptional activity by degrading β-catenin via a proteasome-dependent, but GSK3β-, APC-, AXIN2- and βTrCP-independent, pathway. The data indicate the compounds act at the level of β-catenin to inhibit Wnt/β-catenin/TCF function and highlight a robust strategy for assessing the activity of β-catenin/TCF antagonists.
Keywords: Wnt signaling, β-catenin, T-cell factor (TCF), Small molecule antagonists
The Wnt/β-catenin signaling pathway is a key regulator of cell proliferation, differentiation, migration and apoptosis.1 Activation of the canonical Wnt-signaling pathway by Wnt-ligands leads to an increase in the “free” pool of β-catenin and β-catenin/T-cell factor (TCF) transcriptional activity.2 In the absence of Wnt-ligands, β-catenin forms complexes with other proteins in the cytoplasm including the β-catenin destruction complex consisting of APC (adenomatous polyposis coli), GSK3β (glycogen synthase kinase 3 β), CK1 α (casein kinase 1 α) and AXIN2.3 Upon phosphorylation by GSK3β and CK1 α, β-catenin is mainly ubiquinated by βTrCP (β transducin repeat containing protein) and to a minor degree in an APC-dependent fashion by SIAH (seven in absentia homolog) leading to proteasome dependent degradation of β-catenin.4, 5 Nearly all colorectal cancers (CRCs) and varying subsets of other cancer types have somatic gain-of-function mutations in β-catenin or loss-of-function mutations in key antagonists of β-catenin, namely the APC and AXIN tumor suppressor proteins.6 These defects lead to constitutive β-catenin stabilization resulting in altered transcription of downstream β-catenin/T cell factor (TCF)-regulated target genes including Cyclin D1, Lgr5 and Axin2.7
Inhibition of β-catenin in colon cancer cell lines arrests their growth and highlights the potential of this mechanism for novel cancer therapeutics.8, 9 The search for inhibitors of Wnt/β-catenin signaling has led to the identification of antagonists acting at different levels of the pathway.10, 11 These have derived from both high throughput screening and synthetic campaigns. Calphostin C (1; PKF115-584, Figure 1) and xanthothricin (3; toxoflavin, PKF118-310, Figure 1) have been reported to disrupt β-catenin/TCF4 binding leading to an inhibition of β-catenin transcriptional activity.12 Calphostin C can also inhibit β-catenin signaling through PKC (protein kinase C) in a GSK3β-dependent fashion.13, 14 In contrast XAV939 (2, Figure 1) antagonizes β-catenin levels in a destruction complex dependent fashion by stabilizing AXIN protein levels.15, 16
Figure 1.

Small molecule inhibitors of β-catenin/TCF transcriptional activity and pyrimido[5,4-e][1,2,4]triazine-5,7- dione chemotypes
Antagonists, such as xanthothricin, that act at the level of or downstream of stabilized β-catenin, and thus circumvent the activity of the frequently mutated destruction complex in cancer cells, hold the promise of broader efficacy. Xanthotricin, which was originally isolated from Pseudomonas cocovenenans, was first synthesized in 1961.17 Since then, a number of analogues have been prepared in order to explore the potent antibiotic and pharmacological properties represented by this natural product.18 Many of these analogues have the same methyl substituent found at the N1 position of xanthothricin, in large part due to the ease with which a methyl group can be incorporated via the use of methylhydrazine early in the synthesis.19
Lepourcelet et al. reported that xanthothricin potently antagonizes β-catenin’s binding to TCF4 and β-catenin/TCF-dependent transcription (IC50 of 0.3 μM in cells). They further showed that xanthothricin inhibited β-catenin-dependent Wnt signaling in cells and in Xenopus axis duplication assay.12 A limitation with xanthothricin is that the apparent therapeutic window is small between β-catenin/TCF inhibition and general cytotoxic effects.12 Based on its structural simplicity and low molecular weight, we were interested in utilizing xanthothricin (3) as a lead chemotype to probe SAR expansion. The heterocyclic core of 3 is made up of the pyrimido[5,4-e]-1,2,4-triazine-5,7(1H,6H)-dione ring system. In earlier work, we reported on a novel and efficient route to 3-aryl derivatives of 3 that allows for incorporation of aryl and more elaborate alkyl substituents at the N1 position, thus broadening the scope of accessible analogues.20 We also reported on the generation of compounds within the isomeric fervenulin series (4, Figure 1).21
Here, we report on the utilization of this chemistry toward investigating a small series of 3-aryl analogues of 3 and 4 and from these we have identified four novel Wnt/β-catenin pathway inhibitors within the xanthothricin series. We show that these analogues inhibit Wnt/β-catenin/TCF-regulated genes (Axin2, Lgr5) in rat intestinal epithelial cells (IEC-6) following β-catenin stabilization by Wnt-3a ligand treatment. One of these compounds (compound 8c, Table 1) preferentially affects the growth of colon cancer cell lines (DLD-1 and SW480) with dysregulated β-catenin signaling. We demonstrate that compound 8c inhibits β-catenin transcriptional activity by reducing stabilized β-catenin via a GSK3β-, APC-, AXIN2- and βTrCP- independent but proteasome-dependent pathway. This strongly suggests compound 8c acts at the level of β-catenin to inhibit Wnt/β-catenin/TCF function.
Table 1.
Summary of IC50 values determined by Lgr5 and Axin2 gene expression
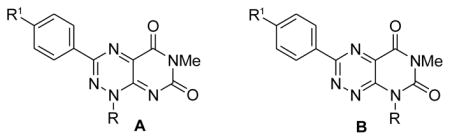
| No. | Core | R | R1 |
Lgr5 IC50 (μM) (Mean +/− SEM) |
Axin2 IC50 (μM) (Mean +/− SEM) |
|---|---|---|---|---|---|
| 1 | calphostin C | 0.39 +/− 0.19 | 0.55 +/− 0.18 | ||
| 2 | XAV939 | 2.9 | 0.64 | ||
| 3 | xanthothricin | 0.29 +/− 0.02 | 0.33 +/− 0.03 | ||
| 8a | A | Me | OMe | 1.53 +/− 0.37 | 2.67 +/− 0.62 |
| 8b | A | Me | OCH2CH2NEt2 | NI up to 0.7 μM | NI up to 0.7 μM |
| 8c | A | Me | Cl | 1.53 +/− 0.42 | 1.77 +/− 0.28 |
| 10a | A | CH2CH2OH | OMe | NI up to 1.2 μM | NI up to 1.2 μM |
| 10b | A | Ph | OMe | 2.1 +/− 0.32 | 2.67 +/− 0.33 |
| 11c | A | Ph | OCH2CH2NEt2 | 0.26 +/− 0.11 | 0.30 +/− 0.11 |
| 20a | B | Me | OCH2CH2NEt2 | NI up to 69 μM | NI up to 69 μM |
| 20b | B | Ph | OCH2CH2NEt2 | 164.7 | PI up to 179 μM |
NI – No inhibition
PI – Partial inhibition
The synthesis of N1-substituted pyrimidotriazinedione derivatives within the xanthothricin series was straightforward and followed previously established procedures.22–24 As shown in Scheme 1, condensation of readily available 3-methyl-6-(1-methylhydrazinyl)pyrimidine-2,4(1H,3H)-dione (5)17 with requisite 4-substituted benzaldehydes led to hydrazone adducts 6a–c in good yields. Each hydrazone was then cyclized by treatment of NaNO2 in acetic acid to a ~1:1 mixture of 3-substituted-5,7-dioxo-1,5,6,7-tetrahydropyrimido[5,4-e][1,2,4]triazine 4-oxide (7) and pyrimido[5,4-e][1,2,4]triazine-5,7(1H,6H)-dione (8). Each mixture of 7a–c was easily converted to desired product 8a–c, respectively, by mild treatment with dithiothreitol (DTT). Additional target derivatives 10a,b and 11c were derived from 6-chloro-3-methyl-5-nitrouracil (9) by our recently published methodology to make compounds with more elaborate N1 substituents.20 The synthesis of N8-substituted pyrimidotriazinedione derivatives (fervenulin series) was achieved by two reaction pathways and is shown in Scheme 2. Heating compound 8a or the 7a/8a mixture in DMF led to a well precedented N1 demethylation reaction to give 12.25, 26 Selective N8 methylation then was carried out with Cs2CO3 as base to give 13. Ether demethylation with BBr3 provided phenol 14a to which was appended an aqueous solubilizing side chain to afford 20a. A series of reactions starting from 1-phenylpyrimidine-2,4,6-trione (15)24 led to known intermediate 18,27 which was then taken to N8 phenyl congener 20b by reactions similar to those described above. Full experimental procedures for compounds outlined in Schemes 1 and 2 are given in Supplemental Materials. We also developed a simple process to make xanthothricin (3; Scheme S3, Supplemental Materials). While not as efficient overall as the best reported synthesis,28 its brevity makes it preferable when smaller quantities of compound are required.
Scheme 1.

Synthesis of analogues in the xanthothricin series. (i) 4-substituted benzaldehyde, EtOH, reflux (31–87% yield); (ii) NaNO2, aq. AcOH, 0 °C – 25 °C (~80% yield); (iii) DTT, EtOH (47 – 87% yield); (iv) see reference 20; (v) for 10b, BBr3, DCM (63% yield); (vi) BrCH2CH2Br, Cs2CO3, DMF (47% yield); (vii) NHMe2, ACN, 80 °C (33% yield). See Supplemental Materials for experimental details.
Scheme 2.

Synthesis of analogues in the fervenulin series. (i) DMF, 90 °C (65 – 73% yield); (ii) for 12 and 16, dimethyl sulfate, Cs2CO3, acetone, rt - 50 °C (84 – 97% yield); (iii) for 14a, BBr3, DCM (65% yield); (iv) POCl3 (48% yield); (v) methylhydrazine, EtOH, reflux (88% yield); (vi) 4-OH-benzaldehyde, EtOH, reflux (64% yield); (vii) NaNO2, aq. AcOH, 0 °C – 25 °C, then DTT, EtOH (44% yield); (viii) ClCH2CH2NEt2. HCl, Cs2CO3, acetone, rt - 50 °C (44 – 63% yield). See Supplemental Materials for experimental details.
All compounds reported in this paper were rigorously purified, and their structural assignments are supported by 1H NMR spectroscopy and mass spectrometry.
Identification of small molecule inhibitors of canonical Wnt – signaling
To establish the induction of Wnt/β-catenin/Tcf-regulated target genes in rat intestinal epithelial cells (IEC-6 cell line), we treated IEC-6 cells for 12 h with 50 ng/mL recombinant mouse Wnt-3a. We then determined the expression levels of the selected Wnt/β-catenin/Tcf-regulated target genes relative to U6 gene expression levels by quantitative PCR using gene-specific primers. The specific candidate Wnt/β-catenin/Tcf-regulated genes studied were Axin2, Lgr5, Bmp4, Nkd1, Edn1, and Irs1 (Table S1, Supplemental Materials). These genes have previously been highlighted as candidate targets of the canonical (β-catenin) Wnt signaling pathway29–34, and we found evidence that the expression of all six genes was induced in IEC-6 cells by recombinant Wnt3a.
We chose the two most potently Wnt-3a-induced target genes, namely Axin2 (10.9-fold +/−0.9 induction) and Lgr5 (9.5-fold +/−1 induction), to determine how previously described Wnt/β-catenin/TCF pathway antagonists as well as our newly designed small molecules affected target gene expression. We treated IEC-6 cells with small molecules at various concentrations for 6 h followed by the addition and incubation of the cells with 50 ng/mL recombinant mouse Wnt-3a for 12 h. The base-line relative levels of Lgr5 (Figure 2B) and Axin2 (Figure 2C) expression, relative to U6 expression after Wnt-3a stimulation was set to 100% and the relative target gene expression for each compound at various concentrations was determined. As expected, we found that previously identified antagonists of β-catenin signaling (calphostin C, XAV939 and xanthothricin) decreased the expression of Lgr5 and Axin2 (Figure 2B and 2C and Table 1). We also found that four of our synthesized pyrimidotriazinedione analogues (8a, 8c, 10b, 11c) inhibited the expression of Lgr5 and Axin2 with varying potencies (Table 1). Within our small series of synthesized compounds, it was clear that inhibition of canonical Wnt-dependent activation of downstream transcriptional targets was associated almost exclusively with the N1-substutited core (xanthothricin series). Addition of solubilizing moieties at R (e.g., 10a) and R1 (e.g., 8b) abolished activity. For the two isomeric compounds (20a, 20b) having N8 substituents (fervenulin series), activity was poor or absent. To determine if compounds of the xanthothricin series have good biopharmaceutical properties such as long biological half-life, we carried out a metabolic stability study. We incubated compound 11c, the most potent inhibitor of Lgr5 and Axin2 gene expression in IEC-6 cells, at 1 μM at 37°C with mouse liver microsomes. Under these conditions compound 11c has a half-life of 34.7 min (Table S2, Supplemental Materials) consistent with good metabolic stability.
Figure 2.

Wnt-3a activation of presumptive Wnt/β-catenin/TCF-regulated target genes and inhibition of Wnt-3a activated target genes Axin2 and Lgr5 in rat intestinal epithelial cells (IEC-6) by small molecules. See Supplemental Materials for experimental details.
A: IEC-6 cells were treated for 12 h with 50 ng/mL recombinant mouse Wnt-3a. The expression levels of the selected Wnt/β-catenin/TCF-regulated target genes (Axin2, Lgr5, Bmp4, Nkd1, Edn1, Irs1) relative to U6 gene expression levels were determined by quantitative PCR using gene specific primers. Each bar represents the mean with standard error mean (SEM) from three independent experiments. B and C: Inhibition of Wnt-3a activated target genes Lgr5 and Axin2 by calphostin C, XAV939, xanthothricin and xanthothricin analogues. The base-line relative levels of Lgr5 (B) and Axin2 expression (C) (relative to U6 expression) after Wnt-3a stimulation was set to 100%. The relative target gene expression for each compound at various concentrations was determined (log M −6 corresponds to 1 μM).
Compound specificity and mechanism of action
We hypothesized that our synthesized compounds inhibited β-catenin/Tcf transcriptional activity by reducing β-catenin protein levels. To address this notion, we therefore treated DLD-1 CRC cells for 20 h with the compounds and analyzed β-catenin protein levels by Western blot. We found that compound 8a and compound 8c reduced total β-catenin protein levels, as well as that of Cyclin D1, which is expressed from a presumptive β-catenin/TCF-regulated gene (Figure 3A).
Figure 3.

Compound specificity assays. See Supplemental Materials for experimental details.
A: Xanthothricin analogues affect total β-catenin protein levels and cyclin D1 protein levels in DLD-1 cells. B, D: DLD-1 and RKO colon cancer cells were seeded in 10% serum containing xanthothricin or compound 8c and surviving colonies present at two weeks after plating were visualized (C, E, respectively). Colony counts from two independent experiments at each concentration done in triplicate are shown (n.s. = not statistically significant; * p < 0.05; *** p < 0.0001).
To determine if these newly identified small molecules preferentially affect the proliferation and survival of CRC cells with β-catenin/TCF defects, we performed colony formation assays with selected colon cancer-derived cell lines. Depletion of β-catenin with shRNA inhibits colony formation of DLD-1 but not RKO cells.15 The DLD-1 cell line, which displays dysregulated β-catenin/TCF transcriptional activity due to bi-allelic APC mutations and the RKO cell line, which has no evidence of β-catenin dysregulation35 were seeded in 10% serum containing xanthothricin (3) or compound 8c, and surviving colonies present at two weeks after plating were visualized (Figures 3B and 3D). Xanthothricin inhibited the growth of colonies in both the RKO and DLD-1 cell lines (Figure 3C), with greater effects on the RKO cell line (i.e., a line which does not display β-catenin dysregulation). In contrast, compound 8c preferentially inhibited the growth of colonies in DLD-1 cells relative to RKO cells (Figure 3E). Hence, the data are consistent with the notion that compound 8c may interfere with Wnt/β-catenin/TCF function in CRC cells.
We therefore sought to determine the effects of compound 8c on the canonical Wnt/β-catenin signaling pathway in DLD-1 cells and assessed the effects of increasing doses of compound 8c on the levels of the “free” signaling-competent pool of β-catenin in DLD-1 cells as well as the total pool of β-catenin. We found that free β-catenin protein levels, and to a lesser extend total β-catenin protein levels, were diminished by compound 8c (Figure 4A). Similarly, in SW480 cells, which have dysregulated β-catenin/TCF transcriptional activity due to APC defects, compound 8c reduced free β-catenin levels and to a lesser extent total β-catenin levels (Figure 4B). To determine if this mechanism is due to a stabilization of AXIN2 protein, akin to how the XAV939 compound acts, we studied AXIN2 protein levels and found a dose-dependent decrease of AXIN2 in response to compound 8c (Figure 4B), suggesting that compound 8c acts via a mechanism that does not require AXIN2 protein stabilization. We then sought to determine if compound 8c exerted the observed effects on β-catenin levels via mechanisms independent of the known activity of GSK3β in regulating the free pool of β-catenin. To do so, we treated HEK293T cells with 5 uM SB216763, a previously described GSK3β antagonist36, and various concentrations of compound 8c. Interestingly, in HEK293T cells, we found that in the presence of GSK3β antagonist and elevated levels of the free pool of β-catenin, compound 8c was able to reduce the levels of free β-catenin and did not affect total β-catenin protein levels (Figure 4C).
Figure 4.

Effect of compound 8c on β-catenin levels. See Supplemental Materials for experimental details.
A–C: Effects of compound 8c on free and total β-catenin protein levels in DLD-1 cells (A), SW480 cells (B) and HEK293T cells (C–E). C treated with 5 μM of the GSK3β antagonist SB216763. Compound 8c degrades β-catenin in a proteasome dependent fashion (D). Compound 8c degrades β-catenin in a βTrCP independent fashion (E).
We next determined if the reduction of free β-catenin levels by compound 8c requires proteasome activity. We treated HEK293T cells simultaneously with compound 8c, the GSK3β-inhibitor SB216762 and the proteasome inhibitor MG-132.37 As previously shown compound 8c reduced free β-catenin protein levels in the presence of SB216763, but this effect was abrogated in the presence of the proteasome inhibitor MG-132 (Figure 4D). This finding demonstrates that compound 8c promotes down-regulation of β-catenin in a proteasome-dependent fashion. The major ubiquitin ligase required for β-catenin ubiquitination in cells are the beta-transducin repeat-containing proteins 1 and 2 (βTrCP1/2). To address if compound 8c requires βTrCP1/2 function we transfected HEK293T cells with dominant negative βTrCP (dnβTrCP) and treated the cells with compound 8c. As previously shown, compound 8c reduced free β-catenin levels in the presence of SB216763. When β-catenin was stabilized by SB216763 treatment and dnβTrCP transfection, compound 8c was still able to reduce the free pool of β-catenin (Figure 4E). This suggests that compound 8c can promote degradation of β-catenin in a βTrCP-independent fashion. Because the only other defined, albeit minor, pathway for regulating the free pool of β-catenin, i.e., Siah1/2-dependent regulation of β-catenin, also has been shown to require a SIAH-APC protein complex, we did not pursue studies to exclude a role for compound 8c in regulating free β-catenin levels via changes in SIAH1/2 levels or function since the effect of compound 8c on β-catenin was independent of APC. In summary, the findings demonstrate that compound 8c is able to promote down-regulation of the free pool of β-catenin in an APC-, GSK3β-, and βTrCP-independent fashion, but in a proteasome-dependent manner.
We used a cell-based screening approach with rat intestinal epithelial cells (IEC-6) to identify novel small molecule antagonists of canonical (β-catenin-dependent) Wnt signaling. We selected one compound (8c) that appeared to preferentially interfere with the oncogenic growth potential of colon cancer cells carrying endogenous gene defects in Wnt/β-catenin/TCF signaling relative to a colon cancer cell line known to lack demonstrable defects in canonical Wnt signaling. We demonstrated that compound 8c inhibits β-catenin transcriptional activity by reducing the stabilized free pool of β-catenin via a GSK3β-, APC-, AXIN2 and βTrCP-independent but proteasome-dependent mechanism.
We did not pursue a Tcf/luciferase-reporter cell-based assay, because our initial studies suggested that some of the xanthothricin analogues we synthesized can non-specifically inhibit firefly luciferase reporter genes expressed under control of other constitutive promoter/enhancer elements (JZ and ERF, unpublished results).38
Our studies and results here on the effects of some previously published compounds on cellular genes regulated by the Wnt/β-catenin/TCF pathway are consistent with previously published findings in the literature. For instance, we found that calphostin C, XAV939 and xanthothricin all decreased the expression of Lgr5 and Axin2 in IEC-6 cells (Figure 2B, Figure 2C and Table 1), with IC50 values similar to those previously determined in TCF-reporter gene assays.12, 15
Under conditions of UV-light and oxygen, xanthothricin was previously reported to generate reactive oxygen species.39 Nucleoredoxin (NRX), a thioredoxin family protein, interacts with Dishevelled (Dvl). Increased NRX protein levels suppress Wnt/ β-catenin signaling.40 We can likely exclude that effects of 8c on β-catenin degradation could occur through generation of reactive oxygen species. The effects of 8c on stabilized β-catenin that we observed were independent of APC and GSK3β, as both exert their function downstream of Dvl. Furthermore we incubated all compounds with cells in the dark.
Considerable efforts have been made to identify drugs and drug candidates that antagonize Wnt/ β-catenin signaling.41 Nevertheless, very few antagonists that act in a destruction complex-independent fashion to reduce stabilized β-catenin protein levels in cells are known with only one such class of compounds, to the best of our knowledge, described in the literature. A recent cell-based screen for Wnt pathway inhibitors identified a series of acyl-hydrazones, such as the FDA-approved drug ciclopiroxoalmine, that appear to promote degradation of stabilized β-catenin in a destruction complex-independent fashion.42 Previous studies reported that xanthothricin inhibits β-catenin/TCF transcriptional activity by disrupting β-catenin/TCF4 binding.12 In contrast to the reported effects of xanthothricin, we show here that compound 8c reduced stabilized β-catenin protein levels and we did not find evidence that it disrupted β-catenin/TCF4 binding (JZ and ERF, unpublished results). These findings suggest perhaps that through modification of the C3 position of xanthothricin, compound 8c may have altered the presumptive β-catenin-TCF4 complex disrupting activity in some fashion to promote the degradation of stabilized β-catenin. Because 8c can promote the degradation of stabilized β-catenin in a destruction complex-independent fashion requiring proteasomal activity independent of the major ubiqutin ligase βTrCP, how then does it promote β-catenin degradation? One possibility is that compound 8c increases the ubiquitination of β-catenin through activation of a still-to-be defined and little used ubiquitin ligation pathway that specifically regulates β-catenin ubiquitination and proteasomal degradation. Another possibility is that compound 8c may increase proteasomal degradation of β-catenin without ubiquitination. For example, dicoumarol-induced p53 degradation by the proteasome occurs independently of ubiquitination.43
In conclusion, our work highlights a robust strategy for assessing the activity of β-catenin/Tcf antagonists. We used a cell-based screening approach with rat intestinal epithelial cells (IEC-6) to identify several novel small molecule antagonists of canonical Wnt signaling. We profiled one compound (8c) that appears to preferentially interfere with the oncogenic growth potential of colon cancer cells carrying endogenous gene defects in Wnt/β-catenin/TCF signaling relative to a colon cancer cell line known to lack demonstrable defects in canonical Wnt signaling. We have also demonstrated that 8c inhibits β-catenin transcriptional activity by reducing the stabilized free pool of β-catenin via a GSK3β- and APC-independent pathway. The findings strongly suggest that 8c acts at the level of β-catenin to inhibit Wnt/β-catenin/TCF function and highlights the potential of carrying out screens for other small molecules likely to have inhibitory effects on downstream Wnt signaling factors and mechanisms in cancer cells. Further work will be required for a more detailed understanding of the means by which compound 8c and related compounds promote β-catenin degradation. Insights into the means by which these promote β-catenin degradation may have important consequences for the targeting of cancer cells with mutational defects in β-catenin or in key destruction components, such as APC and Axin.
Supplementary Material
Table S1. Oligos nucleotide sequences
Table S2. Compound 11c Metabolic Stability in Mouse Liver Microsomes
Acknowledgments
Funding: E.R.F. and H.D.H.S were supported by a Cancer Center Innovation Grant. A.J.T. gratefully acknowledges financial support of the NIGMS (grant number GM007767), the American Chemical Society Division of Medicinal Chemistry, Sheila B. Cresswell, and Fred and Dee Lyons Graduate Fellowships.
References
- 1.Clevers H. Cell (Cambridge, MA, U S) 2006;127:469. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 2.MacDonald BT, Tamai K, He X. Dev Cell. 2009;17:9. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kimelman D, Xu W. Oncogene. 2006;25:7482. doi: 10.1038/sj.onc.1210055. [DOI] [PubMed] [Google Scholar]
- 4.Matsuzawa SI, Reed JC. Mol Cell. 2001;7:915. doi: 10.1016/s1097-2765(01)00242-8. [DOI] [PubMed] [Google Scholar]
- 5.Winston JT, Strack P, Beer-Romero P, Chu CY, Elledge SJ, Harper JW. Genes Dev. 1999;13:270. doi: 10.1101/gad.13.3.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Polakis P. Curr Opin Genet Dev. 2007;17:45. doi: 10.1016/j.gde.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 7.Fearon ER. Annu Rev Pathol: Mech Dis. 2011;6:479. doi: 10.1146/annurev-pathol-011110-130235. [DOI] [PubMed] [Google Scholar]
- 8.Roh H, Green DW, Boswell CB, Pippin JA, Drebin JA. Cancer Res. 2001;61:6563. [PubMed] [Google Scholar]
- 9.van de Wetering M, Sancho E, Verweij C, de Lau W, Oving I, Hurlstone A, van der Horn K, Batlle E, Coudreuse D, Haramis A-P, Tjon-Pon-Fong M, Moerer P, van den Born M, Soete G, Pals S, Eilers M, Medema R, Clevers H. Cell (Cambridge, MA, U S) 2002;111:241. doi: 10.1016/s0092-8674(02)01014-0. [DOI] [PubMed] [Google Scholar]
- 10.Barker N, Clevers H. Nat Rev Drug Discovery. 2006;5:997. doi: 10.1038/nrd2154. [DOI] [PubMed] [Google Scholar]
- 11.Ewan K, Pajak B, Stubbs M, Todd H, Barbeau O, Quevedo C, Botfield H, Young R, Ruddle R, Samuel L, Battersby A, Raynaud F, Allen N, Wilson S, Latinkic B, Workman P, McDonald E, Blagg J, Aherne W, Dale T. Cancer Res. 2010;70:5963. doi: 10.1158/0008-5472.CAN-10-1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lepourcelet M, Chen YNP, France DS, Wang H, Crews P, Petersen F, Bruseo C, Wood AW, Shivdasani RA. Cancer Cell. 2004;5:91. doi: 10.1016/s1535-6108(03)00334-9. [DOI] [PubMed] [Google Scholar]
- 13.Cook D, Fry MJ, Hughes K, Sumathipala R, Woodgett JR, Dale TC. EMBO J. 1996;15:4526. [PMC free article] [PubMed] [Google Scholar]
- 14.Kobayashi E, Nakano H, Morimoto M, Tamaoki T. Biochem Biophys Res Commun. 1989;159:548. doi: 10.1016/0006-291x(89)90028-4. [DOI] [PubMed] [Google Scholar]
- 15.Huang SMA, Mishina YM, Liu S, Cheung A, Stegmeier F, Michaud GA, Charlat O, Wiellette E, Zhang Y, Wiessner S, Hild M, Shi X, Wilson CJ, Mickanin C, Myer V, Fazal A, Tomlinson R, Serluca F, Shao W, Cheng H, Shultz M, Rau C, Schirle M, Schlegl J, Ghidelli S, Fawell S, Lu C, Curtis D, Kirschner MW, Lengauer C, Finan PM, Tallarico JA, Bouwmeester T, Porter JA, Bauer A, Cong F. Nature (London, U K) 2009;461:614. doi: 10.1038/nature08356. [DOI] [PubMed] [Google Scholar]
- 16.Karlberg T, Markova N, Johansson I, Hammarstrom M, Schutz P, Weigelt J, Schuler HJ. Med Chem. 2010;53:5352. doi: 10.1021/jm100249w. [DOI] [PubMed] [Google Scholar]
- 17.Daves GD, Jr, Robins RK, Cheng CC. J Am Chem Soc. 1962;84:1724. [Google Scholar]
- 18.Levenberg B, Linton SN. J Biol Chem. 1966;241:846. [PubMed] [Google Scholar]
- 19.Nagamatsu T, Yamasaki H, Hirota T, Yamato M, Kido Y, Shibata M, Yoneda F. Chem Pharm Bull. 1993;41:362. doi: 10.1248/cpb.41.362. [DOI] [PubMed] [Google Scholar]
- 20.Turbiak AJ, Showalter HDH. Tetrahedron Letters. 2009;50:1996. doi: 10.1016/j.tetlet.2009.02.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Turbiak AJ, Showalter HDH. Synthesis. 2009:4022. [Google Scholar]
- 22.Lacrampe JFA, Connors RW, Ho CY, Richardson A, Freyne EJE, Buijnsters PJJ, Bakker AC. WO2004007498A2. 2004:71.
- 23.Yoneda F, Nagamatsu T. Chem Pharm Bull. 1975;23:2001. doi: 10.1248/cpb.23.2001. [DOI] [PubMed] [Google Scholar]
- 24.Blehaut H, Bellamy F, Matt C, Giraud S, Charre D. WO2010072807A2. 2010:53.
- 25.Yoneda F, Nagamatsu TJ. Heterocycl Chem. 1974;11:271. [Google Scholar]
- 26.Nagamatsu T, Yamasaki HJ. Chem Soc, Perkin Trans 1. 2001:130. [Google Scholar]
- 27.Senda S, Hirota K. Chem Pharm Bull. 1974;22:1459. doi: 10.1248/cpb.22.1459. [DOI] [PubMed] [Google Scholar]
- 28.Black THJ. Heterocycl Chem. 1987;24:1373. [Google Scholar]
- 29.Barker N, van EJH, Jaks V, Kasper M, Snippert H, Toftgard R, Clevers H. Cold Spring Harbor Symp Quant Biol. 2008;73:351. doi: 10.1101/sqb.2008.72.003. [DOI] [PubMed] [Google Scholar]
- 30.Bommer GT, Feng Y, Iura A, Giordano TJ, Kuick R, Kadikoy H, Sikorski D, Wu R, Cho KR, Fearon ER. J Biol Chem. 2010;285:1928. doi: 10.1074/jbc.M109.060319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim JS, Crooks H, Dracheva T, Nishanian TG, Singh B, Jen J, Waldman T. Cancer Res. 2002;62:2744. [PubMed] [Google Scholar]
- 32.Kim TH, Xiong H, Zhang Z, Ren B. Oncogene. 2005;24:597. doi: 10.1038/sj.onc.1208237. [DOI] [PubMed] [Google Scholar]
- 33.Leung JY, Kolligs FT, Wu R, Zhai Y, Kuick R, Hanash S, Cho KR, Fearon ER. J Biol Chem. 2002;277:21657. doi: 10.1074/jbc.M200139200. [DOI] [PubMed] [Google Scholar]
- 34.Van Raay TJ, Fortino NJ, Miller BW, Ma H, Lau G, Li C, Franklin JL, Attisano L, Solnica-Krezel L, Coffey R. J PLoS One. 2011;6:e18650. doi: 10.1371/journal.pone.0018650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sparks AB, Morin PJ, Vogelstein B, Kinzler KW. Cancer Res. 1998;58:1130. [PubMed] [Google Scholar]
- 36.Coghlan MP, Culbert AA, Cross DAE, Corcoran SL, Yates JW, Pearce NJ, Rausch OL, Murphy GJ, Carter PS, Cox LR, Mills D, Brown MJ, Haigh D, Ward RW, Smith DG, Murray KJ, Reith AD, Holder JC. Chem Biol. 2000;7:793. doi: 10.1016/s1074-5521(00)00025-9. [DOI] [PubMed] [Google Scholar]
- 37.Rock KL, Gramm C, Rothstein L, Clark K, Stein R, Dick L, Hwang D, Goldberg AL. Cell (Cambridge, Mass) 1994;78:761. doi: 10.1016/s0092-8674(94)90462-6. [DOI] [PubMed] [Google Scholar]
- 38.Cong F, Cheung AK, Huang SMA. Annu Rev Pharmacol Toxicol. 2012;52:57. doi: 10.1146/annurev-pharmtox-010611-134639. [DOI] [PubMed] [Google Scholar]
- 39.Koh S, Kim H, Kim J, Goo E, Kim YJ, Choi O, Jwa NS, Ma J, Nagamatsu T, Moon JS, Hwang I. Plant Biotechnol J. 2011;9:348. doi: 10.1111/j.1467-7652.2010.00557.x. [DOI] [PubMed] [Google Scholar]
- 40.Funato Y, Michiue T, Asashima M, Miki H. Nat Cell Biol. 2006;8:501. doi: 10.1038/ncb1405. [DOI] [PubMed] [Google Scholar]
- 41.Takahashi-Yanaga F, Kahn M. Clin Cancer Res. 2010;16:3153. doi: 10.1158/1078-0432.CCR-09-2943. [DOI] [PubMed] [Google Scholar]
- 42.Song S, Christova T, Perusini S, Alizadeh S, Bao R-Y, Miller BW, Hurren R, Jitkova Y, Gronda M, Isaac M, Joseph B, Subramaniam R, Aman A, Chau A, Hogge DE, Weir SJ, Kasper J, Schimmer AD, Al-awar R, Wrana JL, Attisano L. Cancer Res. 2011;71:7628. doi: 10.1158/0008-5472.CAN-11-2745. [DOI] [PubMed] [Google Scholar]
- 43.Asher G, Lotem J, Sachs L, Kahana C, Shaul Y. Proc Natl Acad Sci U S A. 2002;99:13125. doi: 10.1073/pnas.202480499. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Oligos nucleotide sequences
Table S2. Compound 11c Metabolic Stability in Mouse Liver Microsomes