

Abstract
Sum-frequency generation (SFG) vibrational spectroscopy is often used to probe the backbone structures and orientations of polypeptides at surfaces. Using the ovispirin-1 polypeptide at the solid/liquid interface of polystyrene, we demonstrate for the first time that SFG can probe the polarization response of a single isotope labeled residue. To interpret the spectral intensities, we simulated the spectra using an excitonic Hamiltonian approach. We show that the polarization dependence of either the label or the unlabeled amide I band alone does not provide sufficient structural constraints to obtain both the tilt and the twist of the ovispirin helix at a solid/liquid interface, but that both can be determined from the polarization dependence of the complete spectrum. For ovispirin, the detailed analysis of the polarized SFG experimental data shows that the helix axis is tilted at roughly 138 degrees from the surface normal, and the transition dipole of the isotope labeled C=O group is tilted at 23 degrees from the surface normal, with the hydrophobic region facing the polystyrene surface. We further demonstrated that the Hamiltonian approach is able to address the coupling effect and the structural disorder. For comparison, we also collected the FTIR spectrum of ovispirin under similar conditions, which reveals the enhanced sensitivity of SFG for structural studies of single monolayer peptide surfaces. Our study provides insight into how structural and environmental effects appear in SFG spectra of the amide I band and establishes that SFG of isotope labeled peptides will be a powerful technique for elucidating secondary structures with residue-by-residue resolution.
Keywords: Amide I, monolayer, Hamiltonian approach, coupling effect, disorder
1. Introduction
Isotope labeling and vibrational spectroscopy provides site-specific structural information on polypeptides and proteins.1,2 Of the intrinsic vibrations inherent to proteins, the amide I band (mainly contributed by the backbone C=O stretching mode) is most often utilized in structural studies because its frequency and line shape are characteristic of the secondary structures and solvent environments of the backbone. Residue specific structural and environmental information can be obtained using isotope labeling. Labeling the backbone carbonyl with 13C, the amide I band is shifted by ~40 cm−1. A shift of ~66 cm−1 can be achieved with 13C=18O labeling. 1,3 Isotope labeling has been used in conjunction with 1D (FTIR) and 2D IR studies to obtain residue-by-residue backbone structural information about soluble proteins,4–6 protein/peptide folding kinetics,7–9 and amyloid aggregation and structure,10, 11 to name only a few studies.
Isotope labeling and vibrational spectroscopy is particularly valuable for studies of membrane proteins since they do not easily crystallize and are difficult to study with solution NMR. Solid-state NMR is a powerful technique but it is challenging to study membrane interactions of proteins and peptides in real time. The samples for solid state NMR studies are pre-mixed lipids and proteins/peptides and usually contain multiple layers of lipids, not a single lipid bilayer. Vibrational spectroscopy has been used to probe the structure of membrane bound alpha-helical bundles12, ion channels4, transmembrane alpha-helices 13–15 and helical dimers 5. In fact, the polypeptide that is the focus of this study, ovispirin, was previously 13C18O labeled for 2D IR experiments on its membrane bound structure. In that experiment, residue-by-residue structural resolution revealed the backbone orientation, tilt and secondary structure of each residue along nearly its entire length. 3 Another approach is to measure the angles of individual transition dipoles relative to the normal of the bilayer, which is done by macroscopically aligning the bilayers on an FTIR sample cell. Polarized light is then used to measure the linear dichroism of the isotope labels to back out the absolute angles. By isotope labeling a series of amino acids, this approach was used to obtain the structure of the CD3-ζ helical bundle 12 and to study the conformation gating of the M2 ion channel from the Influenza viral protein.4
While orientational constraints derived from FTIR spectroscopy are very valuable, as solid state NMR, FTIR dichroism studies cannot be performed on single monolayers because the signal is too small to obtain accurate angles. For adequate signal strength, hundreds or thousands of bilayers are stacked on top of one another. Stacking works well for equilibrated structures, but precludes experiments involving kinetics, drug binding, applied potentials, or systems that cannot be stacked such as solid interfaces. Moreover, since linear dichroism is attenuated by disorder, x-ray-reflectivity must be used to independently assess the disorder of the stacks.16 Thus, it would be quite beneficial to have a technique that is sensitive enough to measure the transition dipole angles of isotope labeled peptides of a single monolayer.
In contrast to FTIR, sum-frequency generation (SFG) vibrational spectroscopy has the sensitivity to observe peptides at sub-monolayer surface coverages. SFG is a second-order nonlinear optical spectroscopy which measures the second order nonlinear optical susceptibility χ(2) of the sample. According to the selection rule of the second order nonlinear optical process, SFG only detects signals from media where inversion symmetry is broken, such as surfaces and interfaces. Since 2003, SFG has been successfully used to study biological molecules with various secondary structures and on different types of surfaces, including α-helices17–20, 310 helices,21, 22 anti-parallel β-sheets23,24 and extended β sheets 25,26. However, there are often many approximations that go into the interpretation of the SFG spectrum of a peptide. For example, to back out the tilt of an alpha-helix at an interface, one often assume that the peptide is rotationally averaged about its helical axis.17–20 This is possible for peptides which insert perpendicularly into the cell membrane because the interior of the lipid bilayer is quite homogeneous (hydrophobic), but this is certainly unlikely for amphipathic and many other types of peptides at a hydrophobic substrate/aqueous solution interface. This assumption is required because there are not enough observables to obtain unique tilt and internal rotation angles. Other assumptions are also common, such as that random coil regions are weak or have different peak centers in SFG spectra and that the vibrational modes follow symmetry rules.17–20 In this paper, we show that a single residue can be resolved using 13C isotope labeling in the 18-residue antibiotic ovispirin at a polystyrene/peptide solution interface. The additional observables that this label provides eliminate the need for rotational averaging. We also learn that coupling to the isotope label may need to be considered. Previously, SFG spectroscopy was used in conjunction with NMR spectroscopy to provide a structural model of the synthetic LKα14 peptide, by measuring isotope labeled side chains. 27, 28 Here, we demonstrate for the first time that SFG spectroscopy can also be used to site-selectively probe the backbone carbonyl groups themselves, thereby providing a more direct measure of secondary structure.
Ovispirin-1 is an ideal target for our initial SFG experiments because its structure has been studied extensively in solution and on model phospholipid membranes. Solution NMR experiments show that in 33% TFE/67% PBS buffer at pH 6.5 ovispirin-1 forms a slightly curved α-helix over residues 4 to 16, with random coil outside of this region.29 Solid-state NMR experiments show that on membrane bilayers, ovispirin is still predominantly alpha-helical, but lies primarily in the plane of the bilayer, with the helix tilted approximately 84° from the surface normal 30. Two-dimensional infrared spectroscopy (2DIR) and molecular dynamics (MD) simulations similarly support the alpha-helical structure and planar orientation, and additionally show that its hydrophobic residues face into the membrane interior, consistent with its amphipathic sequence 3.
However, it is unknown what structure ovispirin will take or how it will interact with a purely hydrophobic surface such as the polystyrene surface we utilize here. As mentioned above, in 33% TFE/67% PBS buffer at pH 6.5 ovispirin-1 forms an α-helix over residues 4 to 16 29. Similarly, our choice of solvent (40% 2,2,2-trifluoroethanol and 60% 10 mM PBS buffer of pH=7.1) should promote helix formation similar to the solution structure (which will be confirmed later), but the tilt relative to the substrate surface may be considerably different than in the membrane bilayer systems, and interaction with the highly hydrophobic surface may perturb the overall peptide secondary structure. In this research, we find that the peptide backbone is tilted 138° with respect to the surface normal and the hydrophobic residues face polystyrene. Thus, the combination of isotope labeling and SFG spectroscopy brings new light to this system and enables many new systems to be studied that were not previously possible.
2. Materials and Methods
SFG theory and experimental details have been reported previously31–35 and will not be repeated in detail here. SFG has been applied to study peptides and proteins at interfaces by several groups.36–39 In our experiment, two laser beams, one visible beam at 532 nm and an infrared beam with tunable frequency (1300 to 4300 cm−1) are overlapped spatially and temporally at the bottom of a right-angled prism.40 The polystyrene (PS) thin film on the prism surface was prepared by spin coating 1 wt% PS solution in toluene on the CaF2 prism surface at 2500 rpm with a spin coater from Specialty coating systems. During every SFG experiment, the PS film was initially in contact with a 2 mL reservoir filled with 40% TFE and 60% 10 mM phosphate buffer (pH=7.1) in H2O or D2O. Then 20 μL ovispirin-1 peptide stock solution (1mg/mL in the same solvent) was injected into the 2 mL reservoir. The final subphase peptide solution concentration in the reservoir was 10 μg/mL and the equilibration time for the peptide-PS interaction was about 1.5 hrs. In order to ensure the homogeneously distributed peptides in the subphase in contact with PS, a magnetic stirrer was used during the data collection process. SFG ssp (s-SFG, s-IR, p-visible) and ppp spectra in the amide I frequency range were collected from ovispirin-1 peptide molecules at the PS/subphase peptide solution interface using a near total reflection geometry.23 The diameter of the overlapped laser beams at the PS/peptide solution interface was ~0.5 mm. Regular ovispirin-1 (with the sequence of KNLRRIIRKIIHIIKKYG) and isotope labeled ovispirin-1 (with the α-carbonyl group of residue 11 Ile being isotope labeled into 13C=16O) were synthesized by Peptide 2.0 Inc. PS, toluene, 2,2,2-trifluoroethanol, PBS, D2O were obtained from Sigma-Aldrich.
A Nicolet 550 spectrometer (Thermo Fisher Scientific. Inc., MA) was used to collect ATR-FTIR spectra of isotope-labeled and regular ovispirin-1 adsorbed on PS surfaces. A thin PS polymer was deposited on the ZnSe crystal surface by casting 1 wt% polymer solution in toluene and then drying it under a nitrogen gas flow. 160 μL 1 mg/mL ovispirin-1 peptide solution in D2O was injected into the ATR-FTIR trough (~1.6 mL) and the final concentration of the ovispirin-1 peptide solution is ~100 μg/mL (ten times larger than that used in the SFG experiment). The spectra with P and S polarizations were collected at ~2 hrs after the injection of the peptide stock solution to the ATR-FTIR trough. The ATR-FTIR sample chamber was purged with nitrogen before and through the measurements.
Spectra were simulated using the transition dipole coupling model and an excitonic Hamiltonian, as described in more detail in the supporting information. Briefly, peptide or ideal helix structures were loaded from a protein databank file. A transition dipole and Raman polarizability were assigned to each amide I local mode. Transition dipole coupling was used to calculate couplings between all pairs of local amide I modes. The resulting Hamiltonian was diagonalized to yield the eigenstates of the system41, whose coefficients were used to take to calculate the normal modes by taking appropriate linear combinations of the local mode of the transition dipoles and Raman polarizabilities. These responses were rotated to the laboratory frame, and summed to give overall intensities for the labeled and unlabelled peaks.42
3. Results and Discussion
In what follows, we begin by presenting the experimental data. The data is then interpreted with detailed orientation analyses. First, we use conventional formulas to determine the tilt of the alpha-helix from the polarization dependence of the unlabeled spectral region of the amide I band, assuming that the helix is isotropic (with a free rotation with the helix principal axis). Second, a similar analysis is applied to the isotope labeled region. But as we show, these two analyses are incongruent. To understand this dilemma, our third analysis uses the ratio of the unlabeled to labeled amide I bands which provides an additional observable that improves the structural analysis, suggesting that the peptide has a preferred orientation on the surface without free rotation. Finally, we highlight that the Hamiltonian approach can address the coupling effect and structural disorder issues.
3.1 Experimental Spectra
SFG spectra in the amide I frequency range (1500–1800 cm−1) were collected from both isotope labeled and unlabeled ovispirin-1 adsorbed at the PS/peptide solution interface. (Figure 1) A strong peak at ~1650 cm−1 was detected from both systems at the interface when a peptide H2O/TFE buffer solution was used to contact PS. With a peptide D2O/TFE buffer solution for a separate experiment, the amide I band appears at 1642 cm−1, with a similar intensity. The 8 cm−1 difference is typical for an amide I band detected from proteins/peptides in H2O and in D2O 43, 44 because the amide I vibrational mode involves the N-H stretch to a small degree. Although SFG signal in this spectral frequency range overlaps with that contributed by the vibrational mode of random coils, we believe here random coils have little contribution to the SFG signal because 1) SFG signal requires the ordering of the chemical groups and thus the amide I signals from the random coils likely cancel with each other. In other words, SFG spectroscopy is much more sensitive to α-helices than random coils.45 [delete: In fact, to the best of our knowledge, amide I signal from a peptide with random coiled structure on the surface has not been observed by SFG spectroscopy.] 2) In both H2O and D2O cases, the main amide I peak has a symmetric feature with a similar bandwidth. If the 1650 cm−1 peak in the H2O case contained two different components with comparable SFG signal intensity contributed from the α-helical structure and the random coiled structure, the deuteration of the peptide solvent would lead to a different spectral feature for the 1642 cm−1 peak,44 which was not observed experimentally here. 3) We used NLOPredict46 developed by Simpson group to estimate the contribution of the α-helical structure and the random coiled structure to the SFG amide I signal and found that the contribution of the random coiled structure is minimal for two typical orientations, namely, with the helical axis perpendicular and parallel to the polymer surface. We believe that other orientations (tilt orientations) should be intermediate to these two extremes, and thus the random coil contribution can be ignored in the analysis (Supporting Information).” For the isotope labeling case in H2O, a well-separated and weak peak at ~1607 cm−1 was also detected, which is the Ile11 isotope labeled amide I band (Figure 1b). Interestingly, the peak center of the isotope labeled C=O appears at ~1607 cm−1, appearing to be a shoulder to the 1642 cm−1 amide I main peak when D2O was used in the solvent (Figure 1d). Previous research on a helical dimer showed that 13C=O Leu in a solvent protected region exhibited a peak at ~1606 cm−1 whereas the 13C=O of a solvent exposed Ala absorbed at ~1585 cm−1. 47 For ovispirin on polystyrene, we find that the 13C=O of Ile11 remains at ~1607 cm−1 regardless of solvent. Thus, Ile11 is not exposed to the solvent, which we discuss below is because this residue faces the polystyrene surface.
Figure 1.


The ssp and ppp SFG amide I spectra of (a) regular ovispirin-1 adsorbed at the PS/peptide solution (with H2O) interface; (b) isotope labeled ovispirin-1 adsorbed at the PS/peptide solution (with H2O) interface; (c) regular ovispirin-1 adsorbed at the PS/peptide solution (with D2O) interface; (d) isotope labeled ovispirin-1 adsorbed at the PS/peptide solution (with D2O) interface.
We fit the SFG spectra shown in Figure 1 to extract out the intensities of the isotope labeled peak, since there exist interferences with the unlabeled amide I band. More details of the spectral fitting can be found in the supporting information. The experimentally deduced χzzz/χxxz ratio after Fresnel coefficients correction (see supporting information) is 2.03±0.03 for the 1650 band in H2O for the regular ovispirin-1. For the isotope labeling case, we fit the spectra considering two peaks with peak centers at 1650 cm−1 and 1607 cm−1, respectively. The fitting result of the 1650 cm−1 peak shows a χzzz/χxxz ratio of 1.95±0.09. For the 1607 cm−1 peak, we obtained an experimentally measured χzzz/χxxz ratio of 3.7±0.2. The spectral fitting results obtained from peptides at the PS/peptide D2O solution interface are similar to the H2O cases (Table 1): 2.08±0.02 (regular) and 1.97±0.06 (isotope labeled) for the 1650 cm−1 signal; 4.1±0.1 for the 1607 cm−1 peak. To take both solvent cases into consideration, we averaged χzzz/χxxz ratios for the main backbone amide I peak of ovispirin-1 (2.00±0.10) and that for the isotope labeled peak (3.85±0.35). Comparing the relative helix and label peak intensities, we find that χzzz (helix)/χzzz (label) = 6.18.
Table 1.
| Amide I | Single residue | |
|---|---|---|
| χzzz/χxxz | χzzz/χxxz | |
| C12 in H2O | 2.03±0.03 | N/A |
| C12 in D2O | 2.08±0.02 | N/A |
| C13 in H2O | 1.95±0.09 | 3.7±0.2 |
| C13 in D2O | 1.97±0.06 | 4.1±0.1 |
3.2 Orientational Analysis of the Helix
Having acquired the data above, the standard approach is to interpret the χzzz/χxxz ratio as a measure of the tilt of the helix at the surface.19,20 For an ideal helix, the transition dipole of each amide I local mode points 42 degrees from the helix axis, there is a 100 degree rotation about this axis from one reside to the next, and each local mode has a Raman tensor associated with it. 19 Using this ideal structure, one generates the normal modes of the helix by diagonalizing a vibrational Hamiltonian built from the local modes of the coupled amide I oscillators. The χzzz/χxxz ratio is then calculated from the transition dipoles of the normal modes (details in the supporting information) as a function of the tilt (θ) and twist (ψ) angles (defined in Figure 2). Doing so for a 13-residue ideal helix produces the χzzz/χxxz ratio shown in Figure 2(a) when ψ is rotationally averaged around the helix axis. Rotationally averaging around ψ is necessary because there is only one experimental observable (the χzzz/χxxz ratio), but two unknowns (θ and ψ). For a perfect and infinitely long helix, one obtains the same θ-dependence whether or not ψ is averaged. For a real helix one can in practice treat the structure as fully symmetric around the helix axis for any length of helix longer than a few turns. Here we chose 13 residues for the helix because that is the length of the ovispirin alpha-helix in solution29. As discussed above, we believe that the peptide retains its α-helical structure at the polymer/solution interface because the SFG spectra are dominated by a single α-helical characteristic peak at ~1650 cm−1. This can be further confirmed by the intensity ratio of the main peak and the signal detected from the isotope labeled unit, which will be discussed in detail in section 3.4. With these considerations in mind, the experimental ratio of χzzz/χxxz = 2.00 gives a helix tilt angle of θ =43 (or 137) +/− 5 degrees relative to the polystyrene surface normal.
Figure 2.

χzzz/χxxz ratios for (a) the unlabeled segment of an ideal alpha helix and (b) the isotope labeled peak, assuming full rotational averaging around the helix axis. (c) Contours indicating ψ/θ pairs giving the experimentally-measured values for the unlabeled and labeled peak χzzz/χxxz ratios. In this figure, the tilt angle θ indicates the angle between the helix axis and the surface normal.
3.3. Orientational Analysis of the Isotope Label
One could apply a similar analysis to that above for just the independent isotope label. In this case, the analysis is much simpler, because one does not need to construct or diagonalize a Hamiltonian. One would use the same transition dipole and Raman polarizability tensors as above to calculate the tilt-angle dependence of the χzzz/χxxz ratio for a single amide-I residue, as shown in Figure 3(a). Here, we have defined θlabel as the angle between the transition dipole and the surface normal and we rotationally average around the label transition dipole, ψlabel. Note, that averaging around ψlabel is not equivalent to averaging around ψ for the helix or around the labeled C=O bond because the rotation axis is different. The tilt-angle dependence for both the ψ-averaged and the individual ψlabel=0 and ψlabel=90 deg. cases are very similar, because the Raman tensor’s major axis points almost directly along the transition dipole with much smaller (though unequal) components along the perpendicular axes (see Supporting Information). The consequence is that there is a maximum of about 10 degrees difference in tilt angles calculated from ψ-fixed or ψ-averaged curves. Thus, while ψ averaging is necessary to eliminate an unknown variable just as for the helix analysis, it does not influence the θlabel more than a few degrees. Using this curve and the experimentally-measured χzzz/χxxz ratio of 3.85 +/− 0.35 for the isotope-labeled peak, we find θlabel = 26 or 154 +/− 4 degrees from the surface normal. In order to do this analysis, we have to use the ψ average because otherwise there are too many unknowns. However, that approximation is not physically reasonable because rotating around the label’s axis swings the entire peptide structure, since ψhelix is not equal to ψlabel. Taking the internal rotational average about ψhelix for the unlabeled band (as done in the last section) makes sense if the helix is long and perfect, because ψ averaging is equivalent to the symmetry modes. Thus, the analysis of a single label by itself makes no physical sense, and what needs to be done is to solve for tilt and twist angles that simultaneously give the correct ratios for the both the labeled and unlabeled peaks.
Figure 3.

Tilt-angle dependence of the χzzz/χxxz ratio for a single amide-I residue, for (a) the ψ-averaged case and two fixed-ψ cases (ψ=0 corresponds to the C(O)N bond lying in the yz plane) and (b) the ψ-averaged case for Gaussian distributions of tilt angles with different full-width-at-half- maxima (indicated). In (a) and (b), the tilt angle θ is defined as the angle between the transition dipole and the surface normal.
While ψ averaging an unlabeled helix is acceptable because of the symmetry, including the isotope label destroys the rotational symmetry, and the rotational average is no longer a valid approximation. This can be shown by calculating the χzzz/χxxz ratio for the label predicted by a fully internally rotationally-averaged helix. When applied to the label, the orientational analysis above predicts that the χzzz/χxxz ratio of the label should match that of the unlabeled amide I band (Figure 2b), because symmetry dictates that in helical molecules the responses of the parallel and perpendicular normal modes (A and E1 modes) are closely related to the parallel and perpendicular components of a single local mode’s response. 52, 53 Thus, for a ψ-averaged helix tilted at 43 (or 137) degrees from the surface normal, one would also predict that a χzzz/χxxz ratio of 2.00 +/− 0.10 would be measured for the label. But the experimentally-measured ratio is actually 3.85 +/− 0.35, which far exceeds the experimental uncertainty. In fact, a χzzz/χxxz ratio of 3.85 +/− 0.35 cannot be matched by a rotationally averaged helix at any tilt angle. That is, the ratios of the unlabeled and labeled amide I bands are incongruous for an analysis that requires rotational averaging around the helix axis.
3.4 Orientational Analysis of the Entire Peptide including Structural Disorder and Coupling
How does one reconcile the ratios of the labeled and unlabeled amide I bands? We need to consider the possibility that the helix is not isotropic about ψhelix. That is to say, the helix does not have a free rotation around the principal axis. In order to determine the orientation of such an α-helix, we need to determine both the tilt angle and the twist angle. We retain a perfect helix with the label uncoupled from the rest of the helix and simultaneously calculate the χzzz/χxxz ratios for both the labeled and unlabeled peaks as a function of tilt and twist angles. Using the two measured χzzz/χxxz ratios for the labeled and unlabeled peaks, we can deduce two unknowns: the tilt angle and the twist angle. As shown in Figure 2(c), we find that our measured χzzz/χxxz ratios are consistent with helix tilt/twist angles of (θ, ψ) = (41°, 5°), (57°, 297°), (123°, 117°), or (138°, 184°) (with around 4° error margins). Homodyne-detected SFG cannot distinguish between the 90+x and 90-x degree tilt angles27, though future phase-sensitive or heterodyne-detected experiments may resolve this difficulty 48–51. However, we can narrow the possibilities using physical intuition and the experimentally observed solvent accessibility of Ile11. Before rotation, our ideal helix is defined such that the hydrophilic region extends from −60 to +120 degrees around the helix axis. Thus the (41°, 5°) orientation corresponds predominantly to having the hydrophilic region of the peptide facing the polystyrene surface, while the (138°, 184°) orientation corresponds to having the hydrophobic region of the peptide facing the polystyrene surface; in the remaining two orientations, the hydrophobic and hydrophilic regions have roughly the same extent of interaction with the surface. Of these tilt-angle possibilities, the (138°, 184°) pair is most physically reasonable, since the solvent dependence of the peak center of the labeled unit indicates that the residue is buried at the hydrophobic interface and the interaction of the hydrophobic side of the peptide with the hydrophobic polystyrene surface should inherently be more favorable than the interaction of the hydrophilic side of the peptide with this surface. The following reasons further support that the (138°, 184°) orientation is the most likely orientation (Figure 4): First, this helix orientation also gives a χzzz (helix)/χzzz (label) ratio of approximately 6.2, consistent with our experimental value of 6.18, a criterion which is not met by the (57°, 297°) and (123°, 117°) orientations (which both yield χzzz (helix)/χzzz (label) ratios of less than 5). Second, at this orientation, the transition dipole of the isotope labeled unit is calculated to be tilted 23 degrees relative to the surface normal, in good agreement with the value calculated from our preceding analysis of the isolated label. To further clarify the final orientation of (138°, 184°), we also present the reference peptide orientation (0°, 0°) along with the final orientation in Figure 4.
Figure 4.
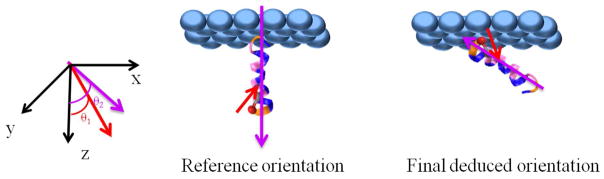
Schematic (right panel) showing the final deduced orientation of ovispirin (tilt angle = 138°, twist angle = 184°) at the polystyrene/water interface. Left panel defines the reference orientation (tilt angle = 0°, twist angle = 0°) where the hydrophilic region (blue) extends from −60 to +120 degrees around the helix axis. θ1 is the angle between the transition dipole of 13C=O chemical group (purple arrow) and the z axis and θ2 is the angle between the peptide helix axis (from N to C terminus) (red arrow) and the z axis. For the reference orientation, θ1=138° and θ2=0° and for the final deduced orientation, θ1=23° and θ2=138°
Thus, analyzing the labeled and unlabeled peaks within the framework of an ideal helix provides insight into the overall orientation of the ovispirin peptide at the solution/polystyrene interface. However, perhaps analyzing the angles alone is not sufficient, because ovispirin is far from an ideal helix at the interface, which we learn from the observations listed below and leads to additional considerations about the interpretation of the spectra. First, comparing the intensities of the labeled and unlabeled amide I bands reveals that only a subset of the residues can contribute to the helical structure. For the (138°, 184°) tilt/twist angles calculated above, in which the angular dependence of the labeled and unlabeled peaks were considered independently, a full 18- residue helix would give a χzzz(helix)/χzzz(label) value of 7.0, which deviates by almost 15% from the experimental value of 6.18. Reducing the length of the helical section to anywhere between 13 to 15 residues gives the χzzz(helix)/χzzz(label) value between 6.1 and 6.3. Further reducing the length of the helical section causes the χzzz(helix)/χzzz(label) ratio to drop far below the experimentally measured value. While this result is consistent with the 13-residue helix analyzed above, it reveals that the entire 18-residue ovispirin peptide cannot form a perfectly ordered helix, because if it did, then the intensity of the isotope label would be much smaller as compared to the unlabeled amide I band (Intensity is proportional to the square of the χ value).
We want to highlight here that the Hamiltonian approach we present in this paper can be used to calculate the coupling between the label and the rest of the peptide, including these couplings in the Hamiltonian disrupts the transition dipoles of the helix, which changes the strength of many of the SFG-active normal modes. Shown in Figure 5(a, b) are spectra for a 13- residue labeled helix at the (138°, 184°) tilt/twist angle. Figure 5(a) shows the response of an isolated label added to that of an un-disrupted helix, while Figure 5(b) shows the spectrum when the isotope label is incorporated into and coupled with the rest of the helix. The stick spectrum shows significant contributions from new SFG-active modes, indicating that the label and the helix cannot be treated as independent vibrational modes. In this regard, 13C=18O isotope labeling is preferable, because the labeled amide group has better decoupling, although both 13C=16O and 13C=18O labels will affect the unlabeled spectrum about the same amount. This effect will be important when the isotope labeled unit is incorporated into different sites of the peptide or more than one isotope labeled units are incorporated.
Figure 5.

Simulated spectra showing (a) a 13-residue ideal helix, with the isotope label calculated independently of (or uncoupled from) the rest of the helix, (b) a 13-residue ideal helix with the isotope label incorporated into and coupled with the other residues in the helix, and (c) a 13- residue ideal helix summed over 50 spectra with 16 cm−1 random disorder in the local mode frequencies. Spectra were simulated using a 15 cm−1 Lorentzian for each mode. Stick spectra (purple) are also included to emphasize how the normal mode structure changes as the couplings are broken.
Second, the Hamiltonian approach can be used to access the effect of the structural disorder. Random disorder along the diagonal of the Hamiltonian occurs in many systems where the individual chromophores have different local mode frequencies due to different local environments in different molecules, for example if the local solvation environment of the amide-I modes varies from amide to amide bond 54, 55. In Figure 5(c), we show the spectrum expected for a 13-residue labeled helix with 16 cm−1 standard deviation random variation in the local mode frequencies added before diagonalizing the Hamiltonian, averaged over 50 different disordered Hamiltonians. 16 cm−1 is about the disorder one expects due to differences in hydrogen bonding for soluble polypeptides. We see that the SFG spectrum of the disordered helix is broader and less intense than that of the ordered helix, because all different members of the ensemble now have slightly different normal modes. The overall calculated tilt angle from the labeled and unlabeled peaks does not change significantly with this added disorder, but the helix/label ratio increases. Thus, disorder that is created by the typical environment surrounding a polypeptide does not change the interpretation of the angular measurements, but does alter the relative intensities of the labeled and unlabeled amide I modes. However, the spectrum shown in Figure 5 (c) is not very similar to the spectra we observed in Figure 1. Therefore in this study, we believe that the effect of the structural disorder is not substantial. At this stage, we cannot quantify this effect in our current measurements because it requires dynamic knowledge of the linewidths, which will be the subject of future work. Nonetheless, the point being made is that disorder breaks the symmetry of the helix, and so in real systems one may no longer think of the normal modes purely as the fully symmetric A and E1 modes especially for a severely disrupted helix.
Interestingly, the tilt angles deduced in this work (138 degrees) indicate that the ovispirin is much more steeply angled away from the plane of the polystyrene surface than it is when adsorbed to a planar lipid bilayer. Using multiple label sites in future experiments will provide residue-by-residue structural information that can be used to obtain a complete backbone structure through constrained molecular dynamics simulations, as has been done with oriented polypeptides and FTIR spectroscopy.56
Last but not least, we want to mention a few other considerations that should be taken into account in future simulations. As revealed by previous NMR, 2D IR, and MD studies, ovispirin exhibits significant structural disorder as well. For example, in the solution NMR studies it is found that the helical portion of the peptide is somewhat curved, and the N- and C-terminal ends disordered. The curve breaks the symmetry of the helix and adds off-diagonal disorder to the Hamiltonian. The disordered ends will have both diagonal and off-diagonal disorder. Therefore such structures may contribute to SFG signals. In the future, we will calculate such contributions and validate the calculation method using multiple label sites in experiments. Also, we calculated the transition dipole and Raman response of a single amide-I mode using values derived from polarized FTIR and Raman experiments23. However, some models use slightly different molecular responses for a single amide-I mode 55,56, which will yield slightly different single-residue tilt angles. We have also assumed a delta-function orientational distribution. If the residues or peptides are instead distributed over a wider range of tilt angles, the tilt-angle dependence flattens out, as shown in Figure 3(b) for a series of Gaussian tilt-angle distributions of varying widths for a single vibrationally isolated label. This flattening of the tilt-angle dependence means that the same experimental χzzz/χxxz ratio yields a center tilt angle closer to the surface normal as the distribution gets broader. Developments underway on modeling SFG spectra of peptides will help to better define these parameters. 57–59
3.5 ATR-FTIR Experiments
For comparison purposes, we also performed ATR-FTIR experiments. The concentration of ovisiprin-1 is 10 times higher because no signal was detected from the isotope labeled group when lower peptide solution concentrations were used. The ATR-FTIR spectra collected from the PS/peptide solution interface are shown in Figure 6. We found that the PS polymer can generate a strong background ATR-FTIR signal at ~1602 cm−1 from one of the benzene ring modes60, appearing as either a positive or negative peak. For both isotope labeled and regular ovispirin-1 samples, there is a broad peak at around ~1647 cm−1 (Figure 6), indicating the existence of both α-helical and random coil components. For isotope labeled sample, besides the negative peak at ~1602 cm−1, an additional peak at ~1612 cm−1 can be assigned to the isotopelabeled C=O chemical group. Data analysis shows that this C=O is ~38 degree vs. the surface normal (Figure 6c) given that the dichroic ratio R of the isotope labeled C=O stretching peak is ~4.13.61 Interestingly the 1602 cm−1 background signal of the PS polymer is silent in SFG spectra because this benzene ring mode is Raman inactive and thus SFG inactive. Besides, ATR-FTIR spectra have multiple contributions from the α-helix, the random coil and the polymer background which made the orientation determination of the single C=O chemical group by ATR-FTIR subject to large errors.
Figure 6.


(A) ATR-FTIR spectra of isotope labeled ovispirin-1 molecules at the PS/peptide solution interface. (B) ATR-FTIR spectra of regular ovispirin-1 molecules at the PS/peptide solution interface. (C) The relationship between the dichroic ratio R of the isotope labeled C=O stretching peak and the tilt angle of the C=O bond direction relative to the surface normal. The dichroic ratio R of the isotope labeled C=O stretching peak detected at the PS/peptide solution interface is shown as a horizontal line.
Compared to ATR-FTIR, SFG studies on the C=O isotope labeled peptides have several advantages: 1) SFG spectra are free of background contribution from the polymer substrate as well as other media in our study. For peptides and proteins, SFG spectra have minimum contributions from random coils. Unlike in IR spectra, where H2O absorption band overlaps with the peptide/protein amide I band, SFG spectra has minimum H2O signal contribution and thus the experiment could be performed in H2O and separate the signals from the isotope unit and the main amide I peak. 2) SFG is more sensitive than ATR-FTIR for detecting well-oriented α-helices, thus can detect signals from peptides from a single monolayer instead of bilayer stacks.
While solid-state NMR (e.g. PISEMA) is a well-established technique in obtaining orientation restraints for membrane bound peptides 62, isotope labeled SFG has some unique advantages: 1. SFG can monitor the interaction process in situ (~several minutes per spectrum) while NMR techniques such as PISEMA suffer from long-time data accumulation (~several hours per experiment), therefore the isotope labeled SFG technique can be used to monitor the dynamics of a single residue in biological processes such as ligand titration, fibril formation, GPCR-G protein interaction, ion channel opening and so on. 2. Only one bilayer is required, which allows very precise difference experiments, such as with ligand binding. 3. A typical solid-state NMR experiment on short peptides requires ~100 μg sample while an SFG experiment only needs ~10μg sample.
For ovisprin-1, we also detected SFG signals from the α-helical backbone and the isotope labeled peptide unit when the peptide associated with a supported lipid bilayer DPPG (dipalmitoylphosphatidyglycerol)/dDPPG (deuterated dipalmitoylphosphatidyglycerol) in contact with the peptide aqueous solution. For ovispirin-1 with residue R4 mutated with 13C=16O incorporated glycine, the SFG ppp spectrum shows a signal contributed from the isotope labeled unit at ~1610 cm−1 (Supporting Information), similar to that shown in Figure 1b. On the other hand, for the ovispirin-1 with R8 mutated with 13C=16O incorporated glycine, the SFG signal detected from the isotope labeled unit shifted to ~1600 cm−1 with a broader width (Supporting Information). The different peak centers and peak widths indicate that R4 and R8 are in different micro-environments. Thus, we expect that this technique is applicable to peptides associated with lipid bilayers or cell membranes. However, since more experimental measurements and theoretical analysis are still under way and such details are beyond the scope of this research, the detailed structure and orientation analysis will be published in the future.
In this paper we demonstrated the approach on a synthesized polypeptide that is isotope labeled using commercially available compounds. But the method is also applicable to proteins using exciting methods developed in the past few years. One set of approaches is native chemical ligation and expressed protein ligation.63, 64 In these techniques, proteins are semi-synthesized from fragments using a thiol-ene chemistry. Thus, one can isotope label a fragment (e.g., a helix instead of a single peptide unit) in the same manner as we did here for ovispirin, and then ligate it to another fragment to form the full protein. Expressed protein ligation has recently been used in conjunction with 2D IR spectroscopy.65 It is also possible to arbitrarily isotope label amino acids in proteins by using a cell free expression system and a stop codon that matches an isotope labeled t-RNA.66 It is now also possible to put in non-natural isotope labels which might be SFG chromophores, such as nitriles, by using a tRNA synthetase pair system, like has been developed for phenylalanine.67 With these new and exciting methods in hand, it is now possible to isotope label and thus monitor the structures of precise locations in nearly arbitrarily sized proteins. It is also important to point out that SFG spectroscopy requires a tiny amount of sample. For example, in the ovispirin-1 case, one SFG experiment only requires 20 μg sample. As a result, experiments are possible even on poorly expressing proteins or systems in which the ligation is inefficient. Furthermore, heterodyne detection can also improve signal-to-noise ratio, 46–49 and thus we believe that in the future, this technique will shed lights on larger protein systems.
4. Conclusion
We have shown that isotopic labeling enables SFG spectroscopy to detect polarized amide I signals from a single peptide unit in an α-helical peptide at buried polymer/solution interface. Such SFG signals can be used to study the microenvironment and interfacial orientation of the isotope labeled residue. If we assume a single distribution, the backbone helix of ovispirin-1 on PS surface is 138 degrees relative to the surface normal with a twist angle of 184 degrees, and the transition dipole of the isotope labeled C=O group is tilted 23 degrees relative to the surface normal. Our result show that using the 13C=O isotope labeling, SFG can be used to obtain residue-specific orientation information. In the future, using multiple label locations will allow us to measure the exact structure and orientation of surface-bound peptides without requiring assumptions about the expected secondary structure. This capability makes SFG a powerful technique for structural analysis and will bring new insight to many biophysical systems.
Supplementary Material
Acknowledgments
This work is supported by the National Institute of Health (1R01GM081655 to ZC and 1R01DK79895 to MTZ). B. D. acknowledges the Barbour Scholarship from the University of Michigan. We thank Ann Marie Woys for providing samples for initial studies.
Footnotes
Supporting Information Available: Detailed SFG data analysis and Hamiltonian approach. This information is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Decatur SM. Elucidation of Residue-Level Structure and Dynamics of Polypeptides via Isotope-Edited Infrared Spectroscopy. Accounts Chem Res. 2006;39:169–175. doi: 10.1021/ar050135f. [DOI] [PubMed] [Google Scholar]
- 2.Ganim Z, Chung HS, Smith AW, Deflores LP, Jones KC, Tokmakoff A. Amide I Two-dimensional Infrared Spectroscopy of Proteins. Accounts Chem Res. 2008;41:432–441. doi: 10.1021/ar700188n. [DOI] [PubMed] [Google Scholar]
- 3.Woys AM, Lin YS, Reddy AS, Xiong W, Pablo JJ, de Skinner JL, Zanni MT. 2D IR Line Shapes Probe Ovispirin Peptide Conformation and Depth in Lipid Bilayers. J Am Chem Soc. 2010;132:2832–8. doi: 10.1021/ja9101776. [DOI] [PubMed] [Google Scholar]
- 4.Manor J, Mukherjee P, Lin YS, Leonov H, Skinner JL, Zanni MT, Arkin IT. Gating Mechanism of the Influenza A M2 Channel Revealed by 1D and 2D IR Spectroscopies. Structure. 2009;17:247–254. doi: 10.1016/j.str.2008.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Remorino A, Korendovych IV, Wu Y, DeGrado WF, Hochstrasser RM. Three-dimensional Structures by Two-dimensional Vibrational Spectroscopy. Science. 2011;332:1206–1209. doi: 10.1126/science.1202997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deng H, Vu DV, Clinch K, Desamero R, Dyer RB, Callender R. Conformational Heterogeneity within the Michaelis Complex of Lactate Dehydrogenase. J Phys Chem B. 2011;115:7670–7678. doi: 10.1021/jp2015929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pozo Ramajo A, Petty SA, Starzyk A, Decatur SM, Volk M. The Alpha-helix Folds More Rapidly at the C-terminus than at the N-terminus. J Am Chem Soc. 2005;127:13784–13785. doi: 10.1021/ja054500+. [DOI] [PubMed] [Google Scholar]
- 8.Smith AW, Lessing J, Ganim Z, Peng CS, Tokmakoff A, Roy S, Jansen TLC, Knoester J. Melting of a β-hairpin Peptide Using Isotope-edited 2D IR Spectroscopy and Simulations. J Phys Chem B. 2010;114:10913–10924. doi: 10.1021/jp104017h. [DOI] [PubMed] [Google Scholar]
- 9.Huang CY, Getahun Z, Zhu Y, Klemke JW, DeGrado WF, Gai F. P. Helix Formation via Conformation Diffusion Search. P Natl Acad Sci USA. 2002;99:2788–2793. doi: 10.1073/pnas.052700099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Petty SA, Decatur SM. Intersheet Rearrangement of Polypeptides During Nucleation of β-sheet Aggregates. P Natl Acad Sci USA. 2005;102:14272–14277. doi: 10.1073/pnas.0502804102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim YS, Liu L, Axelsen PH, Hochstrasser RM. Two-dimensional Infrared Spectra of Isotopically Diluted Amyloid Fibrils from Abeta40. P Natl Acad Sci USA. 2008;105:7720–7725. doi: 10.1073/pnas.0802993105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin YS, Shorb JM, Mukherjee P, Zanni MT, Skinner JL. Empirical Amide I Vibrational Frequency Map: Application to 2D-IR Line Shapes for Isotope-edited Membrane Peptide Bundles. J Phys Chem B. 2009;113:592–602. doi: 10.1021/jp807528q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alfieri KN, Vienneau AR, Londergan CH. Using Infrared Spectroscopy of Cyanylated Cysteine to Map the Membrane Binding Structure and Orientation of the Hybrid Antimicrobial Peptide CM15. Biochemistry. 2011;50:11097–11108. doi: 10.1021/bi200903p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Earnest TN, Herzfeld J, Rothschild KJ. Polarized Fourier Transform Infrared Spectroscopy of Bacteriorhodopsin. Transmembrane Alpha Helices are Resistant to Hydrogen/deuterium Exchange. Biophys J. 1990;58:1539–1546. doi: 10.1016/S0006-3495(90)82498-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beevers AJ, Kukol A. Secondary Structure, Orientation, and Oligomerization of Phospholemman, a Cardiac Transmembrane Protein. Protein Sci. 2006;15:1127–1132. doi: 10.1110/ps.051899406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Manor J, Khattari Z, Salditt T, Arkin IT. Disorder Influence on Linear Dichroism Analyses of Smectic Phases. Biophys J. 2005;89:563–571. doi: 10.1529/biophysj.104.058842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen X, Wang J, Boughton AP, Kristalyn CB, Chen Z. Multiple Orientation of Melittin Inside a Single Lipid Bilayer Determined by Combined Vibrational Spectroscopic Studies. J Am Chem Soc. 2007;129:1420–1427. doi: 10.1021/ja067446l. [DOI] [PubMed] [Google Scholar]
- 18.Boughton AP, Yang P, Tesmer VM, Ding B, Tesmer JJG, Chen Z. Heterotrimeric G protein β1γ2 Subunits Change Orientation upon Complex Formation with G Protein-coupled Receptor Kinase 2 (GRK2) on a Model Membrane. P Natl Acad Sci USA. 2011;108:E667–673. doi: 10.1073/pnas.1108236108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nguyen KT, Clair SV, Le YeS, Chen Z. Orientation Determination of Protein Helical Secondary Structures Using Linear and Nonlinear Vibrational Spectroscopy. J Phys Chem B. 2009;113:12169–12180. doi: 10.1021/jp904153z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ding B, Chen Z. Molecular Interactions between Cell Penetrating Peptide Pep-1 and Model Cell Membranes. J Phys Chem B. 2012;116:2545–2552. doi: 10.1021/jp209604m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ye S, Nguyen KT, Boughton AP, Mello CM, Chen Z. Orientation Difference of Chemically Immobilized and Physically Adsorbed Biological Molecules on Polymers Detected at the Solid/liquid Interfaces in situ. Langmuir. 2010;26:6471–6477. doi: 10.1021/la903932w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ye S, Li H, Wei F, Jasensky J, Boughton AP, Yang P, Chen Z. Observing a Model Ion Channel Gating Action in Model Cell Membranes in Real Time in situ: Membrane Potential Change Induced Alamethicin Orientation Change. J Am Chem Soc. 2012;134:6237–43. doi: 10.1021/ja2110784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nguyen KT, King JT, Chen Z. Orientation Determination of Interfacial β-sheet Structures in situ. J Phys Chem B. 2010;114:8291–8300. doi: 10.1021/jp102343h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang J, Chen X, Clarke ML, Chen Z. Detection of Chiral Sum Frequency Generation Vibrational Spectra of Proteins and Peptides at Interfaces in situ. P Natl Acad Sci USA. 2005;102:4978–83. doi: 10.1073/pnas.0501206102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fu L, Ma G, Yan ECY. In situ Misfolding of Human Islet Amyloid Polypeptide at Interfaces Probed by Vibrational Sum Frequency Generation. J Am Chem Soc. 2010;132:5405–5412. doi: 10.1021/ja909546b. [DOI] [PubMed] [Google Scholar]
- 26.Fu L, Xiao D, Wang Z, Batista VS, Yan ECY. Chiral Sum Frequency Generation for In Situ Probing Proton Exchange in Antiparallel β-Sheets at Interfaces. J Am Chem Soc. 2013;135:3592–3598. doi: 10.1021/ja3119527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weidner T, Breen NF, Li K, Drobny GP, Castner DG. Sum Frequency Generation and Solid-state NMR Study of the Structure, Orientation, and Dynamics of Polystyrene-adsorbed Peptides. P Natl Acad Sci USA. 2010;107:13288–13293. doi: 10.1073/pnas.1003832107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weidner T, Apte JS, Gamble LJ, Castner DG. Probing the Orientation and Conformation of α-Helix and β-Strand Model Peptides on Self-Assembled Monolayers Using Sum Frequency Generation and NEXAFS Spectroscopy. Langmuir. 2010;26:3433–3440. doi: 10.1021/la903267x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sawai MV, Waring AJ, Kearney WR, McCray PB, Forsyth WR, Lehrer RI, Tack BF. Impact of Single-residue Mutations on the Structure and Function of Ovispirin/novispirin Antimicrobial Peptides. Protein Eng. 2002;15:225–232. doi: 10.1093/protein/15.3.225. [DOI] [PubMed] [Google Scholar]
- 30.Yamaguchi S, Huster D, Waring A, Lehrer RI, Kearney W, Tack BF, Hong M. Orientation and Dynamics of an Antimicrobial Peptide in the Lipid Bilayer by Solid-state NMR Spectroscopy. Biophys J. 2001;81:2203–2214. doi: 10.1016/S0006-3495(01)75868-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mermut O, Phillips DC, York RL, McCrea KR, Ward RS, Somorjai G. In situ Adsorption Studies of a 14-amino Acid Leucine-lysine Peptide onto Hydrophobic Polystyrene and Hydrophilic Silica Surfaces Using Quartz Crystal Microbalance, Atomic Force Microscopy, and Sum Frequency Generation Vibrational Spectroscopy. J Am Chem Soc. 2006;128:3598–3607. doi: 10.1021/ja056031h. [DOI] [PubMed] [Google Scholar]
- 32.Perry A, Neipert C, Space B. Theoretical Modeling of Interface Specific Vibrational Spectroscopy: Methods and Applications to Aqueous Interfaces. Chem Rev. 2006;106:1234–1258. doi: 10.1021/cr040379y. [DOI] [PubMed] [Google Scholar]
- 33.Liu J, Conboy JC. Direct Measurement of the Transbilayer Movement of Phospholipids by Sum-frequency Vibrational Spectroscopy. J Am Chem Soc. 2004;126:8376–8377. doi: 10.1021/ja048245p. [DOI] [PubMed] [Google Scholar]
- 34.Ma G, Chen X, Allen HC. Dangling OD Confined in a Langmuir Monolayer. J Am Chem Soc. 2007;129:14053–14057. doi: 10.1021/ja075806e. [DOI] [PubMed] [Google Scholar]
- 35.Roeters SJ, van Dijk CN, Torres-Knoop A, Backus EHG, Campen Rk, Bonn M, Woutersen S. Determining in situ Protein Conformation and Orientation from the Amide-I Sum-frequency Generation Spectrum: Theory and Experiment. J Phys Chem A. 2013 doi: 10.1021/jp401159r. Article ASAP. [DOI] [PubMed] [Google Scholar]
- 36.Sagle LB, Cimatu K, Litosh VA, Liu Y, Flores SC, Chen X, Yu B, Cremer PS. Methyl Groups of Trimethylamine N-oxide Orient Away from Hydrophobic Interfaces. J Am Chem Soc. 2011;133:18707–18712. doi: 10.1021/ja205106e. [DOI] [PubMed] [Google Scholar]
- 37.Chen X, Flores SC, Lim SM, Zhang Y, Yang T, Kherb J, Cremer PS. Specific Anion Effects on Water Structure Adjacent to Protein Monolayers. Langmuir. 2010;26:16447–16454. doi: 10.1021/la1015862. [DOI] [PubMed] [Google Scholar]
- 38.Engel MFM, vandenAkker CC, Schleeger M, Velikov KP, Koenderink GH, Bonn M. The Polyphenol EGCG Inhibits Amyloid Formation Less Efficiently at Phospholipid Interfaces than in Bulk Solution. J Am Chem Soc. 2012;134:14781–14788. doi: 10.1021/ja3031664. [DOI] [PubMed] [Google Scholar]
- 39.Li H, Ye S, Wei F, Ma S, Luo Y. In situ Molecular-level Insights into the Interfacial Structure Changes of Membrane-associated Prion Protein Fragment [118–135] Investigated by Sum Frequency Generation Vibrational Spectroscopy. Langmuir. 2012;28:16979–16988. doi: 10.1021/la302655p. [DOI] [PubMed] [Google Scholar]
- 40.Wang J, Even MA, Chen X, Schmaier AH, Waite JH, Chen Z. Detection of Amide I Signals of Interfacial Proteins in situ Using SFG. J Am Chem Soc. 2003;125:9914–9915. doi: 10.1021/ja036373s. [DOI] [PubMed] [Google Scholar]
- 41.Hamm P, Zanni M. Concepts and Methods of 2D Infrared Spectroscopy. Cambridge University Press; Cambridge, U.K: 2011. [Google Scholar]
- 42.Laaser JE, Zanni MT. Extracting Structural Information from the Polarization Dependence of One- and Two-Dimensional Sum Frequency Generation Spectra. J Phys Chem A. 2013 doi: 10.1021/jp307721y. Article ASAP. [DOI] [PubMed] [Google Scholar]
- 43.Tamm LK, Tatulian SA. Infrared Spectroscopy of Proteins and Peptides in Lipid Bilayers. Q Rev Biophys. 1997;30:365–429. doi: 10.1017/s0033583597003375. [DOI] [PubMed] [Google Scholar]
- 44.Starzyk A, Barber-Armstrong W, Sridharan M, Decatur SM. Spectroscopic Evidence for Backbone Desolvation of Helical Peptides by 2,2,2-trifluoroethanol: an Isotope-edited FTIR Study. Biochemistry. 2005;44:369–376. doi: 10.1021/bi0481444. [DOI] [PubMed] [Google Scholar]
- 45.Nguyen KT, Soong R, Lm SC, Waskell L, Ramamoorthy A, Chen Z. Probing the Spontaneous Membrane Insertion of a Tail-Anchored Membrane Protein by Sum Frequency Generation Spectroscopy. J Am Chem Soc. 2010;132:15112–15115. doi: 10.1021/ja106508f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moad AJ, Moad CW, Perry JM, Wampler RD, Goeken GS, Begue NJ, Shen T, Heiland R, Simpson GJ. NLOPredict : Visualization and Data Analysis Software for Nonlinear Optics. J Comp Chem. 2007;28:1996–2002. doi: 10.1002/jcc.20706. [DOI] [PubMed] [Google Scholar]
- 47.Manas ES, Getahun Z, Wright WW, DeGrado WF, Vanderkooi JM. Infrared Spectra of Amide Groups in α-Helical Proteins: Evidence for Hydrogen Bonding between Helices and Water. J Am Chem Soc. 2000;122:9883–9890. [Google Scholar]
- 48.Ji N, Ostroverkhov V, Chen CY, Shen RY. Phase-sensitive Sum-frequency Vibrational Spectroscopy and its Application to Studies of Interfacial Alkyl Chains. J Am Chem Soc. 2007;129:10056–10057. doi: 10.1021/ja071989t. [DOI] [PubMed] [Google Scholar]
- 49.Stiopkin IV, Jayathilake HD, Bordenyuk AN, Benderskii A. Heterodyne-Detected Vibrational Sum Frequency Generation Spectroscopy. V. J Am Chem Soc. 2008;130:2271–2275. doi: 10.1021/ja076708w. [DOI] [PubMed] [Google Scholar]
- 50.Nihonyanagi S, Yamaguchi S, Tahara T. Direct Evidence for Orientational Flip-flop of Water Molecules at Charged Interfaces: a Heterodyne-detected Vibrational Sum Frequency Generation Study. J Chem Phys. 2009;130:204704. doi: 10.1063/1.3135147. [DOI] [PubMed] [Google Scholar]
- 51.Laaser JE, Xiong W, Zanni MT. Adding a dimension to the infrared spectra of interfaces using heterodyne detected 2D sum-frequency generation (HD 2D SFG) spectroscopy. J Phys Chem B. 2011;115:2536–2546. doi: 10.1073/pnas.1115055108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Higgs PW. The Vibration Spectra of Helical Molecules: Infra-Red and Raman Selection Rules, Intensities and Approximate Frequencies. Proc R Soc Lond. 1953;220:472–485. [Google Scholar]
- 53.Moffitt W. Optical Rotatory Dispersion of Helical Polymers. J Chem Phys. 1956;25:467. [Google Scholar]
- 54.Choi JH, Hahn S, Cho M. Vibrational Spectroscopic Characteristics of Secondary Structure Polypeptides in Liquid Water: Constrained MD Simulation Studies. Int J Quantum Chem. 2005;104:616–634. doi: 10.1002/bip.20583. [DOI] [PubMed] [Google Scholar]
- 55.Mukherjee P, Kass I, Arkin IT, Zanni MT. Structural disorder of the CD3ζ transmembrane domain studied with 2D IR spectroscopy and molecular dynamics simulations. J Phys Chem B. 2006;110:24740–24749. doi: 10.1021/jp0640530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kuko1 A, Torres J, Arkin IT. A Structure for the Trimeric MHC Class II-associated Invariant Chain Transmembrane Domain. J Mol Biol. 2002;320:1109– 1117. doi: 10.1016/s0022-2836(02)00563-6. [DOI] [PubMed] [Google Scholar]
- 57.Wang L, Middleton CT, Singh S, Reddy AS, Woys AM, Strasfeld DB, Marek P, Raleigh DP, Pablo JJD, Zanni MT, Skinner JL. 2DIR Spectroscopy of Human Amylin Fibrils Reflects Stable β-Sheet Structure. J Am Chem Soc. 2011;133:16062–16071. doi: 10.1021/ja204035k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pieniazek PA, Tainter CJ, Skinner JL. Interpretation of the Water Surface Vibrational Sum-frequency Spectrum. J Chem Phys. 2011;135:044701. doi: 10.1063/1.3613623. [DOI] [PubMed] [Google Scholar]
- 59.Skinner JL, Pieniazek PA, Gruenbaum SM. Vibrational Spectroscopy of Water at Interfaces. Accounts Chem Res. 2012;45:93–100. doi: 10.1021/ar200122a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Krimm S. Infrared Spectra of High Polymers. Fortschr Hochpolym-Forsh Bd. 1960;2:241–254. [Google Scholar]
- 61.Hubner W, Mantsch HH. Orientation of Specifically 13C=O Labeled Phosphatidylcholine Multilayers from Polarized Attenuated Total Reflection FT-IR Spectroscopy. Biophys J. 1991;59:1261–1272. doi: 10.1016/S0006-3495(91)82341-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ramamoorthy A, Wei Y, Lee D-K. PISEMA Solid-State NMR Spectroscopy. Annu Rep NMR Spectrosc. 2004;52:1–52. [Google Scholar]
- 63.Flavell RR, Muir TW. Expressed Protein Ligation (EPL) in the Study of Signal Transduction, Ion Conduction, and Chromatin Biology. Acc Chem Res. 2009;42:107–116. doi: 10.1021/ar800129c. [DOI] [PubMed] [Google Scholar]
- 64.Dawson PE, Muir TW, Clark-Lewis I, Kent SB. Synthesis of Proteins by Native Chemical Ligation. Science. 1994;266:776–779. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- 65.Moran SD, Decatur SM, Zanni MT. Structural and Sequence Analysis of the Human γD-crystallin Amyloid Fibril Core Using 2D IR Spectroscopy, Segmental 13C Labeling, and Mass Spectrometry. J Am Chem Soc. 2012;134:18410–18416. doi: 10.1021/ja307898g. [DOI] [PubMed] [Google Scholar]
- 66.Wang L, Schultz PG. Expanding the Genetic Code. Angew Chem Int Ed Engl. 2004;44:34–66. doi: 10.1002/anie.200460627. [DOI] [PubMed] [Google Scholar]
- 67.Taskent-Sezgin H, Chung J, Patsalo V, Miyake-Stoner SJ, Miller AM, Brewer SH, Mehl RA, Greene DF, Raleigh DP, Carrico I. Interpretation of p-cyanophenylalanine Fluorescence in Proteins in Terms of Solvent Exposure and Contribution of Side-chain Quenchers: a Combined Fluorescence, IR and Molecular Dynamics Study. Biochemistry. 2009;48:9040–9046. doi: 10.1021/bi900938z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.