

Abstract
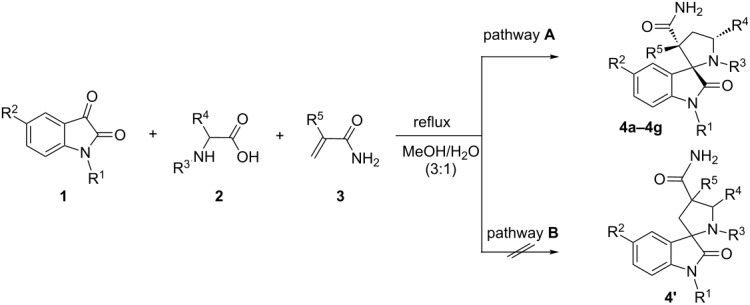
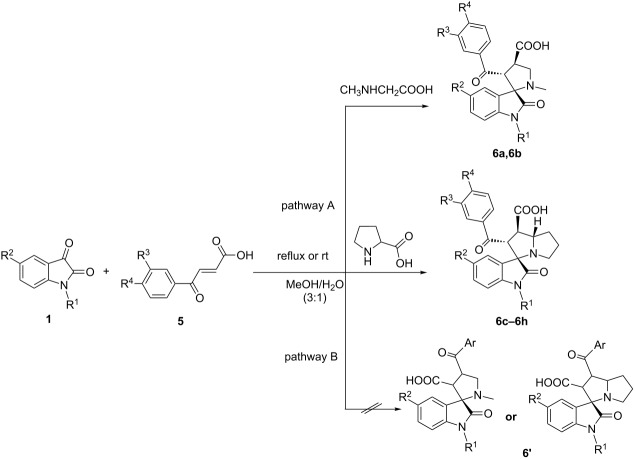
The regioselective three-component condensation of azomethine ylides derived from isatins and α-amino acids with acrylamides or aroylacrylic acids as dipolarophiles has been realized through a one-pot 1,3-dipolar cycloaddition protocol. Decarboxylation of 2'-aroyl-2-oxo-1,1',2,2',5',6',7',7a'-octahydrospiro[indole-3,3'-pyrrolizine]-1'-carboxylic acids is accompanied by cyclative rearrangement with formation of dihydropyrrolizinyl indolones.
Keywords: acrylamides, aroylacrylic acids, azomethine ylide, cyclative rearrangement, cycloaddition, multicomponent, spirooxindoles
Introduction
The design of new spirocyclic compounds is intriguing due to their unique non-planar structure and great potential for binding to biomolecules due to their inherent rigid chiral structure. The spirooxindolo pyrrolidine and pyrrolizidine frameworks form core units of many naturally occurring molecules possess significant pharmacological activities. Among them are alkaloids such as horsfiline from Horsfieldia superbа [1–3], elacomine from Elaeagnus commutatа [4], mitraphylline from Uncaria tomentosa [5] and spirotryprostatines A and B from the secondary metabolites of Aspergillus fumigatus [6–8]. In particular, oxindole derivatives are well known as powerful anti-tumor agents due to their kinase inhibitory properties, especially as tyrosine kinase inhibitors [9–10]. The multicomponent 1,3-dipolar cycloaddition of azomethine ylides, generated in situ via decarboxylative condensation of isatins and α-amino acids with olefinic and acetylenic dipolarophiles, represents a key approach for the regio- and stereoselective construction of a variety of complex spirooxindoles. Recently, this route has become significant in combinatorial chemistry due to its process simplicity, mild conditions, atomic economy and extension of the scope of substrates. A large number of focused libraries of spirooxindolo pyrrolidines and pyrrolizidines containing a wide set of natural and nonnatural α-amino acids [11–13], more than fifteen isatins [14], and 1,3-dipolarophiles, e.g. α,β-unsaturated ketones [15–17], maleimides [18–19], benzo[b]thiophene-1,1-dioxide [20], bis(arylmethylidene)acetones and -cycloalkanones [21–22], 1,4-naphthoquinone [23], arylidenemalonodinitriles [24], arylidenerhodanines [25–26], α,β-unsaturated lactones [27], nitrostyrenes [28], acrylic and propiolic esters [29], acrylonitriles [30] and arylidene-1,3-dimethylpyrimidine-2,4,6-triones [31] have been reported. However, the molecular diversity of suitable building blocks for construction of spirooxindoles is by far not exhausted with the above mentioned substances. Our interest to spirooxindoles is inspired by the search of new antidiabetic substances that might inhibit 11β-hydroxysteroid dehydrogenase type I (11β-HSD1) in metabolically relevant tissues such as liver and adipose tissue. Recent studies have demonstrated that 11β-HSD1 is a novel molecular target for treating the “metabolic syndrome” and type 2 diabetes mellitus, and that compounds inhibiting the activity of this enzyme provide promising opportunities for the development of therapeutic interventions [32–33]. Among the large class of 11β-HSD1 inhibitors there are compounds containing a pyrrolidine-2-one as a part of the spirocyclic system [34].
In the present work we report the synthesis of spirooxindolo pyrrolidines and pyrrolizidines by utilizing a 1,3-dipolar cycloaddition of hitherto uninvestigated acrylamides and aroylacrylic acids with azomethine ylides, generated in situ via decarboxylative condensation of isatins and N-substituted α-amino acids (sarcosine, proline and thiazolidine-4-carboxilic acid) in a three-component fashion.
Results and Discussion
The three-component condensation of equimolar amounts of isatins 1, α-amino acids 2 and acrylamides 3 in boiling aqueous methanol (1:3) afforded the spirooxindoles 4a–4g in moderate to excellent yields (Table 1). The reaction times largely depend on the reactivity of the employed α-amino acid. The longest reaction time (7 h) was found for sarcosine, while the fastest reaction (40 min) was found for proline as a substrate (Table 1, entries 1 and 3).
Table 1.
Three-component synthesis of spirooxindoles 4a–4g.
 | ||||||||
| entry | compound | R1 | R2 | R3 | R4 | R5 | yield (%) | time |
| 1 | 4a | H | Br | CH3 | H | H | 31 | 7 h |
| 2 | 4b | H | NO2 | CH2CH2CH2 | H | 85 | 3 h | |
| 3 | 4c | H | NO2 | CH2CH2CH2 | CH3 | 37 | 40 min | |
| 4 | 4d | H | Br | CH2SCH2 | H | 42 | 1 h | |
| 5 | 4e | H | NO2 | CH2SCH2 | H | 60 | 6 h | |
| 6 | 4f | 4-CH2C6H4Cl | H | CH2SCH2 | H | 38 | 2 h | |
| 7 | 4g | CH3 | Br | CH2CH2CH2 | CH3 | 58 | 2 h | |
The 1,3-dipolar cycloaddition of unsymmetrical dipolarophiles such as acrylamides can occur via the two pathways A and B leading to the formation of the regioisomers 4 and 4’. In our case, spirooxindol 4 is exclusively formed. All new cycloadducts obtained by the above method were characterized by mass spectrometry, 1H and 13C NMR, and elemental analyses. The regiochemical outcome of the cycloaddition was unambiguously confirmed by NOE experiments in 1H NMR as well as later by a single crystal X-ray structure analysis of the cycloadduct 4a.
The 1H NMR spectra of compounds 4b–4d have two multiplets at 4.07–3.72 ppm for 7a’-CH and 3.50–3.35 ppm for 2’-CH (compound 4b) or 6’-CH (compound 4d) and a singlet at 1.45 ppm for 2’-CCH3 of compound 4c. The relative stereochemistry of compounds 4b–4d was established by NOE cross peaks between 7a’-CH and 2’(6’-CH) and 2’-CCH3. Also, multiplets for 7a’-CH and 2’-(6’-CH) and singlet for 2’-CCH3 show correlation signals to the neighboring methylene groups. Additionally, the absence of the NOE cross peak of 4-CH of the isatin nucleus and 2’(6’-CH) or 2’-CCH3 of the pyrrolizidine moiety was indicative for the assigned relative stereochemistry. Therefore, the relative stereochemistry could be as shown in Figure 1.
Figure 1.

The NOE correlations of the signals in 1H NMR spectra of compounds 4b–4d.
The NH-proton of the oxindole moiety appeared as a singlet between 10.38–10.86 ppm. The 13C NMR spectra of compounds 4a–4g showed characteristic peaks at 71–73 ppm due to the spiro carbon nucleus.
The structure of compound 4a was determined by an X-ray diffraction study of a single crystal and supports the structure deduced from NMR spectroscopy (Figure 2).
Figure 2.

Molecular structure of spirooxindole 4a according to X-ray diffraction data.
Dipolarophiles, such as aroylacrylic acids 5, can also be successfully used in this three-component reaction. The cycloaddition of dipolarophiles 5 with non-stabilized azomethine ylides generated from isatins 1 and sarcosine/proline has led to spiropyrrolidines 6a,6b and spiropyrrolizidines 6c–6h in moderate to good yields. In this reaction also two regioisomers can be expected, but in all experiments solely the regioisomer 6 is isolated without detectable trace amounts of other isomers.
The higher reactivity of aroylacrylic acids induces remarkable rate acceleration and decreases the reaction time to only 10–15 min in a boiling mixture of methanol and water. The low to moderate yields of the target compounds 6c–6h can be explained by considerable resinification of the reaction mixture and by formation of byproducts. To suppress these negative adverse processes we carried out the reaction under stirring at room temperature. The results are shown in Table 2.
Table 2.
Synthesis of spirooxindoles 6a–6h from the three-component reaction.
 | |||||||||
| entry | compound | R1 | R2 | R3 | R4 | reflux | room temperature (rt) | ||
| yield (%) | time | yield (%) | time | ||||||
| 1 | 6a | H | Br | Cl | Cl | 50 | 6 h | – | – |
| 2 | 6b | CH3 | Br | H | Br | 34 | 8 h | – | – |
| 3 | 6c | H | H | Cl | Cl | 27 | 30 min | 67 | 12 h |
| 4 | 6d | CH3 | CH3 | Cl | Cl | 30 | 1 h | – | – |
| 5 | 6e | H | H | H | Br | 15 | 1 h | 57 | 25 min |
| 6 | 6f | H | H | H | NO2 | 74 | 10 min | 76 | 3 h |
| 7 | 6g | H | CH3 | H | NO2 | 30 | 20 min | 74 | 3 h |
| 8 | 6h | H | Br | H | NO2 | 50 | 15 min | 76 | 5 h |
All compound structures are fully supported by spectroscopic data and elemental analysis as illustrated for compound 6c. The 1H NMR spectrum of compound 6c shows a doublet at 4.66 ppm (J =11.4 Hz) for 2’-CH and two multiplets at 3.77–3.93 ppm for 7a’-CH and 3.60–3.43 ppm for 1’-CH. The stereochemistry of compound 6c was assigned by NOE cross peaks between 7a’-CH and 2’-CH and as well as the neighboring 7’-CH2 with a multiplet at 1.91–2.12 ppm. Although a week NOE correlation was found between 1'-CH and 2'-CH, the trans-configuration of the mentioned protons is predetermined by the trans-configuration of the initial aroyl acrylic acid. Also, a NOE correlation is found between signals of 2’-CH and doublet at 7.30 ppm (J =1.8 Hz) for 2-CH of the aroyl acrylic acid moiety. In addition, the absence of NOE cross peaks between 4-CH of the isatin core and 2’-CH of the pyrrolizidine fragment supports the assignment. The NH-proton of the oxindole moiety and the 1’-COOH proton of the pyrrolidine/pyrrolizidine ring give singlets at 10.25 and 12.67 ppm, respectively. Therefore, the correct stereochemistry can be drawn as shown in Figure 3. The 13C NMR spectrum of compound 6c shows a characteristic peak at 73 ppm due to the spiro carbon nucleus.
Figure 3.
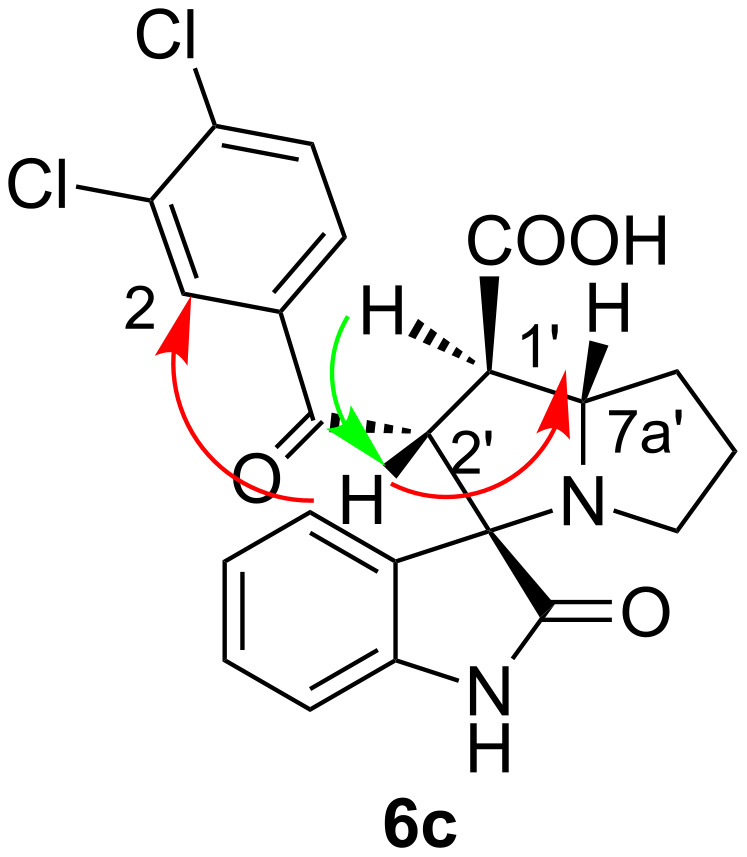
The NOE correlations of the signals in 1H NMR spectrum of compound 6c.
A single crystal X-ray study of compound 6a provided a conclusive support for the assigned structure (Figure 4). Interesting feature of this structure is a pincers-like conformation of the molecule. The substituent at the C9 atom has equatorial orientation (the N2–C7–C9–C14 torsion angle is 122.7(2)°) and its carbonyl group is almost coplanar to the C9–C10 endocyclic bond (the C10–C9–C14–O4 torsion angle is 10.2(4)°). Such an orientation of this substituent creates conditions for appearance of intramolecular stacking interactions between the aromatic rings of the indole fragment and the aryl substituent (angle between planes of aromatic rings is 22.9° and the shortest distance between carbon atoms (C6…C15) is 3.04 Å).
Figure 4.

Molecular structure of spirooxindole 6a observed in crystal phase as solvate with methanol according to X-ray diffraction data.
The mechanism of the azomethine ylide formation by a decarboxylative route has been repeatedly described by a number of authors and is depicted in Scheme 1 [35–36]. The reaction between isatin and the α-amino acid affords the azomethine ylide, which regioselectively adds to the C=C bond of acrylamide or aroylacrylic acid.
Scheme 1.

The mechanism of the regioselective synthesis of compounds 4 and 6.
Since the stereochemistry of the cycloadducts 4a and 6a was clarified by a single-crystal X-ray analysis, the structures of the reacting systems – the azomethine ylide and dipolarophiles (acrylamide and benzoylacrylic acid) – were investigated computationally. The geometrical structures of all possible conformers of the reacting systems were optimized using M06-2X [37] theory with the cc-pVTZ basis set [38] in the GAUSSIAN09 program [39]. The character of stationary points on the potential energy surface was verified by calculations of vibrational frequencies within the harmonic approximation, using analytical second derivatives at the same level of theory. All stationary points possess zero imaginary frequencies. It was found that the acrylamide conformer I was more stable than conformer II by 1.24 kcal/mol. The most stable conformation of benzoylacrylic acid possesses the benzoyl and carboxylic groups trans to each other (Figure 5).
Figure 5.

Conformations of acrylamide and benzoylacrylic acid.
The atom charges for the analysis of the Fukui function indices were calculated within the Natural Bonding Orbitals theory [40] with the NBO 5.0 program [41], that revealed the most reactive sites of the reagents. The reaction proceeds regioselectively with the addition of the most nucleophilic methylene group carbon of the azomethine ylide to the most electrophilic sites of the acrylamide and benzoylacrylic acid, which affords only one stereoisomer of cycloadducts 4 and 6 stereoselectively despite the presense of several stereocenters in the molecules (Figure 6).
Figure 6.

The Fukui function indices of acrylamide, azomethine ylide and benzoylacrylic acid.
For assigning structures of byproducts we carried out the reaction of isatins 1, aroylacrylic acids 5 and proline in a boiling mixture of EtOH and water, which resulted in the formation and isolation of compounds 7a–7c (Scheme 2). The unexpected structure of rearranged product 7a was confirmed by 1H, 13C and 2D NMR spectroscopy (Table 3).
Scheme 2.

The synthesis of compounds 7a–7c.
Table 3.
13C and 1H spectral data for compound 7a.
| entry | functional group | 13C | 1H | ||
| δ, ppm | δ, ppm | multiplicity | J, Hz | ||
| 1 | 1-NH | – | 10.59 | s | – |
| 2 | 2-CO | 178.09 | – | – | – |
| 3 | 3-CH | 45.33 | 4.56 | s | – |
| 4 | 3a-C (oxindole) | 134.06 | – | – | – |
| 5 | 4-CH (oxindole) | 127.55 | 7.05 | s | – |
| 6 | 5-C (oxindole) | 113.56 | – | – | – |
| 7 | 6-CH (oxindole) | 130.77 | 7.33 | dd | 8.1; 2.2 |
| 8 | 7-CH (oxindole) | 111.57 | 6.80 | d | 8.1 |
| 9 | 7a-C (oxindole) | 142.21 | – | – | – |
| 10 | 5-C | 124.67 | – | – | – |
| 11 | 6-C | 120.38 | – | – | – |
| 12 | 7-CH | 99.69 | 5.30 | s | – |
| 13 | 7a-C | 138.34 | – | – | – |
| 14 | 1-CH2 | 24.43 | 2.85–2.63 | m | – |
| 15 | 2-CH2 | 27.31 | 2.43–2.27 | m | – |
| 16 | 3-CH2 | 46.26 | 4.20–4.02, 3.90–3.70 | m | – |
| 17 | 1-C Ar | 129.50 | – | – | – |
| 18 | 2-CH Ar | 129.87 | 7.82 | d | 1.8 |
| 19 | 3-C Ar | 133.12 | – | – | – |
| 20 | 4-C Ar | 131.63 | – | – | – |
| 21 | 5-CH Ar | 130.95 | 7.65 | d | 8.1 |
| 22 | 6-CH Ar | 128.44 | 7.55 | dd | 8.2; 1.8 |
The main feature of the 13C spectra of compounds 7a–7c is the absence of the signal of the 3C-spiro nucleus. The 1H NMR spectrum of compound 7a displays a singlet at 5.30 ppm for the 7-CH of the dihydropyrrolizinyl moiety, which shows a H,H-NOESY correlation with a singlet at 4.56 ppm (3-CH of the oxindole ring) and HMBCs with 7a-C at 138.34 ppm. The singlet at 4.56 ppm of 3-CH of the oxindole ring shows H,H-COSY and H,H-NOESY correlations with a singlet at 7.05 ppm of 4-CH (oxindole ring) and HMBCs with 2-CO at 178.09 ppm, 4-C at 127.55 ppm and 6-C at 120.38 ppm (Figure 7). The NH proton of the oxindole ring gives a singlet at 10.59 ppm.
Figure 7.

The selected COSY, NOESY and HMBC correlations of the signals in the 1H and 13C NMR spectra of compound 7a.
The tentative mechanism for the formation of 7a is outlined in Scheme 3. First, the initially formed spiropyrrolizidine undergoes decarboxylation via ring opening of the spiro cycle. The subsequent enolization of the intermediate leads to the formation of the dihydropyrrolizinyl oxindole system.
Scheme 3.

Tentative reaction mechanism for the decarboxylative cyclative rearrangement of the initial three-component product.
Conclusion
The 1,3-dipolar cycloaddition of azomethine ylides generated in situ from isatins and sarcosine or cyclic amino acids to acrylamides or aroylacrylic acids afforded regio- and stereoselectively the spirooxindoles 4 and 6 in moderate to good yields. The selectivity of the three-component condensation of isatins and α-amino acids with aroylacrylic acids can be controlled by the reaction temperature and the reaction medium. While spiro cycloadducts can be obtained in methanol dihydropyrrolizinyl oxindoles are formed in aqueous ethanol media at higher temperatures. Therefore, reactions involving aroylacrylic acids as substrates can afford the product in a regiocontrolled manner.
Experimental
Reagents and analytics: The 1H NMR spectra were recorded on Varian Mercury VX-200 (200 MHz) and Bruker Avance DRX-500 (500 MHz) instruments in DMSO-d6 with TMS as an internal standard. The 13C NMR spectra were recorded on a Bruker Avance DRX-500 (125 MHz) and Bruker AM-300 (75 MHz) instruments in DMSO-d6 with TMS as an internal standard. The COSY, NOESY, HSQC, and HMBC spectra were recorded using the standard procedure with gradient separation of the signal. The mass spectra were recorded on a Varian 1200L GC–MS instrument, ionization by EI at 70 eV. Elemental analysis was carried out on an EA 3000 Eurovector elemental analyzer. Melting points were determined on a Kofler hot bench. The progress of reactions and also the purity of the obtained compounds were monitored by TLC on Silufol UV-254 plates with acetone/heptane (4:1) as an eluent. Commercially available reagents and solvents were used without further purification. The aroylacrylic acids 5 were prepared according to the previously reported procedure [42].
General procedure for the synthesis of spirooxindoles 4a–4g from the three-component reaction of isatins, sarcosine or cyclic α-amino acids and acrylamides: A mixture of isatin (1.0 mmol), α-amino acid (1.0 mmol) and acrylamide (1.0 mmol) in 4.0 mL aqueous methanol (1:3) was heated in an oil bath to reflux temperature for 40 min to 7 hours. The resulting precipitates were collected by filtration and washed with cold methanol to give the analytically pure products 4. 4a: colorless solid, 31%, mp 260–262 °C; 1H NMR (200 MHz, DMSO-d6) δ 10.42 (s,1H, 1-NH), 7.32 (dd, J = 8.2, 1.8 Hz, 1H, 6-CH), 7.21 (d, J = 1.8 Hz, 1Н, 4-СН), 7.11 (s, 1Н, NH-amide), 6.74 (s, 1Н, NH-amide), 6.70 (d, J = 8.1 Hz, 1Н, 7-СН), 3.11–2.99 (m, 2Н, 4’-СН2), 2.99–2.89 (m, 1Н, 3’-СН), 2.38–2.26 (m, 1Н, 5’-СН2), 2.13–1.98 (m, 1Н, 5’-СН2), 1.91 (s, 3Н, 1’-NСН3); 13C NMR (75 MHz, DMSO-d6) δ 178.03 (2-CO), 170.35 (CONH2), 142.38, 131.75, 129.77, 127.23, 112.27, 111.06, 72.26 (C-spiro), 56.51, 48.56, 34.38, 25.44; MS (m/z) (%): 325/323 (M+, 19/20), 295/292 (59/78), 280/278 (97/100), 252/250 (54/38), 131/129 (15/43), 57 (78); anal. calcd for C13H14BrN3O2 (324.17): C 48.17, H 4.35, N 12.96; found: C 48.19, H 4.40, N 12.99.
General procedure for the synthesis of spirooxindoles 6a–6h from the three-component reaction of isatins, sarcosine or proline and aroylacrylic acids: A mixture of isatin (1.0 mmol), α-amino acid (1.0 mmol) and aroylacrylic acid (1.0 mmol) in 4.0 mL aqueous methanol (1:3) was heated in an oil bath to reflux temperature for about 20 min or stirred at room temperature for 25 min to 12 hours. The resulting precipitates were collected by filtration and washed with cold methanol to give the analytically pure products 6. 6a: colorless solid, 50%, mp 240–242 °C; 1H NMR (200 MHz, DMSO-d6) δ 12.81 (s, 1H, 4’-COOH), 10.65 (s, 1H, 1-NH), 7.63 (d, J = 8.4 Hz, 1H, 5-CH (dichlorobenzoyl)), 7.44 (s, 1H, 2-CH (dichlorobenzoyl)), 7.36 (d, J = 8.4 Hz, 1H, 6-CH (dichlorobenzoyl)), 7.21 (d, J = 8.1 Hz, 1H, 6-CH), 6.96 (s, 1H, 4-CH), 6.44 (d, J = 8.4 Hz, 1H, 7-CH), 4.51 (d, J = 9.2 Hz, 1H, 3’-CH), 3.99 (q, J = 8.4 Hz, 1H, 4’-CH), 3.32–3.12 (m, 2H, 5’-CH2), 1.96 (s, 3H, 1’-NCH3); 13C NMR (125 MHz, DMSO-d6) δ 195.09 (CO-benzoyl), 177.42 (2-CO), 173.16 (4’-COOH), 141.26, 136.44, 132.08, 131.70, 130.95, 130.32, 129.02, 128.55, 128.20, 127.13, 113.55, 111.33, 72.27 (C-spiro), 56.54, 54.66, 42.96, 34.40; anal. calcd for C20H15BrСl2N2O4 (498.15): C 48.22, H 3.04, N 5.62; found: C 48.17, H 3.10, N 5.67.
General procedure for synthesis of compounds 7a–7c from the three-component reaction of isatins, proline and aroylacrylic acids: A mixture of isatin (1.0 mmol), proline (1.0 mmol) and aroylacrylic acid (1.0 mmol) in 4.0 mL aqueous ethanol (1:3) was heated in an oil bath to reflux temperature for 15 min. The resulting precipitates were collected by filtration and washed with cold ethanol to give analytically pure products 7. 7a: orange powder, 22%, mp 215–216 °C. 1H (500 MHz, DMSO-d6) and 13C NMR (125 MHz, DMSO-d6) data are given in Table 3. MS (m/z) (%): 462 (M+, 52), 435 (45), 405 (8), 353 (12), 317 (18), 289 (52), 208 (30), 173 (33), 127 (46), 75 (30), 41 (100); anal. calcd for C21H15BrСl2N2O (462.17): C 54.57; H 3.27; N 6.06; found: C 54.48; H 3.19; N 6.10.
Experimental part of X-ray diffraction study
The colourless crystals of 4a (C13H14N3O2Br) are triclinic. At 293 K, a = 6.9659(3), b = 7.5441(4), c = 13.4092(5) Å, α = 90.994(4)°, β = 90.947(4)°, γ = 106.218(5)°, V = 676.36(5) Å3, Mr = 324.18, Z = 2, space group P , dcalc = 1.592 g/сm3, μ(Mo Kα) = 3.040 mm−1, F(000) = 328. Intensities of 6411 reflections (3942 independent, Rint = 0.018) were measured on an «Xcalibur-3» diffractometer (graphite monochromated Mo Kα radiation, CCD detector, ω-scaning, 2Θmax = 60°).
, dcalc = 1.592 g/сm3, μ(Mo Kα) = 3.040 mm−1, F(000) = 328. Intensities of 6411 reflections (3942 independent, Rint = 0.018) were measured on an «Xcalibur-3» diffractometer (graphite monochromated Mo Kα radiation, CCD detector, ω-scaning, 2Θmax = 60°).
The colourless crystals of 6a (C21H19N2O5BrCl2) are triclinic. At 293 K, a = 8.8751(7), b = 10.5764(9), c = 12.352(1) Å, α = 75.858(5)°, β = 84.297(5)°, γ = 76.237(5)°, V = 1091.0(2) Å3, Mr = 530.19, Z = 2, space group P, dcalc= 1.614 g/сm3, μ(Mo Kα) = 2.165 mm−1, F(000) = 536. Intensities of 15460 reflections (3842 independent, Rint = 0.043) were measured on an «Xcalibur-3» diffractometer (graphite monochromated Mo Kα radiation, CCD detector, ω-scaning, 2Θmax = 50°).
The structures were solved by direct methods using the SHELXTL package [43]. The absorption correction was performed using the multi-scan method (Tmin = 0.582, Tmax = 0.751 for 4a and Tmin = 0.563 Tmax = 0.671 for 6a). Position of the hydrogen atoms were located from electron density difference maps and refined by “riding” model with Uiso = nUeq of the carrier atom (n = 1.5 for methyl and hydroxy groups and n = 1.2 for other hydrogen atoms). Full-matrix least-squares refinement of the structures against F2 in anisotropic approximation for non-hydrogen atoms using 3908 (4a), 3801 (6a) reflections was converged to: wR2 = 0.111 (R1 = 0.046 for 2795 reflections with F>4σ(F), S = 1.049) for structure 4a and wR2 = 0.110 (R1 = 0.043 for 2435 reflections with F>4σ(F), S = 1.055) for structure 6a. The final atomic coordinates, and crystallographic data for molecules 4a and 6a have been deposited at the Cambridge Crystallographic Data Centre, 12 Union Road, CB2 1EZ, UK (fax: +44-1223-336033; e-mail: deposit@ccdc.cam.ac.uk) and are available on request quoting the deposition numbers CCDC 964021 for 4a and CCDC 972494 for 6a).
Supporting Information
Spectroscopic and analytical data.
X-ray diffraction data description for compounds 4a and 6a.
Crystallographic information file for compound 4a.
Crystallographic information file for compound 6a.
This article is part of the Thematic Series "Multicomponent reactions II".
References
- 1.Jossang A, Jossang P, Hadi H, Sevenet A T, Bodo B. J Org Chem. 1991;56:6527–6530. doi: 10.1021/jo00023a016. [DOI] [Google Scholar]
- 2.Jones K, Wilkinson J. J Chem Soc, Chem Commun. 1992:1767–1769. doi: 10.1039/C39920001767. [DOI] [Google Scholar]
- 3.Palmisano G, Annuziata R, Papeo G, Sisti M. Tetrahedron: Asymmetry. 1996;7:1–4. doi: 10.1016/0957-4166(95)00406-8. [DOI] [Google Scholar]
- 4.James M N G, Williams G J B. Can J Chem. 1972;50:2407–2412. doi: 10.1139/v72-386. [DOI] [Google Scholar]
- 5.García Prado E, García Gimenez M D, De la Puerta Vázquez R, Espartero Sánchez J L, Saenz Rodríguez M T. Phytomedicine. 2007;14:280–284. doi: 10.1016/j.phymed.2006.12.023. [DOI] [PubMed] [Google Scholar]
- 6.Cui C-B, Kakeya H, Osada H. Tetrahedron. 1996;52:12651–12666. doi: 10.1016/0040-4020(96)00737-5. [DOI] [Google Scholar]
- 7.Sebahar P R, Williams R M. J Am Chem Soc. 2000;122:5666–5667. doi: 10.1021/ja001133n. [DOI] [Google Scholar]
- 8.Meyers C, Carreira E M. Angew Chem, Int Ed. 2003;42:694–696. doi: 10.1002/anie.200390192. [DOI] [PubMed] [Google Scholar]
- 9.Connell R D. Expert Opin Ther Pat. 2003;13:737–749. doi: 10.1517/13543776.13.6.737. [DOI] [Google Scholar]
- 10.Abadi A H, Abou-Seri S M, Abdel-Rahman D E, Klein C, Lozach O, Meijer L. Eur J Med Chem. 2006;41:296–305. doi: 10.1016/j.ejmech.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 11.Lashgari N, Ziarani G M. ARKIVOC. 2012;(i):277–320. [Google Scholar]
- 12.Gothelf K V, Jørgensen K A. Chem Rev. 1998;98:863–910. doi: 10.1021/cr970324e. [DOI] [PubMed] [Google Scholar]
- 13.Coldham I, Hufton R. Chem Rev. 2005;105:2765–2810. doi: 10.1021/cr040004c. [DOI] [PubMed] [Google Scholar]
- 14.Fokas D, Ryan W J, Casebier D S, Coffen D L. Tetrahedron Lett. 1998;39:2235–2238. doi: 10.1016/S0040-4039(98)00234-2. [DOI] [Google Scholar]
- 15.Abdel-Aziz S El-Ahl. Heteroat Chem. 2002;13:324–329. doi: 10.1002/hc.10038. [DOI] [Google Scholar]
- 16.Hazra A, Paira P, Sahu Kr B, Naskar S, Saha P, Paira R, Mondal S, Maity A, Luger P, Weber M, et al. Tetrahedron Lett. 2010;51:1585–1588. doi: 10.1016/j.tetlet.2010.01.052. [DOI] [Google Scholar]
- 17.Babu S R, Raghunathan R. Tetrahedron Lett. 2008;49:4618–4620. doi: 10.1016/j.tetlet.2008.05.089. [DOI] [Google Scholar]
- 18.Azizian J, Asadi A, Jadidi K. Synth Commun. 2001;31:2727–2733. doi: 10.1081/SCC-100105318. [DOI] [Google Scholar]
- 19.Karthikeyan K, Saranya N, Kalaivani A, Perumal P T. Synlett. 2010:2751–2754. doi: 10.1055/s-0030-1258810. [DOI] [Google Scholar]
- 20.Lakshmi N, Thirumurugan V P, Jayakumar C, Perumal P T. Synlett. 2010:955–961. doi: 10.1055/s-0029-1219550. [DOI] [Google Scholar]
- 21.Chen G, He H-P, Ding J, Hao X-J. Heterocycl Commun. 2009;15:355–360. doi: 10.1515/HC.2009.15.5.355. [DOI] [Google Scholar]
- 22.Raj A A, Raghunathan R. Tetrahedron. 2001;57:10293–10298. doi: 10.1016/S0040-4020(01)01042-0. [DOI] [Google Scholar]
- 23.Gangaru B, Yuvaraj A, Chandrasekar B, Chandrasekara S, Paramasivan P T. Eur J Med Chem. 2012;51:79–91. doi: 10.1016/j.ejmech.2012.02.024. [DOI] [PubMed] [Google Scholar]
- 24.Dandia A, Jain A K, Bhati D S. Tetrahedron Lett. 2011;52:5333–5337. doi: 10.1016/j.tetlet.2011.08.014. [DOI] [Google Scholar]
- 25.Ponnala S, Sahu D P, Kumar R, Maulik P R. J Heterocycl Chem. 2006;43:1635–1640. doi: 10.1002/jhet.5570430631. [DOI] [Google Scholar]
- 26.Shvets A A, Kurbatov S V. Chem Heterocycl Compd. 2009;45:866–867. doi: 10.1007/s10593-009-0344-1. [DOI] [Google Scholar]
- 27.Naga Siva Rao J, Raghunathan R. Tetrahedron Lett. 2012;53:854–858. doi: 10.1016/j.tetlet.2011.12.025. [DOI] [Google Scholar]
- 28.Poornachandran M, Raghunathan R. Synth Commun. 2007;37:2507–2517. doi: 10.1080/00397910701462575. [DOI] [Google Scholar]
- 29.Grigg R. Bull Soc Chim Belg. 1984;93:593–603. doi: 10.1002/bscb.19840930707. [DOI] [Google Scholar]
- 30.Faraji L, Arvinnezhad H, Alikami N, Jadidi K. Lett Org Chem. 2010;7:472–474. doi: 10.2174/157017810791824946. [DOI] [Google Scholar]
- 31.Huang Z, Zhao Q, Chen G, Wang H, Lin W, Xu L, Liu H, Wang J, Shi D, Wang Y. Molecules. 2012;17:12704–12717. doi: 10.3390/molecules171112704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lipson V V, Zamigajlo L L, Petrova O N. Ukr Bioorg Acta. 2011;9(2):3–13. [Google Scholar]
- 33.Fotsch C, Wang M. J Med Chem. 2008;51:4851–4857. doi: 10.1021/jm800369f. [DOI] [PubMed] [Google Scholar]
- 34.Yao W, He C, Zhuo J, Xu M, Zhang C, Gian D, Burns D, Metcalf B, inventors. Lactam compounds and their use as pharmaceuticals. WO2006053024. 2006 May 18;
- 35.Girgis A S, Stawinski J, Ismail N S M, Faraga H. Eur J Med Chem. 2012;47:312–322. doi: 10.1016/j.ejmech.2011.10.058. [DOI] [PubMed] [Google Scholar]
- 36.Kia Y, Osman H, Kumar R S, Murugaiyah V, Basiri A, Perumal S, Razak I A. Bioorg Med Chem Lett. 2013;23:2979–2983. doi: 10.1016/j.bmcl.2013.03.027. [DOI] [PubMed] [Google Scholar]
- 37.Zhao Y, Truhlar D G. Theor Chem Acc. 2008;120:215–241. doi: 10.1007/s00214-007-0310-x. [DOI] [Google Scholar]
- 38.Kendall R A, Dunning T H, Jr, Harrison R J. J Chem Phys. 1992;96:6796–6806. doi: 10.1063/1.462569. [DOI] [Google Scholar]
- 39.Gaussian 09. Wallingford, CT: Gaussian, Inc.; 2010. [Google Scholar]
- 40.Weinhold F. In: Encyclopedia of Computational Chemistry. Schleyer P v R, Allinger N L, Clark T, et al., editors. Chichester, UK: John Wiley & Sons; 1998. pp. 1792–1811. [Google Scholar]
- 41.Glendening, E. D., Badenhoop, J. K., Reed, A. E., Carpenter, J. E., Bohmann, J. A., Morales, C. M., Weinhold, F. NBO 5.0. Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, 2001.
- 42.Messer R, Schmitz A, Moesch L, Häner R. J Org Chem. 2004;69:8558–8560. doi: 10.1021/jo048351. [DOI] [PubMed] [Google Scholar]
- 43.Sheldrick G M. Acta Crystallogr, Sect A. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Spectroscopic and analytical data.
X-ray diffraction data description for compounds 4a and 6a.
Crystallographic information file for compound 4a.
Crystallographic information file for compound 6a.