

Abstract
Excessive glucocorticoid exposure during chronic stress causes synapse loss and learning impairment. Under normal physiological conditions, glucocorticoid activity oscillates in synchrony with the circadian rhythm. Whether and how endogenous glucocorticoid oscillations modulate synaptic plasticity and learning is unknown. Here we show that circadian glucocorticoid peaks promote postsynaptic dendritic spine formation in the mouse cortex after motor skill learning, whereas troughs are required for stabilizing newly formed spines that are important for long-term memory retention. Conversely, chronic and excessive exposure to glucocorticoids eliminates learning-associated new spines and disrupts previously acquired memories. Furthermore, we show that glucocorticoids promote rapid spine formation through a non-transcriptional mechanism by means of the LIM kinase–cofilin pathway and increase spine elimination through transcriptional mechanisms involving mineralocorticoid receptor activation. Together, these findings indicate that tightly regulated circadian glucocorticoid oscillations are important for learning-dependent synaptic formation and maintenance. They also delineate a new signaling mechanism underlying these effects.
Glucocorticoids (CORT) are hormones that respond to stress by coordinating diverse physiological processes aimed at maintaining homeostasis1,2. Many lines of evidence indicate that prolonged secretion of glucocorticoids during chronic stress disrupts learning and memory3–5, but transient release at the time of learning may promote it5–7. Notably, under normal physiological conditions, glucocorticoid activity oscillates in synchrony with circadian rhythms: diurnal peaks and troughs are closely coupled with the active and inactive phases of the circadian cycle1,2. Whether and how circadian glucocorticoid oscillations contribute to learning and memory processes is unknown.
Many lines of evidence suggest that learning-dependent remodeling of synaptic connections is important in learning and memory8–12. For example, motor learning induces the formation of persistent postsynaptic dendritic spines, and the survival of these spines is strongly correlated with behavioral performance after learning10. Recent studies indicate that glucocorticoids have rapid effects on dendritic spine development and plasticity in the mouse somatosensory cortex13. Together, these studies raise the possibility that circadian glucocorticoid oscillations may affect learning and memory by regulating synaptic formation and maintenance.
Previous studies have shown that prolonged exposure to glucocorticoids causes postsynaptic dendritic spine loss and branch atrophy in diverse cortical and subcortical areas14–18. Classically, these effects were presumed to be mediated by transcriptional mechanisms, which typically act relatively slowly by regulating gene expression19–21. Recent reports indicate that rapid, non-transcriptional mechanisms may also contribute. For example, glucocorticoids influence synaptic glutamate release and receptor trafficking through non-transcriptional mechanisms22–25 and rapidly modulate the function of inhibitory interneurons in the prefrontal cortex through non-transcriptional regulation of endocannabinoid signaling25–27. However, it remains unclear whether non-transcriptional mechanisms mediate glucocorticoid effects on learning-related synaptic plasticity.
In this study, we examined the effects of oscillating glucocorticoid activity, glucocorticoid deprivation and prolonged glucocorticoid exposure on learning-associated dendritic spine remodeling using transcranial two-photon microscopy. We found that circadian glucocorticoid oscillations were important for the formation and maintenance of new spines after learning, acting in part through a newly identified, non-transcriptional signaling pathway. Our findings indicate that oscillating levels of glucocorticoid activity balance spine formation, pruning and maintenance, whereas states of prolonged glucocorticoid exposure disrupt these processes.
Results
Circadian peaks promote spine formation after learning
Experience-dependent dendritic spine remodeling involves two distinct processes: first, learning induces spine formation, and subsequently, a fraction of new spines is selectively stabilized and a subset of pre-existing spines is pruned10,28. We began by investigating how circadian glucocorticoid oscillations might affect the process of spine formation in a rotarod motor skill-learning model10. We assessed how learning-associated spine formation varied with naturally occurring circadian peaks and troughs and with exogenous manipulations of the diurnal glucocorticoid rhythm (Fig. 1a; see Online Methods). Two-day spine formation in layer V pyramidal cells was quantified by imaging the forelimb area of motor cortex 1 d before and immediately after two sessions of motor training in transgenic mice overexpressing yellow fluorescent protein in cortical layer V pyramidal cells (YFP-H line; see Online Methods). To minimize the potential effects of training on diurnal corticoster-one rhythms, mice were first habituated to a constant, low-speed (15 r.p.m.) rotating rod for 3 d and then trained on the accelerating rod for 2 d. We validated manipulations of circulating glucocorticoids by ELISA quantification of plasma corticosterone in blood obtained from a separate cohort of mice approximately 45 min after training under identical conditions (Fig. 1b; see also Supplementary Fig. 1 for more data on effects of training and habituation).
Figure 1.

Circadian glucocorticoid peaks promote spine formation after learning. (a) Experimental procedure. Blue arrows: formed on day 2, persisting on day 7. Red arrows: formed on day 2, eliminated on day 7. White arrows: eliminated on day 2. Scale bar, 2 μm. (b) ELISA quantification of plasma corticosterone (cort) (F4,34 = 41.0, P < 0.001). *P < 0.05 versus circadian peak–trained group after Holm-Bonferroni correction. (c) Effects of circadian cort on training-induced spine formation (F6,33 = 42.2, P < 0.001). Spine formation rates represent the number of spines formed during the 2-d training period, as a percentage of the total number quantified at day 0 baseline. *P < 0.05 (corrected) versus untrained control. **P < 0.001 versus untrained control and peak-trained group. (d) Effects of circadian cort on spine survival on day 7 (F6,28 = 9.40, P < 0.001). Spine survival represents the number of spines formed during the 2-d training period and persisting on day 7, as a percentage of the total number quantified at baseline. *P < 0.05 (corrected) versus untrained control. (e) Corresponding cort effects on rotarod performance on day 7 (F5,55 = 2.84, P = 0.02). Day 7 performance is expressed as percent change in each subject's baseline performance on day 1. *P < 0.05 (corrected) versus day 1 baseline. (f) Survival of spines that formed during the training period predicted memory retention on day 7 (r = 0.86, P < 0.001). Error bars, s.e.m. See Supplementary Table 1 for statistics and details.
Training increased spine formation when it coincided with the circadian glucocorticoid peak. In contrast, training during the circadian trough had no significant effect on spine formation in habituated mice (Fig. 1c; for effects on filopodia, see Supplementary Fig. 2). Four lines of evidence indicate that elevated glucocorticoid exposure immediately after training was important for learning-related spine formation (Fig. 1c). First, a corticosterone injection (2.5 mg per kilogram body weight) immediately after training during the trough increased spine formation to a value comparable to that observed in mice that were trained during the circadian peak. Second, administration of the same corticosterone injection immediately after training during the peak further increased spine formation. Third, corticosterone specifically enhanced learning-induced spine formation and not merely formation in general: 2-d formation was significantly greater when corticosterone was administered immediately after training than when an identical dose was applied 8 h before each training session. And fourth, these effects were not merely an artifact of exogenous corticosterone treatment: suppression of endogenous glucocorticoid activity by dexamethasone (see Online Methods) was sufficient to block the formation of new spines. Together, these findings suggest that the circadian glucocorticoid peak is important physiologically for generating new spines after learning.
Previous studies have shown that long-term retention of improvements in motor performance correlates with the survival of learning-related spines10. To evaluate the long-term impact of glucocorticoids on spine formation and memory retention, we tested and reimaged the same mice on day 7, after a 5-d period without further rotarod training (Fig. 1a). Spines that formed during the 2-d training period persisted in greater numbers in the corticosterone-exposed groups (Fig. 1d), with correspondingly enhanced retention of the learned motor skill (Fig. 1e). The persistence of these training-related spines strongly predicted performance on day 7 (r = 0.86, P < 0.001; Fig. 1f and Supplementary Figs. 3 and 4). Notably, mice that were trained during the circadian trough failed to retain the learned motor skill. This lack of improvement may have been due in part to prior habituation29, as modest but significant increases in performance (P = 0.05) and spine formation (P = 0.03) were observed in a group of trough-trained mice that were not habituated (Supplementary Fig. 5). Together, these experiments show that elevated glucocorticoid secretion during the circadian peak facilitates the formation of enduring new spines after learning and enhances long-term memory retention.
Circadian glucocorticoid troughs stabilize new spines
Although the results above highlight the importance of the circadian glucocorticoid peak in experience-dependent spine formation, it is not known whether the circadian glucocorticoid oscillation influences spine maintenance. Learning-related new spines are initially highly unstable: most new spines will be pruned within days after their formation, but a subset will be selectively stabilized over time10, and most of those that survive will contain functional synapses30,31. To investigate whether glucocorticoid oscillations affect this stabilization process in the days after learning, we trained mice during the circadian peak for 2 d, administered corticosterone injections (2.5 mg kg−1 intraperitoneally (i.p.) once daily) to disrupt the circadian trough for 3 d after training and then examined spine remodeling and performance on day 7 (Fig. 2a). Disruption of the diurnal trough reduced the survival of learning-related new spines on day 7 (Fig. 2b) and impaired the rotarod performance (Fig. 2c,d). By contrast, corticosterone administered during the circadian peak did not affect spine retention or performance, indicating a specific function of the circadian trough beyond merely reducing cumulative exposure. We also found that the circadian trough was critical only during the first few days after training: disruption of the trough in the second week after learning did not affect the survival of new spines or performance on day 14 (Fig. 2b–d). These results show that a subset of newly formed spines is progressively stabilized through a process that requires intact circadian troughs. They also indicate that the impact of glucocorticoids on learning-related remodeling evolves over time: whereas elevated glucocorticoid secretion enhances spine formation immediately after training, the stabilization of these newly formed spines depends on reduced glucocorticoid secretion in the days that follow.
Figure 2.

Circadian glucocorticoid troughs preserve newly formed spines. (a) Schematic of experimental procedure. (b) Disruption of the circadian trough reduced the survival of spines that formed during the training period (F4,18 = 5.02, P = 0.007). Corticosterone (cort) that was administered during the second week (days 11–13) had no effect on spine survival, indicating that new spines require circadian troughs only during a critical period after their formation. New spine survival represents the number of spines formed during the 2-d training period and persisting on day 7, as a percentage of the total number of spines at baseline. *P < 0.05 (corrected) versus untrained control. (c) Manipulations of the circadian trough had corresponding effects on motor skill memory retention (F3,35 = 4.24, P = 0.01). Rotarod performance is depicted as a percent change in each group's baseline performance on day 1. *P < 0.05 (corrected) versus day 1 baseline. (d) Survival of spines that formed during the 2-d training period predicted retention of the learned skill on day 7 (r = 0.89, P < 0.001). Error bars, s.e.m. See Supplementary Table 2 for statistics and details.
Previous work has shown that the stabilization of a subset of new spines is associated with the pruning of a corresponding set of preexisting spines10. To better understand the impact of glucocorticoid oscillations on learning-induced synaptic pruning, we studied the elimination of spines that were established before training (Fig. 3a). As consistent with previous work10, we found that learning increased spine elimination 7 d but not 2 d after the first training session (Fig. 3b and Supplementary Fig. 6). Notably, disruption of the trough on days 4, 5 and 6 not only reduced the survival of learning-related spines (Fig. 2b) but also interfered with learning-induced pruning of preexisting spines (Fig. 3b). Spine pruning, in turn, was highly correlated with long-term memory retention on day 7 (Fig. 3c; r = 0.83, P < 0.001). Together, these results show that glucocorticoids selectively stabilize a subset of learning-related spines during the circadian trough while pruning a corresponding set of pre-existing synapses. Disruption of the circadian trough during a critical period after learning interferes with this stabilization and pruning process.
Figure 3.
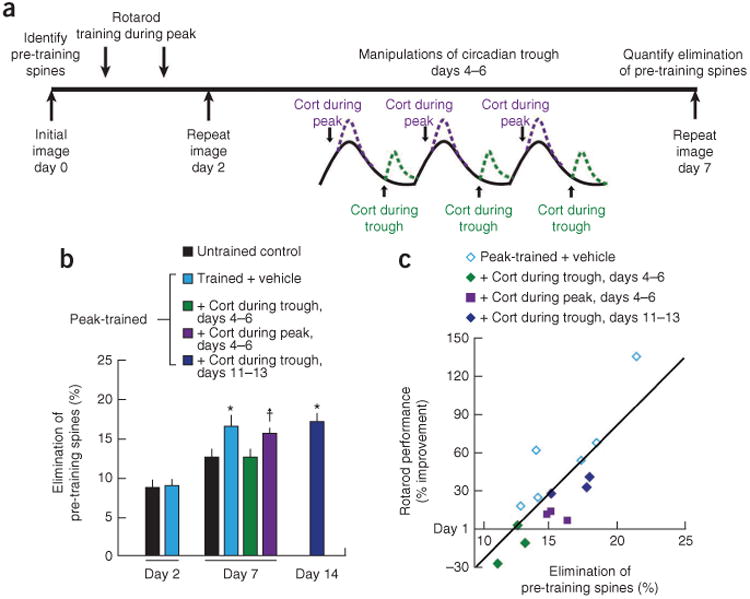
Disruption of glucocorticoid troughs reduces learning-dependent spine pruning. (a) Schematic of experimental procedure. (b) Learning caused a delayed increase in the elimination of pre-existing spines, which represents the number of spines that were present before training (day 0) and eliminated on day 2 or 7, expressed as a percentage of the total number of spines at baseline. Whereas training had no immediate impact on spine elimination on day 2 (F1,12 = 0.03, P = 0.87), spine elimination was significantly elevated on day 7 (F4,15 = 3.56, P = 0.03). Learning-induced spine pruning required an intact glucocorticoid trough 4–6 d after training. *P < 0.05 versus untrained control after Holm-Bonferroni correction. †P < 0.05 uncorrected, P < 0.10 corrected. (c) Elimination of pre-training spines was correlated with retention of the learned motor skill on day 7 (r = 0.83, P < 0.001). Cort, corticosterone. Error bars, s.e.m. See Supplementary Table 3 for statistics and details.
Chronic glucocorticoids cause spine loss and memory deficit
The results above delineate important roles for circadian glucocorticoid peaks and troughs in generating and preserving new spines after learning. On the one hand, transient disruption of the trough during a critical period after learning increased the elimination of new spines (Fig. 2b) but not pre-existing ones (Fig. 3b). On the other hand, long-term exposure to high levels of glucocorticoids has been linked to widespread spine loss and dendritic retraction in hippocampus and prefrontal cortex, as well as to learning and memory deficits3,13–18. This raises the possibility that chronic glucocorticoid excess may disrupt both learning-related spines and pre-existing synapses, even long after learning occurs. To test this, we trained mice for 2 d and, after a week of rest, administered daily, high-dose (15 mg kg−1) corticosterone injections for 10 d, beginning during the second week after training (Fig. 4a). Whereas transient disruption of the circadian trough with low-dose corticosterone (2.5 mg kg−1 i.p.) had no effect on learning-related spines during this period (Fig. 2b), prolonged and excessive glucocorticoid exposure eliminated them (Fig. 4b) and caused corresponding deficits in memory retention (Fig. 4c).
Figure 4.

Chronic glucocorticoid exposure causes spine loss and memory impairment. (a) Experimental procedure. (b) Prolonged corticosterone (cort) exposure disrupted the survival of learning-related spines that were present for at least 1 week (F1,5 = 54.2, P < 0.001). New spine survival represents the number of spines formed during the 2-d training period and persisting on day 20, expressed as a percentage of the total number of spines at baseline. (c) Prolonged cort exposure was associated with corresponding deficits in retention of the motor skill (F1,16 = 42.2, P < 0.001). (d) Elimination rates for spines present before training also increased (F1,5 = 66.6, P < 0.001). Elimination rates describe the number of spines that were present before training (day 0) and eliminated on day 20, expressed as a percentage of the total number of spines at baseline. (e) Chronic cort (F1,5 = 125.9, P < 0.001) but not transient cort or learning (F4,19 = 2.07, P = 0.13), caused significant spine loss. Net change in spine number represents the combined effects of elimination and formation on total spine number, relative to the small (4–5%) rate of net spine loss observed in untrained controls (ctrl). (f) The survival of new spines formed during training was strongly correlated with the elimination of pre-existing spines (r = 0.91, P < 0.001), but this balance was disrupted after chronic cort exposure (red). *P < 0.05 (corrected) versus vehicle-treated control. Error bars, s.e.m. See Supplementary Table 4 for statistics and details.
Notably, chronic glucocorticoid excess also increased the elimination of spines that were present before training (Fig. 4d). Consequently, chronic glucocorticoid exposure caused a substantial net loss of cortical spines (Fig. 4e). In contrast, neither transient alterations in glucocorticoid activity nor learning caused significant, long-term changes in spine number (Fig. 4e). And even though new spines formed at varying rates across all other experimental groups in this study, their long-term survival was always balanced by the pruning of a small, highly correlated quantity of pre-existing connections (Fig. 4f: r = 0.91, P < 0.001), thereby mitigating the impact of learning on total spine number. Prolonged glucocorticoid hyperactivity—a model of chronic stress—disrupted this balance, eliminating both recently formed spines and a substantial proportion of spines formed early in development.
Distinct mechanisms for formation and pruning
Our findings indicate that circadian oscillations in glucocorticoid activity are important for learning-related spine formation and pruning, but the mechanisms by which glucocorticoids regulate these processes are unclear. Classically, it was thought that glucocorticoids act relatively slowly by regulating gene expression through nuclear corticosteroid receptors, but recent reports indicate that they may also act rapidly through non-transcriptional mechanisms23–27. To better understand how glucocorticoids regulate spine remodeling, we began by investigating the time course of cortical spine formation and elimination hours after corticosterone exposure (15 mg kg−1 i.p.). Corticosterone increased both spine formation (Fig. 5a) and elimination (Fig. 5b), but effects on formation were more rapid. Spine formation was significantly increased by 1 h after injection, whereas effects on elimination were not significant until 5 h and continued to accumulate over the course of 24 h.
Figure 5.

Glucocorticoids promote spine formation and pruning through distinct signaling pathways. (a) Corticosterone (cort) increased spine formation rapidly (F1,40 = 124.0, P < 0.001). (b) Cort caused a delayed increase in spine elimination (F1,40 = 102.3, P < 0.001). (c) Glucocorticoids promote spine formation rapidly through a GR-dependent, non-transcriptional mechanism. Direct cortical application of cort caused rapid increases in spine formation (t = 14.6, P < 0.001), with comparable effects evident after cortical application of cort plus actinomycin D (50 μg ml−1; t = 18.8, P < 0.001) or a membrane-impermeant cort-BSA conjugate (t = 4.83, P < 0.001). Co-administration of the GR antagonist (antag) mifepristone (100 μM) blocked this effect. (d) Mifepristone (20 mg kg−1 i.p.) administered immediately after training reduced learning-dependent spine formation (t = 8.46, P < 0.001). (e) Glucocorticoids cause a delayed increase in spine elimination through a MR-dependent, transcriptional mechanism. A selective MR antagonist (spironolactone, 10 μM) reduced 24-h spine elimination (t = 6.19, P < 0.001), whereas a selective MR agonist (aldosterone, 10 μM) had the opposite effect (t = 18.3, P < 0.001). This effect was blocked by cotreatment with either actinomycin D or anisomycin, consistent with a transcriptional mechanism of action. (f) Spironolactone administered during the circadian trough on days 4, 5 and 6 after training interfered with learning-induced spine elimination (t = 4.84, P = 0.002). *P < 0.05 versus corresponding control. Error bars, s.e.m. See Supplementary Table 5 for statistics and details.
The relatively rapid increase in spine formation after corticosterone exposure suggests a non-transcriptional mechanism. To test this, we measured spine turnover after direct application of corticosterone to the cortex (10 μM; see also Online Methods) and after cotreatment with actinomycin D, an inhibitor of transcription. Corticosterone rapidly enhanced spine formation, but not elimination, just 20 min after exposure (Fig. 5c and Supplementary Fig. 7), whereas actinomycin D had no effect. We observed similar effects after treatment with a membrane-impermeant glucocorticoid (corticosterone–bovine serum albumin (BSA) conjugate) that cannot access the nuclear receptors that regulate transcription (Fig. 5c). Because circadian glucocorticoid oscillations cause cycles of activation and deactivation of the type II corticosteroid receptor (glucocorticoid receptor; GR)1,32, we reasoned that spine formation during the circadian peak may depend on GR activity. Indeed, we found that cotreatment with corticosterone and a GR antagonist (mifepristone) blocked the enhancement of spine formation (Fig. 5c). To evaluate the role of this GR-dependent, non-transcriptional pathway during learning, we administered mifepristone (20 mg kg−1 i.p.) immediately after training during the circadian peak. Mifepristone interfered with learning-induced spine formation (Fig. 5d). Together, these findings suggest that glucocorticoids promote learning-related spine formation rapidly through a GR-dependent, non-transcriptional pathway.
In contrast, glucocorticoid effects on spine elimination were relatively delayed and accumulated slowly over the course of 24 h (Fig. 5b), pointing to a transcriptional mechanism of action. We also found that the enhancement of spine elimination by glucocorticoids was not affected by cotreatment with mifepristone (Supplementary Fig. 8), suggesting that glucocorticoid effects on spine pruning may be mediated through the type I corticosteroid receptor (mineralocorticoid receptor; MR). To test this, we measured spine elimination rates 24 h after direct cortical application of aldosterone, a selective MR agonist, or spironolactone, a selective MR antagonist. Whereas spironolactone significantly reduced 24-h spine elimination rates, aldosterone increased spine pruning, and this effect was blocked by cotreatment with an inhibitor of transcription (actinomycin D) or translation (anisomycin; Fig. 5e). To determine whether this MR-dependent transcriptional mechanism also affects learning-related spine pruning, we used spironolactone (20 mg kg−1 i.p. daily) to block MR-activity during the trough on days 4, 5 and 6 after training. We found that spironolactone caused a significant reduction in learning-related spine pruning (Fig. 5f). These findings suggest that glucocorticoids enhance learning-related spine elimination through an MR-dependent, transcriptional mechanism that is distinct from the non-transcriptional mechanism that promotes spine formation.
Corticosterone promotes spine formation via LIMK1-cofilin
Previous studies have extensively characterized the effects of glucocorticoids on gene expression linked to synaptic plasticity and loss19–21. However, relatively few studies have investigated non-transcriptional mechanisms of action. To explore potential non-transcriptional pathways underlying glucocorticoid effects on spine formation, we obtained cortical biopsies 20–25 min after direct cortical application of corticosterone and examined changes in protein expression. We observed rapid increases in expression of the phosphorylated forms of LIM kinase-1 (LIMK1) and its substrate cofilin (Fig. 6a–c) that correlated with expression levels of phospho-GR, a marker of GR activity (Supplementary Fig. 9). Comparable effects occurred after coadministration of corticosterone and actinomycin D (Fig. 6b,c), indicating a non-transcriptional process. Phosphorylated cofilin stabilizes actin polymers and has been linked to spine growth, whereas the dephosphorylated form has opposing effects33. Thus, the effect of glucocorticoids on spine formation is likely mediated through the LIMK1-cofilin pathway.
Figure 6.

Glucocorticoids promote spine formation rapidly through non-transcriptional regulation of LIMK1-cofilin activity. (a) Expression of phospho-LIMK1 (pLIMK1; upper panels) and phospho-cofilin (pCofilin; lower panels) in cortical lysates 20 min after corticosterone (cort). (b) Cort rapidly increased the expression of phospho-LIMK1 in vivo (F4,68 = 2.74, P = 0.036). This effect was blocked by mifepristone but not by actinomycin D. (c) Similar effects of cort on pCofilin (F4,68 = 6.95, P < 0.001). (d) Cortical pyramidal cells in culture were transfected with a construct encoding GFP plus an interfering RNA (shRNA) specific for GR (lower panels) or a scrambled control construct (upper panels). Phospho-GR (pGR; left), pLIMK1 (middle) and pCofilin (right) immunofluorescence (white) and GFP expression in the same frame (green insets), which identifies pyramidal cells, are depicted after treatment with cort (1 μM) or vehicle for 20 min. White insets show 3× magnifications of boxed representative dendritic segments. Scale bar, 10 μm. (e) Cort increased dendritic and somatic pGR, pLIMK, and pCofilin immunofluorescence after transfection with a scrambled RNA construct (P < 0.001) but not the GR-specific shRNA construct (P > 0.21). A.u., arbitrary units. (f) Direct cortical application of cort (10 μM) increased spine formation within 20 min in wild-type (WT) controls, but not in Limk1 knockout mice (main effect of cort: F1,11 = 58.7, P < 0.001; interaction between genotype and cort: F1,11 = 71.1, P < 0.001). Spine formation at 24 h in vehicle-treated Limk1 knockouts was also reduced (t = 5.34, P < 0.001 versus wild-type). *P < 0.05 versus corresponding control; NS, not significant. Error bars, s.e.m. See Supplementary Table 6 for statistics and details.
To determine whether glucocorticoids mediate this effect through mechanisms that require neuronal GR signaling, we studied protein phosphorylation in a cortical culture system, transfecting neurons with a GFP–interfering RNA construct specific for GR. Whereas corticosterone increased dendritic expression of phospho-GR, phosphocofilin and phospho-LIMK1 in cells transfected with a scrambled control construct, there was no effect after transfection with the interfering RNA (Fig. 6d,e). This suggests that glucocorticoids modulate spine growth on cortical pyramidal cells through the LIMK1 and cofilin pathway and direct effects on neuronal GRs.
To determine whether glucocorticoid effects on spine formation require LIMK1, we tested the effects of direct cortical application of corticosterone (10 μM) in LIMK1 knockout (Limk1−/−) mice, which show abnormal spine morphology and learning impairments34. Limk1−/− mice are known as a model of Williams syndrome, a neurodevelopmental disorder characterized by learning disability and abnormal LIMK1 expression34,35. We found that spine formation was significantly reduced in Limk1−/− mice compared to wild-type controls, and corticosterone had no effect on spine formation in Limk1−/− (Fig. 6f). Together with the data showing the efficacy of a membrane-impermeant glucocorticoid (corticosterone-BSA) and the lack of an actinomycin D effect, these results show that the rapid effect of glucocorticoids on spine formation occurs through a non-transcriptional pathway that requires LIMK1 activity.
Discussion
Glucocorticoid secretion varies phasically with stressful environmental triggers and tonically with the circadian rhythm1,2. It is well established that excessive glucocorticoid exposure causes dendritic atrophy and spine loss, with complex effects on learning and memory3–7,14–18. Here we have identified new functions for circadian glucocorticoid oscillations in forming and consolidating stable structural correlates of learning experiences. Specifically, our findings indicate that circadian glucocorticoid peaks are important for forming new spines after learning, whereas troughs are required for stabilizing a subset of new spines during a critical period after their formation. On the one hand, learning-induced spine remodeling and memory are enhanced when learning occurs during the circadian peak or coincides with elevated glucocorticoid secretion and when subsequent glucocorticoid troughs remain intact. On the other hand, disruption of the glucocorticoid trough reduces the survival of learning-associated new spines and impairs memory retention, and prolonged, excessive exposure leads to a substantial loss of spines formed early in life. These findings suggest that loss of the glucocorticoid oscillation may contribute to learning and other cognitive deficits in neuropsychiatric disease and chronic stress states1,2,36. Furthermore, alterations of the glucocorticoid milieu may contribute to pathophysiology in many immunological and neurological diseases, for which long-term maintenance on synthetic glucocorticoids and related receptor modulators are often mainstays of treatment.
Our findings also underscore the importance of the timing of training relative to the endogenous glucocorticoid rhythm for optimal synaptic remodeling and maintenance. This effect may account in part for the observation that motor skill learning in humans varies with the time of day37,38. Studies in both rodents and humans have identified circadian variation in other forms of cognition, including working memory, associative learning, declarative memory and fear conditioning39–41. Future work will be required to determine whether and how circadian glucocorticoid oscillations regulate spine remodeling in these contexts.
Our results define a new, non-transcriptional pathway by which glucocorticoids promote spine formation after learning. Specifically, they indicate that GR-dependent, non-transcriptional regulation of LIMK1 and cofilin is critical in generating new spines. These signaling mechanisms may generate new spines through direct effects on the dendritic cytoskeleton, by modulating neuronal network activity, or by some combination of the two. Previous studies indicate that spine formation can occur within minutes in brain slice cultures42. Thus, the non-transcriptional mechanisms described here could facilitate the process of new spine formation after learning. In addition, deficits in linking this pathway to experience-dependent plasticity may contribute to spine abnormalities and learning disability in Williams syndrome, a genetic neurodevelopmental disorder caused by the deletion of LIMK1 and other genes on the long arm of chromosome 7 (refs. 34,35). The LIMK1 and cofilin signaling pathway may likewise contribute to cortical spine abnormalities in stress-related neuropsychiatric diseases. Future imaging studies in awake animals will be required to characterize the time course of spine formation minutes to hours after training, to better understand the action of this non-transcriptional pathway in learning-dependent spine formation under normal and disease conditions.
Our results also show that glucocorticoids promote spine elimination through MR-dependent transcriptional mechanisms, which regulate the expression of dozens of genes linked to synaptic plasticity20. On the one hand, spine pruning may serve to compensate for the addition of new synapses after training. On the other hand, it may also contribute actively to learning by fine-tuning connections in neuronal circuits, as has been demonstrated in the developing nervous system and in other forms of behavioral learning12,43,44. Future studies are needed to elucidate the function of spine elimination in learning and the involvement of other mechanisms in this process. For example, histone deacetylases are sensitive to stress45,46 and are established regulators of synapse number47. They may influence synapse loss in part through interactions with mitogen enhancer factor 2, leading to transcription of Arc, Syngap1 and other genes implicated in synaptic plasticity48,49. Furthermore, glucocorticoid secretion exhibits ultradian oscillations that are superimposed on the circadian rhythm, leading to high-frequency fluctuations in transcription that may interact with circadian rhythms to regulate both spine formation and elimination21,50. Amid these complexities, our findings highlight a central, glucocorticoid-regulated pathway that is important for remodeling excitatory synapses in the cortex after learning. Future use of genetic approaches to activate and inactivate MR and GR pathways rapidly in specific cell types will aid in further dissecting the underlying mechanisms.
Methods
Methods and any associated references are available in the online version of the paper.
Supplementary Material
Acknowledgments
We thank B.S. McEwen and Gan laboratory members for critical comments on this manuscript. This work was supported by grants from the US National Institutes of Health (R01 NS047325 and a subcontract of R01MH085324) to W.-B.G. C.L. is supported by grants from the US National Institute of Mental Health (K99 MH097822) and the DeWitt Wallace Reader's Digest Foundation at Weill Cornell Medical College. F.J. and M.V.C. are supported by grants from the US National Institutes of Health (MH086651 and NS21072).
Footnotes
Author Contributions: C.L. and W.-B.G. conceived the project and designed all experiments. C.L. collected, quantified and analyzed the in vivo spine remodeling data. J.M.C. and F.J. contributed to the design of the cortical culture experiments, and J.M.C. carried them out and analyzed the results. C.L. obtained cortical biopsies and plasma samples. F.J. and M.V.C. contributed to the biochemical analysis of the samples. Z.J. developed and shared the LIMK1 knockout mouse. C.L. and W.-B.G. wrote the manuscript.
Competing Financial Interests: The authors declare no competing financial interests.
Note: Supplementary information is available in the online version of the paper.
References
- 1.de Kloet ER, Vreugdenhil E, Oitzl MS, Jöels M. Brain corticosteroid receptor balance in health and disease. Endocr Rev. 1998;19:269–301. doi: 10.1210/edrv.19.3.0331. [DOI] [PubMed] [Google Scholar]
- 2.McEwen BS. Protective and damaging effects of stress mediators. N Engl J Med. 1998;338:171–179. doi: 10.1056/NEJM199801153380307. [DOI] [PubMed] [Google Scholar]
- 3.Lupien SJ, et al. Cortisol levels during human aging predict hippocampal atrophy and memory defcits. Nat Neurosci. 1998;1:69–73. doi: 10.1038/271. [DOI] [PubMed] [Google Scholar]
- 4.de Quervain DJF, Roozendaal B, McGaugh JL. Stress and glucocorticoids impair retrieval of long-term spatial memory. Nature. 1998;394:787–790. doi: 10.1038/29542. [DOI] [PubMed] [Google Scholar]
- 5.Bangasser DA, Shors TJ. The hippocampus is necessary for enhancements and impairments of learning following stress. Nat Neurosci. 2007;10:1401–1403. doi: 10.1038/nn1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shors TJ, Weiss C, Thompson RF. Stress-induced facilitation of classical conditioning. Science. 1992;257:537–539. doi: 10.1126/science.1636089. [DOI] [PubMed] [Google Scholar]
- 7.Quirarte GL, Roozendaal B, McGaugh JL. Glucocorticoid enhancement of memory storage involves noradrenergic activation in the basolateral amygdala. Proc Natl Acad Sci USA. 1997;94:14048–14053. doi: 10.1073/pnas.94.25.14048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bailey CH, Kandel ER. Structural changes accompanying memory storage. Annu Rev Physiol. 1993;55:397–426. doi: 10.1146/annurev.ph.55.030193.002145. [DOI] [PubMed] [Google Scholar]
- 9.Engert F, Bonhoeffer T. Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature. 1999;399:66–70. doi: 10.1038/19978. [DOI] [PubMed] [Google Scholar]
- 10.Yang G, Pan F, Gan WB. Stably maintained dendritic spines are associated with lifelong memories. Nature. 2009;462:920–924. doi: 10.1038/nature08577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu TH, et al. Rapid formation and selective stabilization of synapses for enduring motor memories. Nature. 2009;462:915–919. doi: 10.1038/nature08389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lai CSW, Franke TF, Gan WB. Opposite effects of fear conditioning and extinction on dendritic spine remodelling. Nature. 2012;483:87–91. doi: 10.1038/nature10792. [DOI] [PubMed] [Google Scholar]
- 13.Liston C, Gan WB. Glucocorticoids are critical regulators of dendritic spine development and plasticity in vivo. Proc Natl Acad Sci USA. 2011;108:16074–16079. doi: 10.1073/pnas.1110444108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wellman CL. Dendritic reorganization in pyramidal neurons in medial prefrontal cortex after chronic corticosterone administration. J Neurobiol. 2001;49:245–253. doi: 10.1002/neu.1079. [DOI] [PubMed] [Google Scholar]
- 15.Radley JJ, et al. Repeated stress induces dendritic spine loss in the rat medial prefrontal cortex. Cereb Cortex. 2006;16:313–320. doi: 10.1093/cercor/bhi104. [DOI] [PubMed] [Google Scholar]
- 16.Liston C, et al. Stress-induced alterations in prefrontal cortical dendritic morphology predict selective impairments in perceptual attentional set-shifting. J Neurosci. 2006;26:7870–7874. doi: 10.1523/JNEUROSCI.1184-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dias-Ferreira E, et al. Chronic stress causes frontostriatal reorganization and affects decision-making. Science. 2009;325:621–625. doi: 10.1126/science.1171203. [DOI] [PubMed] [Google Scholar]
- 18.Watanabe Y, Gould E, McEwen BS. Stress induces atrophy of apical dendrites of hippocampal CA3 pyramidal neurons. Brain Res. 1992;588:341–345. doi: 10.1016/0006-8993(92)91597-8. [DOI] [PubMed] [Google Scholar]
- 19.Morsink MC, et al. Acute activation of hippocampal glucocorticoid receptors results in different waves of gene expression throughout time. J Neuroendocrinol. 2006;18:239–252. doi: 10.1111/j.1365-2826.2006.01413.x. [DOI] [PubMed] [Google Scholar]
- 20.Datson NA, van der Perk J, De Kloet ER, Vreugdenhil E. Identifcation of corticosteroid-responsive genes in rat hippocampus using serial analysis of gene expression. Eur J Neurosci. 2001;14:675–689. doi: 10.1046/j.0953-816x.2001.01685.x. [DOI] [PubMed] [Google Scholar]
- 21.Stavreva DA, et al. Ultradian hormone stimulation induces glucocorticoid receptormediated pulses of gene transcription. Nat Cell Biol. 2009;11:1093–1102. doi: 10.1038/ncb1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yuen EY, et al. Acute stress enhances glutamatergic transmission in prefrontal cortex and facilitates working memory. Proc Natl Acad Sci USA. 2009;106:14075–14079. doi: 10.1073/pnas.0906791106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karst H, et al. Mineralocorticoid receptors are indispensable for nongenomic modulation of hippocampal glutamate transmission by corticosterone. Proc Natl Acad Sci USA. 2005;102:19204–19207. doi: 10.1073/pnas.0507572102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lösel R, Wehling M. Nongenomic actions of steroid hormones. Nat Rev Mol Cell Biol. 2003;4:46–56. doi: 10.1038/nrm1009. [DOI] [PubMed] [Google Scholar]
- 25.Popoli M, Yan Z, McEwen BS, Sanacora G. The stressed synapse: the impact of stress and glucocorticoids on glutamate transmission. Nat Rev Neurosci. 2011;13:22–37. doi: 10.1038/nrn3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Di S, Malcher-Lopes R, Halmos KC, Tasker JG. Nongenomic glucocorticoid inhibition via endocannabinoid release in the hypothalamus: a fast feedback mechanism. J Neurosci. 2003;23:4850–4857. doi: 10.1523/JNEUROSCI.23-12-04850.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hill MN, et al. Recruitment of prefrontal cortical endocannabinoid signaling by glucocorticoids contributes to termination of the stress response. J Neurosci. 2011;31:10506–10515. doi: 10.1523/JNEUROSCI.0496-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hübener M, Bonhoeffer T. Searching for engrams. Neuron. 2010;67:363–371. doi: 10.1016/j.neuron.2010.06.033. [DOI] [PubMed] [Google Scholar]
- 29.Roozendaal B, Okuda S, Van der Zee EA, McGaugh JL. Glucocorticoid enhancement of memory requires arousal-induced noradrenergic activation in the basolateral amygdala. Proc Natl Acad Sci USA. 2006;103:6741–6746. doi: 10.1073/pnas.0601874103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lohmann C, Bonhoeffer T. A role for local calcium signaling in rapid synaptic partner selection by dendritic flopodia. Neuron. 2008;59:253–260. doi: 10.1016/j.neuron.2008.05.025. [DOI] [PubMed] [Google Scholar]
- 31.Knott GW, Holtmaat A, Wilbrecht L, Welker E, Svoboda K. Spine growth precedes synapse formation in the adult neocortex in vivo. Nat Neurosci. 2006;9:1117–1124. doi: 10.1038/nn1747. [DOI] [PubMed] [Google Scholar]
- 32.Reul JMHM, de Kloet ER. Two receptor systems for corticosterone in rat brain: microdistribution and differential occupation. Endocrinology. 1985;117:2505–2511. doi: 10.1210/endo-117-6-2505. [DOI] [PubMed] [Google Scholar]
- 33.Gu J, et al. ADF/coflin-mediated actin dynamics regulate AMPA receptor traffcking during synaptic plasticity. Nat Neurosci. 2010;13:1208–1215. doi: 10.1038/nn.2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meng Y, et al. Abnormal spine morphology and enhanced LTP in LIMK-1 knockout mice. Neuron. 2002;35:121–133. doi: 10.1016/s0896-6273(02)00758-4. [DOI] [PubMed] [Google Scholar]
- 35.Bellugi U, Lichtenberger L, Mills D, Galaburda A, Korenberg JR. Bridging cognition, the brain and molecular genetics: evidence from Williams syndrome. Trends Neurosci. 1999;22:197–207. doi: 10.1016/s0166-2236(99)01397-1. [DOI] [PubMed] [Google Scholar]
- 36.Holsboer F. The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology. 2000;23:477–501. doi: 10.1016/S0893-133X(00)00159-7. [DOI] [PubMed] [Google Scholar]
- 37.Miller NL, Tvaryanas AP, Shattuck LG. Accommodating adolescent sleepwake patterns: the effects of shifting the timing of sleep on training effectiveness. Sleep. 2012;35:1123–1136. doi: 10.5665/sleep.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Atkinson G, Reilly T. Circadian variation in sports performance. Sports Med. 1996;21:292–312. doi: 10.2165/00007256-199621040-00005. [DOI] [PubMed] [Google Scholar]
- 39.Ruby NF, et al. Hippocampal-dependent learning requires a functional circadian system. Proc Natl Acad Sci USA. 2008;105:15593–15598. doi: 10.1073/pnas.0808259105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wright KP, Hull JT, Hughes RJ, Ronda JM, Czeisler CA. Sleep and wakefulness out of phase with internal biological time impairs learning in humans. J Cogn Neurosci. 2006;18:508–521. doi: 10.1162/jocn.2006.18.4.508. [DOI] [PubMed] [Google Scholar]
- 41.Chaudhury D, Colwell CS. Circadian modulation of learning and memory in fear-conditioned mice. Behav Brain Res. 2002;133:95–108. doi: 10.1016/s0166-4328(01)00471-5. [DOI] [PubMed] [Google Scholar]
- 42.Dailey ME, Smith SJ. The dynamics of dendritic structure in developing hippocampal slices. J Neurosci. 1996;16:2983–2994. doi: 10.1523/JNEUROSCI.16-09-02983.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Buffelli M, et al. Genetic evidence that relative synaptic effcacy biases the outcome of synaptic competition. Nature. 2003;424:430–434. doi: 10.1038/nature01844. [DOI] [PubMed] [Google Scholar]
- 44.Colman H, Nabekura J, Lichtman JW. Alterations in synaptic strength preceding axon withdrawal. Science. 1997;275:356–361. doi: 10.1126/science.275.5298.356. [DOI] [PubMed] [Google Scholar]
- 45.Tsankova NM, et al. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci. 2006;9:519–525. doi: 10.1038/nn1659. [DOI] [PubMed] [Google Scholar]
- 46.Renthal W, et al. Histone deacetylase 5 epigenetically controls behavioral adaptations to chronic emotional stimuli. Neuron. 2007;56:517–529. doi: 10.1016/j.neuron.2007.09.032. [DOI] [PubMed] [Google Scholar]
- 47.Guan JS, et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459:55–60. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Flavell SW, Greenberg ME. Signaling mechanisms linking neuronal activity to gene expression and plasticity of the nervous system. Annu Rev Neurosci. 2008;31:563–590. doi: 10.1146/annurev.neuro.31.060407.125631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Flavell SW, et al. Activity-dependent regulation of MEF2 transcription factors suppresses excitatory synapse number. Science. 2006;311:1008–1012. doi: 10.1126/science.1122511. [DOI] [PubMed] [Google Scholar]
- 50.McNally JG, Muller WG, Walker D, Wolford R, Hager GL. The glucocorticoid receptor: rapid exchange with regulatory sites in living cells. Science. 2000;287:1262–1265. doi: 10.1126/science.287.5456.1262. [DOI] [PubMed] [Google Scholar]
- 51.Feng G, et al. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- 52.Yang G, Pan F, Parkhurst CN, Grutzendler J, Gan WB. Thinned-skull cranial window technique for long-term imaging of the cortex in live mice. Nat Protoc. 2010;5:201–208. doi: 10.1038/nprot.2009.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li CX, Waters RS. Organization of the mouse motor cortex studied by retrograde tracing and intracortical microstimulation (ICMS) mapping. Can J Neurol Sci. 1991;18:28–38. doi: 10.1017/s0317167100031267. [DOI] [PubMed] [Google Scholar]
- 54.Campolongo P, et al. Endocannabinoids in the rat basolateral amygdala enhance memory consolidation and enable glucocorticoid modulation of memory. Proc Natl Acad Sci USA. 2009;106:4888–4893. doi: 10.1073/pnas.0900835106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Karssen AM, Meijer OC, Berry A, Pinol RS, de Kloet ER. Low doses of dexamethasone can produce a hypocorticosteroid state in the brain. Endocrinology. 2005;146:5587–5595. doi: 10.1210/en.2005-0501. [DOI] [PubMed] [Google Scholar]
- 56.Binder DK, Papadopoulos MC, Haggie PM, Verkman AS. In vivo measurement of brain extracellular space diffusion by cortical surface photobleaching. J Neurosci. 2004;24:8049–8056. doi: 10.1523/JNEUROSCI.2294-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jeanneteau F, Garabedian MJ, Chao MV. Activation of Trk neurotrophin receptors by glucocorticoids provides a neuroprotective effect. Proc Natl Acad Sci USA. 2008;105:4862–4867. doi: 10.1073/pnas.0709102105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Buitrago MM, Schulz JB, Dichgans J, Luft AR. Short and long-term motor skill learning in an accelerated rotarod training paradigm. Neurobiol Learn Mem. 2004;81:211–216. doi: 10.1016/j.nlm.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 59.Jeanneteau F, Deinhardt K, Miyoshi G, Bennett AM, Chao MV. The MAP kinase phosphatase MKP-1 regulates BDNF-induced axon branching. Nat Neurosci. 2010;13:1373–1379. doi: 10.1038/nn.2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jiang M, Chen G. High Ca2+-phosphate transfection effciency in low-density neuronal cultures. Nat Protoc. 2006;1:695–700. doi: 10.1038/nprot.2006.86. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.