

Abstract
The increasing availability and use of predictive models to facilitate informed decision making highlights the need for careful assessment of the validity of these models. In particular, models involving biomarkers require careful validation for two reasons: issues with overfitting when complex models involve a large number of biomarkers, and inter-laboratory variation in assays used to measure biomarkers. In this paper we distinguish between internal and external statistical validation. Internal validation, involving training-testing splits of the available data or cross-validation, is a necessary component of the model building process and can provide valid assessments of model performance. External validation consists of assessing model performance on one or more datasets collected by different investigators from different institutions. External validation is a more rigorous procedure necessary for evaluating whether the predictive model will generalize to populations other than the one on which it was developed. We stress the need for an external dataset to be truly external, that is, to play no role in model development and ideally be completely unavailable to the researchers building the model. In addition to reviewing different types of validation, we describe different types and features of predictive models and strategies for model building, as well as measures appropriate for assessing their performance in the context of validation. No single measure can characterize the different components of the prediction, and the use of multiple summary measures is recommended.
Introduction
Risk prediction tools combining biomarkers with other risk factors are increasingly being proposed for a variety of purposes in the management of individual cancer patients. Many of these tools are available as user-friendly calculators on the internet and easily found and accessed by patients. They facilitate informed decision making between doctor and patient on whether or not to pursue more invasive diagnostic testing, likelihood of progression of disease or outcome to specific therapies. Some examples include the Prostate Cancer Prevention Trial (PCPT) calculator for predicting the probability of prostate cancer (1), the Gail model which gives the lifetime risk of breast cancer (2), the many nomograms for predicting disease progression in prostate cancer (3), and Oncotype DX for predicting risk of recurrence in breast cancer based on gene expression data from 21 genes (4). Models which use serial measurements of biomarkers include ROCA for predicting ovarian cancer based on serial measurements of CA125 (5), and a web based calculator for predicting prostate cancer recurrence using serial PSA values following radiation therapy, available at psacalc.sph.umich.edu. Combined with the recent explosion of new high dimensional panels of markers, including those proposed by genomic, proteomic and other -omic fields, the uncontrolled proposal and mass posting of risk prediction tools raises deep concerns whether these tools have in fact been suitably validated on external populations of the type intended for use. Indeed there is an “emerging breed of forensic statisticians” (6) debunking the claims of some of these tools; recent analyses of a few high-profile risk prediction tools have demonstrated lack of reproducibility (7, 8). This article educates and/or reminds practitioners of the essential ingredients for proper validation of cancer risk prediction tools, summarized in Table 1. Throughout we use methods and results from the PCPT and a lung cancer gene expression study predicting survival time from gene expression data (9) to illustrate steps in the validation process.
Table 1.
Components of biomarker-based model validation
|
What is a Model?
In this article we do not distinguish between prognostic and predictive models (10), terminology that is becoming familiar to oncologists and is described in more detail in the articles by George (11) and Simon (12) in this issue. Prognostic models evaluate general risk whereas predictive models assess who may respond to a certain therapy. In statistical terms the distinction is that predictive models include an interaction between biomarker and treatment. Despite this distinction, the general principles behind validation of both types of models remain the same.
There is vast statistical literature on how to build predictive models (e.g. 13, 14). Multivariable models allowing simultaneous association of biomarkers and predictors with clinical outcome, such as logistic regression for presence/absence of disease or Cox regression with survival, are common building blocks of biomarker-based risk prediction tools. More algorithmic statistical models, such as support vector machines or neural networks, can also be used.
Some models have a simple structure, combining information from a few variables in a transparent way. At the opposite end of the spectrum are complex “black box” models, e.g. a web-based calculator with a large number of input variables. Some models give a simple prediction such as high versus low risk, more refined models give a continuous score; even more desirable is when the continuous score is on an interpretable scale such as a probability of recurrence. The PCPT calculator uses a multivariable logistic regression model and four variables (PSA, digital rectal examination, history of prostate cancer in a first-degree relative, history of a prior negative biopsy with prostate cancer status on biopsy) to provide an estimated risk of prostate cancer ranging from 0 to 100%. Other examples of simple models are the nomogram models of Kattan et al. (3) that provide an easily interpretable prediction. The graphical approach of the nomogram explicitly shows how three clinical measures (PSA, clinical stage, and Gleason Score) are scored and summed to yield a predicted 5-year cancer free survival probability. An example of a more complex model comes from the lung cancer gene expression study where expression values from 13,838 genes are combined in a weighted linear sum, with the weights for each gene estimated using a penalized Cox model. Prediction is a continuous score, with higher scores indicating increased hazard of death, but the actual score does not have an easy interpretation (i.e. it is not a probability).
However appealing the idea of transparency, simplicity of the model is not necessarily a virtue; performance of the model is more important, and simplicity over complexity should not be the primary consideration in the model building process. Proper validation of a reasonable model is more important than the pursuit of an ideal model, which may never be found in any case. In the lung cancer gene expression study the chosen model (using 13,838 genes) had slightly better performance in validation datasets than models based on significantly reduced numbers of genes, and the model based on the single best gene performed very poorly. However, one benefit to simple models is a logistical one; there may be reduced expense and more focused quality control if the number of biomarkers in the model is small.
In building a model, there is sometimes the perception that newer technologies, such as neural networks, offer a better approach. While such models are certainly more complex, involving combinations of interactions and non-linearities, there is also more danger of over-fitting and illogical resulting models (15). Whether a model fits better will depend on the context and the available data. In a review of 28 studies directly comparing neural networks to traditional regression modeling, Sargent et al. (16) found that neural networking was not a better approach in more than half the studies. Whatever method is proposed it is imperative that sufficient details are provided to both understand how it was developed and so that others could implement it themselves.
Models Involving Biomarkers
Models must be built based on good statistical principles, and to this end the resulting model should fit the data used to develop it. However, if the predictive model involves a panel of biomarkers then the problem associated with building models with a high number of variables can arise, in which the model overfits the data. Often there is little a priori knowledge about which biomarkers to include in the model and the temptation is to throw them all into an automated procedure such as stepwise regression. This often leads to the model capturing not only real patterns but also idiosyncratic features of the particular dataset, resulting in poor performance in external validation. Better methods for handling datasets with large numbers of variables exist, including penalized regression methods such as ridge regression and LASSO (14). These can be very useful with large numbers of input variables and can provide more reliable results and help avoid overfitting. However, even with more advanced statistical methods for model building validation of a prognostic model is necessary to separate true associations from noise.
In addition to using caution to avoid over-fitting, models that include biomarker data can also suffer from low power because of misunderstanding about what drives the power to detect significant effects. Sample size requirements for validating a new biomarker are demanding and even more demanding for showing it to be beneficial above and beyond existing markers. It is not the number of measurements per subject that drives power (e.g. number of genes measured) but the number of subjects. Obtaining tons of measures on a small set of patients will likely not yield significant results, especially for predictive models associated with a specific therapy as defined above. Such models by definition include interaction terms and thus large numbers of subjects are required to find any significant interaction effects with treatment.
In our opinion there is too much emphasis on the specific variables that are included in a prediction model. For example for Oncotype DX or the lung cancer gene expression study one may ask, “Why these genes?” or, “Why these weights?” In situations where there are a lot of biomarkers to choose from it will frequently be the case that many subsets of variables give very similar predictions, thus too much sanctity should not be placed on the specific model. It is not at all surprising in gene expression studies that models built from different datasets for the same goal have contained very different sets of genes (17).
Models involving biomarkers as input variables raise unique issues due to technological advances and assay inconsistencies, discussed in the article by Owzar et al. in this issue (18), Hammond et al. (19) and the REMARK guidelines (20). Assays to measure biomarkers evolve over time; thus measurements from a new assay cannot be substituted into a model built using measures from an earlier assay unless the two assays are highly correlated. If correlation is weak then the performance of the predictive model will likely be worse even if the new assay provides more accurate measurement. Even when changes in technology do not interfere, any number of factors may cause an assay to be systematically higher or lower or more variable in one lab compared to another.
One caveat in prognostic model building is that small p-values can be misleading. An often disappointing result of multivariable risk models combining new biomarkers with established ones is that the proposed biomarkers may appear statistically significant in a multivariable model, i.e. have small p-values, but then may not increase the prognostic ability of the model as a whole. It can be demonstrated statistically that in most cases encountered in clinical prediction very high odds ratios or hazard ratios are required to have an impact on measures of predictive ability (21). For a binary outcome (e.g. diseased/not diseased) Pepe et al. (22) show that a biomarker with strong predictive ability that correctly classifies 80% of diseased subjects and only misclassifies 10% of non-diseased subjects would yield an odds ratio of 36.0 – well above odds ratios commonly encountered in practice.
What is Validation?
In biomarker research validation means different things to different people. In this issue, Chau et al. (23) discuss analytical validation of a biomarker assay itself and describe a “fit-for-purpose” approach in which all aspects of validation are incorporated. This article concerns a different form of validation: validation of statistical models based on biomarkers. Altman and Royston (24) distinguish between two types of validated models: statistically validated and clinically validated. While both types involve evaluation of model performance, statistical validation focuses on aspects such as goodness-of-fit whereas clinical validation places the prediction in context to evaluate performance (i.e. is the prediction accurate enough for its purpose) and might also involve considerations of costs (25). In this paper we focus on statistical validation.
Furthermore, we draw distinction between validating population versus patient-level predictions. For example, the Gail model did very well overall in predicting how many breast cancers would occur (observed/expected ratio 1.03), and in general prognostic classification schemes, for example using stage, do reasonably well at this. However, individual predictions are more difficult and variable, consequently influencing utility (26). Our focus will be on validating models at individual level predictions.
Validation requires a comparison of model prediction with a true outcome which must be ascertainable or observable in the future. For example, the true outcome (“gold standard”) might be whether the patient has cancer, requiring an invasive procedure to ascertain, or that a cancer will eventually recur, requiring often lengthy follow-up to observe. In the PCPT the true outcome was prostate cancer status obtained via biopsy. In the lung cancer gene expression study the true outcome was survival time. Some outcomes may not be ascertainable and are thus not suited for validation. For example, Sorlie et al. (27) used gene expression data to cluster breast cancer patients into subtypes; the true group membership of subjects in a subtype is not ascertainable and so this outcome cannot be validated. Validation also requires that the input data, prediction, and the truth all be available for the subjects. Furthermore, the size of the validation dataset will be crucial to the ability to validate a model, with larger datasets preferable.
Internal Validation
In general there are two forms of validation. The first, internal validation, is performed in the context of an individual study, for example by splitting the study dataset into one dataset to train or build the model (training set) and one dataset to test performance (test set, also called the validation set). The appealing feature of internal validation is its convenience, as it does not require collection of data beyond the original study. The considerable disadvantage of the training-testing split is reduced efficiency resulting from using only a fraction of the data in model building. Results may depend on the particular split of the data, thus more sophisticated methods than simply splitting the data into training and testing datasets may be preferred.
For internal validation to be a valid procedure the testing dataset must be completely untouched and no aspect of testing data may play a role in model development. Even seemingly innocuous uses of testing data in model building can invalidate the internal validation attempts. For example, Potti et al. (28) employed separate training and testing gene expression datasets for predicting response to chemotherapy. However, the authors used the combined data to create gene clusters and used these clusters to aid in model building using training data. Even though outcomes from the testing data were not used in model building, their strategy allowed information to “leak” from the testing data and invalidated the assessment of the model on the testing data (29). An example of strict adherence to the training/testing split is the use of an “honest broker” to provide the model builder(s) only the required subset of data at each step of validation. The lung cancer gene expression study utilized this approach, with the broker first providing data from the training set. After a model was chosen he provided the gene expression data for the testing datasets, and only after individual predictions were made for each subject was the outcome data for the testing datasets’ subjects made available.
An extension of the simple training-testing split is to split the data into training and test groups a large number of times. Commonly used examples of this approach include leave-one-out, k-fold and repeated random-split cross-validation. With leave-one-out cross-validation each observation in turn serves as the test set with the remaining data used as the training set; there are therefore as many testing-training splits as there are observations. For k-fold cross-validation the data are divided into k subsets with each subset serving as the test set for the remaining k-1 subsets pooled together. For repeated random-split cross-validation the procedure of splitting the data into training and testing sets is randomly repeated many times. With cross-validation the model is refit to each training set, evaluated on the corresponding test set, and validation results are reported as the average performance over all test sets. This allows for nearly unbiased estimates of model performance, furthermore sample size is not sacrificed when leave-one-out cross-validation is used.
In developing the PCPT trial, 4-fold cross-validation was performed. The 5519 participants used for the analysis were split into four subsets of size 1380, 1380, 1380, and 1379, stratified by prostate cancer status so that the percentage of cancer cases ranged from 20 to 23% in each subset (1). On each possible grouping of three of the subsets the entire model selection process used for the development of the overall PCPT Risk Model was performed to yield an optimal model that was then validated on the single subset left out. The results from the four testing-training data splits were averaged to obtain a single measure of model performance.
In the lung cancer gene expression study data came from four institutions; data from two institutions were pooled to create the training dataset (N=256) and data from the other two institutions served as separate external testing datasets (N=104 and N=82). There were qualitative differences in some aspects of the gene expression data for one of the external datasets, so this was viewed as a more challenging, but realistic, way to assess the performance of the model. The model was built with the training data and an internal validation (repeated random-split cross validation) was used to evaluate its performance. In the random splits 200 of the 256 samples were used as the training data and the procedure was repeated 100 times. Finally the two remaining datasets were used for an external validation (see section “External Validation”).
An advantage of cross-validation to assess performance is that the final model can be built using all the data and is more efficient than a single testing-training split. A disadvantage is that it is cumbersome and may be impossible to implement. To properly undertake cross-validation every aspect of the model building process must be repeated independently, including all data preprocessing, selection of important variables and estimation, as was done for the PCPT calculator and for the lung cancer gene expression study. Since aspects of model building may be subjective, automating this process can be difficult. Cross-validation permits an honest assessment of the variability of the internal validation procedure, but the method does produce a large number of different risk prediction models, since the multivariable models are bound to differ among training sets, and each of these models may differ from the final model. Alternatives to cross-validation include bootstrapping methods (13, 30).
A pitfall for internal validation is that due to small sample sizes it may be tempting to forego the split and simply evaluate the risk prediction tool with the data used to develop it. This validation will be highly biased in an over-optimistic direction, especially when a large number of biomarkers are involved. Even when an internal validation is properly done the operating characteristics of the risk prediction tool may be over-optimistic relative to validation on a completely external dataset.
External Validation
External validation on a different dataset provided by a different study circumvents these issues. Validation on heterogeneous external datasets allows for evaluation of the generalizability of the risk prediction tool to wider populations than originally reported. For example, the Gail model for breast cancer recurrence was developed using data from the Breast Cancer Detection Demonstration Project (BCDDP) and was subsequently externally validated using data from the Breast Cancer Prevention Trial placebo arm (31). Even when external validation uses a dataset that appears to arise from a similar population, it is bound to differ due to, for example, different geographic locations or different clinical practices. It is likely that the model's operating characteristics will be diminished compared to internal validity assessments, but still may be good enough to declare the model useful. For example, external validation for the PCPT Risk Calculator yielded estimates of model performance that were lower than those initially reported using internal validation (32); more detail is given in the section “Sensitivity, Specificity, ROC Curves and AUC.”
One caveat with external validation is that investigators only get one shot at evaluating a model on an external dataset. While it may be very instructive to use external validation results to improve the model, it is not legitimate to subsequently reevaluate the model on this external dataset.
Metrics of Validation
At the heart of the issue of validation is the statistical challenge of comparing predictions from the model with true outcomes. While validity of a model could be couched as a yes/no determination (i.e. valid/not valid), this is not the best approach as most models would be deemed invalid, even potentially useful ones. It is useful to think in terms of degrees of validity, in particular to assess whether a model is useful, if so how useful, and lastly is it as good as advertised. Whether a model performs well enough for its intended application is a question of clinical validity and is not addressed here. In terms of statistical validity, the agreement between prediction and true outcomes is usually summarized by a few measures, discussed below.
Sensitivity, Specificity, ROC Curves and AUC
With binary outcomes and predictions, results can be displayed in a 2x2 table that cross-classifies true status (e.g. subject has the disease, yes/no) with predicted status (e.g. subject is predicted to have disease, yes/no), summarizing the performance of the prognostic model. From this one can calculate several simple measures that regularly appear in the medical literature and are thus familiar and easy to understand. Sensitivity is the proportion of the true positive outcomes (e.g. truly diseased subjects) that are predicted to be positive. Specificity is the proportion of the true negative outcomes (e.g. truly disease-free subjects) that are predicted to be negative. Since both of these measures are simple proportions, standard errors and confidence intervals can easily be calculated and should be reported.
If prediction is instead on a continuous scale, such as a predicted probability or a risk score, then for any given cut-point of the continuous scale one could create the 2x2 table and calculate sensitivity and specificity. As part of the validation of the PCPT calculator is was applied to a cohort from San Antonio (32). Table 2 shows how the sensitivities and specificities change as cut-offs for both PCPT risk and PSA are varied. As expected there is a continuum of change, with the sensitivity decreasing as the specificity increases with different cutoffs.
Table 2.
Sensitivities and Specificities of PCPT calculator applied to San Antonio cohort. Sensitivities of PSA cutoffs and PCPT risk cutoffs chosen to obtain same specificity as PSA cutoffs. Reprinted from Urology, Vol. 68(6), Dipen J. Parekh, Donna Pauler Ankerst, Betsy A. Higgins, Javier Hernandez, Edith Canby-Hagino, Timothy Brand, Dean A. Troyer, Robin J. Leach and Ian M. Thompson, External validation of the Prostate Cancer Prevention Trial risk calculator in a screened population, 1152–5, Copyright (2006), with permission from Elsevier.
| PSA Cutoff (ng/ml) | PCPT Risk Cutoff (%) | PSA and PCPT Risk Specificity (%) | Sensitivity (%),PSA | Sensitivity (%),PCPT Risk |
|---|---|---|---|---|
| 1.0 | 20.5 | 27.5 | 90.5 | 89.9 |
|
| ||||
| 1.5 | 25.5 | 40.3 | 84.5 | 82.4 |
|
| ||||
| 2.0 | 27.5 | 45.6 | 79.1 | 76.4 |
|
| ||||
| 2.5 | 29.0 | 51.7 | 68.9 | 69.6 |
| 3.0 | 33.0 | 63.4 | 61.5 | 55.4 |
|
| ||||
| 4.0 | 36.5 | 73.8 | 39.9 | 48.6 |
|
| ||||
| 6.0 | 46.0 | 88.6 | 15.5 | 20.3 |
|
| ||||
| 8.0 | 55.0 | 95.3 | 9.5 | 11.5 |
|
| ||||
| 10.0 | 59.0 | 97.3 | 4.7 | 8.8 |
The overall performance of the model can be summarized with a Receiver Operating Characteristic (ROC) curve. For each possible cut-point the resulting sensitivity and specificity are indicated as a point on a graph. This is illustrated in Figure 1 for both PCPT risk and PSA. The overall strength of association is summarized by the area underneath the curve (AUC, often called the concordance C statistic). An AUC of 0.5 (50%) indicates no association between prediction and true outcome and a value of 1.0 (100%) indicates perfect association. In general, AUCs below 0.6 are not considered medically useful while values of 0.75 are. However, there are no absolute rules about how large the AUC must be for the predictive model to be useful, and what represents large enough will be context-dependant. Finally, standard errors should be calculated to capture uncertainty in the estimate of AUC; one can and should give confidence intervals. For the PCPT risk calculator in figure 1, the AUC is 65.5%, with 95% confidence interval 60.2% to 70.8%.
Figure 1.
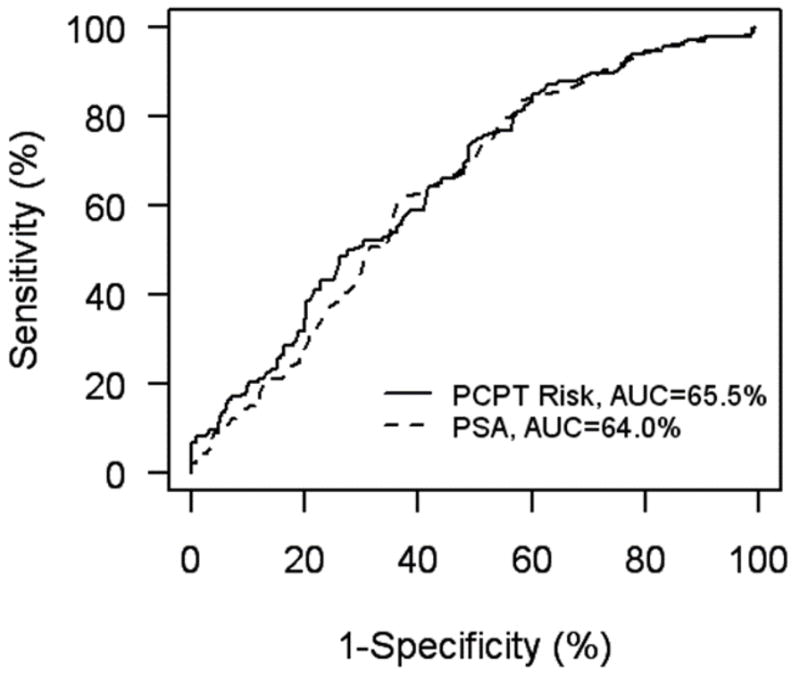
ROC curves for PCPT risk calculator and PSA applied to the San Antonio cohort. Adapted from Urology, Vol. 68(6), Dipen J. Parekh, Donna Pauler Ankerst, Betsy A. Higgins, Javier Hernandez, Edith Canby-Hagino, Timothy Brand, Dean A. Troyer, Robin J. Leach and Ian M. Thompson, External validation of the Prostate Cancer Prevention Trial risk calculator in a screened population, 1152–5, Copyright (2006), with permission from Elsevier.
The closeness of the AUC calculated from the external dataset to the AUC estimated from the dataset that was used to build the model is a measure of the validity of the model. For the PCPT, the original internally validated estimate of AUC was 70.2%. The PCPT was later externally validated on the significantly younger and more ethnically diverse San Antonio population (AUC=65.5%, N=446) and on two observational cohorts, one by Johns Hopkins University (AUC=66%, N=4,672) and another at the University of Chicago (AUC=67%, N=1,108) (32,33,34). External validation thus resulted in a reduction of approximately 4 percentage points from the internal validation.
ROC curves are invariant to monotonic transformations, for example, if all predictions were multiplied a constant factor the ROC curve would remain unchanged. This property can have some unexpected results. If the continuous prediction has an interpretable scale, such as the probability of recurrence, this scale is ignored by the ROC curve thus it is feasible to see a high AUC when the predicted probabilities are systematically biased. For example, if risk predictions for the PCPT were all multiplied by 0.5, which would clearly give underestimates of risk, the AUC would remain unchanged compared to the original. ROC curves help describe classification performance, are useful for comparing risk tools and can even be used to determine which biomarkers to include in the tool; however, they are only part of the assessment. A valid model should also be calibrated, that is for example a model of cancer risk should return computed risks that agree with actual observed proportions of cancer in a population of individuals with these computed risks. For example, if a model predicts that 20% of subjects will have an event within 3 years, but 40% actually do, then the model is not calibrated, regardless of how good measures such as the AUC may be. For the PCPT calculator applied to the San Antonio cohort the observed prostate cancer rates increased with increasing PCPT risk: 15.7%,39.0%, 48.8% and 100% for PCPT risk calculator values of less than 25%, 25% to 50%, 50% to 75% and greater than 75%, respectively. This demonstrates that the PCPT risk calculator is reasonably well calibrated.
When the true outcome is a censored event, i.e. has not yet occurred at the time of analysis, the calculation and interpretation of an ROC curve is harder. It is not legitimate to ignore the follow-up time and simply use dead or alive at last contact as a binary outcome. For the lung cancer gene expression study the authors demonstrate how the ROC curve can be calculated for the outcome dead or alive at three years (9). Time-dependant ROC curves have been recently developed for situations where the true outcome in the validation dataset is a censored event, such as a survival time (35).
Positive and Negative Predicted Values
From the perspective of the patient, he or she would like to know the probability that his particular prediction is correct. For a 2x2 table the positive predicted value (PPV) measures the proportion of times the true outcome is positive amongst those for whom it was predicted to be positive. Similarly negative predictive value (NPV) measures the proportion of true negatives out of all who tested negative. When reporting PPV and NPV one must define the population to which it applies. For example, the PPV for a predictive model in lung cancer screening is likely to be larger in a population of heavy smokers compared to a population of non-smokers. In the context of external validation, if a predictive model claims to have a certain PPV then one aspect of validation is to determine whether this PPV is seen in the external dataset. Although PPV and NPV are defined for binary outcome and prediction, they can be extended to the situation where the prediction is a continuous scale (36, 37).
When prediction is binary but the true outcome is a censored event time it is standard practice to draw a Kaplan-Meier estimate of the time-to-event distribution for each predicted group. The PPV and NPV can be read off these graphs at any desired follow-up time. A good separation between the Kaplan-Meier curves is required for the model to be useful; the relative hazard summarizes the predictive difference between groups. If the prediction has quantitative summaries associated with it, e.g. the probability of recurrence in each group, then accuracy of these claims can be assessed on external data by comparison to estimates of recurrence probability calculated from Kaplan-Meier plots (38).
When true outcome is a censored event and the prediction is a continuous score we recommend dividing the data into at least three equal sized groups for the purpose of drawing Kaplan-Meier estimates. The reason more than two groups is preferred is because this allows an assessment of a “dose-response” relationship, which is a desirable characteristic of a predictive model. Equally sized groups are needed for objectivity. Even though cut-points may not be round numbers this avoids the pitfall of seeking out the best cut-points, which is well known to give inflated measures of the performance (39).
Other Measures
When the true outcome is binary and the prediction a probability, other measures of predictive accuracy have been suggested. The mean squared error, called the Brier score in this context (40, 41), measures the distances between true outcomes (0=no event, 1=event) and predictions (probabilities ranging from 0 to 1) for a particular test dataset. A number of adaptations and generalizations of the Brier score have been proposed, including handling the situation of censored event time data (41, 42, 43). These scores are useful for comparing two competing models.
Cut-points
A common but much overemphasized practice in predictive modeling is to seek optimal cut-points or thresholds in the prediction. While cut-points are needed in the final stages of a model to provide guidelines for medical decision making, models that provide a continuous score provide potentially useful information, particularly for subjects near the threshold. It seems unrealistic to think risk levels are truly discrete; more plausible is a continuum of risk, and for this reason we prefer models providing a continuous score. Similarly, when subject characteristics are measured on a continuous scale, such as is often the case for biomarkers, it is implausible that there exists a fixed threshold at which risk abruptly changes. It is most efficient to keep biomarkers in the model as continuous variables if the assay measures them as such. Only at the final stage when the model is built from all the biomarkers might it be necessary to introduce cut-points to aid in classifying people into distinct groups.
Discussion/Conclusion
The challenges of validating a biomarker-based predictive model are considerable, and attention to the planned validation should be delivered as early as possible in the design phase of building the risk prediction tool. External validation using data from a completely different study provides the highest irrefutable evidence that a tool validates. The more external validations the better, particularly when they come from more heterogeneous populations that put a stress on the generalizability of the risk tool. Internal validation is more convenient and perhaps the only option for introducing the risk tool in a timely fashion, but is no substitute for external validation. For this reason the details of the prediction tools should be published and made available for others to attempt external validation. A variety of statistical summaries, such as the AUC, PPV, and Brier score can be used to summarize the performance characteristics of a risk prediction tool; in practice no one measure is enough, and the use of multiple summaries characterizing different components of prediction is recommended.
References
- 1.Thompson IM, Ankerst DP, Chi C, et al. Assessing prostate cancer risk: Results from the Prostate Cancer Prevention Trial. J Natl Cancer Inst. 2006;98:529–34. doi: 10.1093/jnci/djj131. [DOI] [PubMed] [Google Scholar]
- 2.Gail MH, Brinton LA, Byar DP, et al. Projecting individualized probabilities of developing breast cancer for white females who are being examined annually. J Natl Cancer Inst. 1989;81:1879–86. doi: 10.1093/jnci/81.24.1879. [DOI] [PubMed] [Google Scholar]
- 3.Kattan MW, Eastham JA, Stapleton AMF, Wheeler TM, Scardino PT. A preoperative nomogram for disease recurrence following radical prostatectomy for prostate cancer. J Natl Cancer Inst. 1998;90:766–71. doi: 10.1093/jnci/90.10.766. [DOI] [PubMed] [Google Scholar]
- 4.Paik S, Shak S, Tang G, et al. A multi-gene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med. 2004;351:2817–26. doi: 10.1056/NEJMoa041588. [DOI] [PubMed] [Google Scholar]
- 5.Skates SJ, Pauler DK, Jacobs IJ. Screening based on the risk of cancer calculation from Bayesian hierarchical change point and mixture models of longitudinal markers. J Am Stat Assoc. 2001;96:429–39. [Google Scholar]
- 6.Ransohoff DF. Lessons from controversy: ovarian cancer screening and serum proteomics. J Natl Cancer Inst. 2005;97:315–9. doi: 10.1093/jnci/dji054. [DOI] [PubMed] [Google Scholar]
- 7.Baggerly KA, Morris JS, Edmonson SR, Coombes KR. Signal in noise: evaluating reported reproducibility of serum proteomic tests for ovarian cancer. J Natl Cancer Inst. 2005;97:307–9. doi: 10.1093/jnci/dji008. [DOI] [PubMed] [Google Scholar]
- 8.Ioannidis JPA. Microarrays and molecular research: Noise discovery? Lancet. 2005;365:454–5. doi: 10.1016/S0140-6736(05)17878-7. [DOI] [PubMed] [Google Scholar]
- 9.Shedden K, Taylor JMG, Enkemann SA, et al. Gene expression-based survival prediction in lung adenocarcinoma: A multi-site, blinded validation study. Nat Med. 2008 doi: 10.1038/nm.1790. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hayes DF, Bast RC, Desch CE, et al. Tumor marker utility grading system: a framework to evaluate clinical utility of tumor markers. J Natl Cancer Inst. 1996;20:1456–66. doi: 10.1093/jnci/88.20.1456. [DOI] [PubMed] [Google Scholar]
- 11.George SL. Statistical issues arising in the application of genomics and biomarkers in clinical trials. Clin Cancer Res. 2008;18 in press. [Google Scholar]
- 12.Simon R. Using genomics in clinical trial design. Clin Cancer Res. 2008;18 doi: 10.1158/1078-0432.CCR-07-4531. in press. [DOI] [PubMed] [Google Scholar]
- 13.Harrell FE. Regression modeling strategies with applications to linear models, logistic regression, and survival analysis. New York: Springer-Verlag; 2001. [Google Scholar]
- 14.Hastie T, Tibshirani R, Friedman J. The elements of statistical learning;data-mining, inference, and prediction. New York: Springer; 2001. [Google Scholar]
- 15.Schwarzer G, Vach W, Schumacher M. On the misuses of artificial neural networks for prognostic and diagnostic classification in oncology. Stat Med. 2000;19:541–61. doi: 10.1002/(sici)1097-0258(20000229)19:4<541::aid-sim355>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 16.Sargent DJ. Comparison of artificial neural networks with other statistical approaches: results from medical data sets. Cancer. 2001;91:1636–42. doi: 10.1002/1097-0142(20010415)91:8+<1636::aid-cncr1176>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 17.Ein-Dor L, Kela I, Getz G, Givol D, Domany E. Outcome signature genes in breast cancer: Is there a unique set? Bioinformatics. 2005;21:171–8. doi: 10.1093/bioinformatics/bth469. [DOI] [PubMed] [Google Scholar]
- 18.Owzar K, Barry WT, Jung S-H, Sohn I, George SL. Statistical challenges in pre-processing in microarray experiments in cancer. Clin Cancer Res. 2008;18 doi: 10.1158/1078-0432.CCR-07-4532. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hammond ME, Fitzgibbons PL, Compton CC, Grignon DJ, Page DL, Fielding LP, Bostwick D, Pajak TF. College of American Pathologists Conference XXXV: solid tumor prognostic factors-which, how and so what? Summary document and recommendations for implementation. Cancer Committee and Conference Participants. Arch Pathol Lab Med. 2000;124:958–65. doi: 10.5858/2000-124-0958-COAPCX. [DOI] [PubMed] [Google Scholar]
- 20.McShane LM, Altman DG, Sauerbrei W, et al. Reporting recommendations for tumor marker prognostic studies (REMARK) J Natl Can Inst. 2005;97:1180–4. doi: 10.1093/jnci/dji237. [DOI] [PubMed] [Google Scholar]
- 21.Ware JH. The limitations of risk factors as prognostic tools. N Engl J Med. 2006;355:2615–17. doi: 10.1056/NEJMp068249. [DOI] [PubMed] [Google Scholar]
- 22.Pepe MS, Janes H, Longton G, Leisenring W, Newcomb P. Limitations of the odds ratio in gauging the performance of a diagnostic, prognostic, or screening marker. Am J Epidemiol. 2004;159:882–890. doi: 10.1093/aje/kwh101. [DOI] [PubMed] [Google Scholar]
- 23.Chau CH, McLeod H, Figg WD. Validation of analytical methods for biomarkers employed in drug development. Clin Cancer Res. 2008;18 in press. [Google Scholar]
- 24.Altman DG, Royston P. What do we mean by validating a prognostic model? Stat Med. 2000;19:453–73. doi: 10.1002/(sici)1097-0258(20000229)19:4<453::aid-sim350>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 25.Greenland S. The need for reorientation towards cost-effective prediction: Comments on “Evaluating the added predicted ability of a new marker: From area under the ROC curve to reclassification and beyond” by M. J Pencina et al. Stat Med. 2008;27:199–206. doi: 10.1002/sim.2995. [DOI] [PubMed] [Google Scholar]
- 26.Gail MH, Costantino JP, Bryant J, et al. Weighing the risks and benefits of tamoxifen treatment for preventing breast cancer. J Natl Cancer Inst. 1999;91:1829–46. doi: 10.1093/jnci/91.21.1829. [DOI] [PubMed] [Google Scholar]
- 27.Sorlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci. 2001;98:10869–74. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Potti A, Dressman HK, Bild A, et al. Genomic signatures to guide the use of chemotherapeutics. Nat Med. 2007;12:1294–1300. doi: 10.1038/nm1491. [DOI] [PubMed] [Google Scholar]
- 29.Coombes KR, Wang J, Baggerly KA. Microarrays: retracing steps. Nat Med. 2007;13:1276–7. doi: 10.1038/nm1107-1276b. [DOI] [PubMed] [Google Scholar]
- 30.Efron B, Tibshirani R. Improvements on cross-validation: The. 632+ bootstrap method. J Am Stat Assoc. 1997;92:548–60. [Google Scholar]
- 31.Constantino JP, Gail MH, Pee D, et al. Validation studies for models projecting the risk of invasive and total breast cancer incidence. J Natl Cancer Inst. 1999;91:1541–8. doi: 10.1093/jnci/91.18.1541. [DOI] [PubMed] [Google Scholar]
- 32.Parekh DJ, Ankerst DP, Higgins BA, et al. External validation of the Prostate Cancer Prevention Trial risk calculator in a screened population. Urology. 2006;68:1152–5. doi: 10.1016/j.urology.2006.10.022. [DOI] [PubMed] [Google Scholar]
- 33.Han M, Humphreys EB, Hernandez DJ, Partin AW, Roehl KA, Catalona WJ. AUA abstract 1875: Comparison between the prostate cancer risk calculator and serum PSA. J Urol. 2007;177:624. [Google Scholar]
- 34.Hernandez DJ, Han M, Humphreys EB, Mangold LA, Brawer MK, Taneja SS, et al. AUA abstract 1874: External validation of the prostate cancer risk calculator. J Urol. 2007;177:623. [Google Scholar]
- 35.Heagerty PJ, Zheng Y. Survival model predictive accuracy and ROC curves. Biometrics. 2005;61:92–105. doi: 10.1111/j.0006-341X.2005.030814.x. [DOI] [PubMed] [Google Scholar]
- 36.Moskowitz CS, Pepe MS. Quantifying and comparing the predictive accuracy of continuous prognostic factors for binary outcomes. Biostatistics. 2004;5:113–27. doi: 10.1093/biostatistics/5.1.113. [DOI] [PubMed] [Google Scholar]
- 37.Pepe MS, Feng Z, Huang Y, et al. UW Biostatistics Working Paper Series. 2006. Integrating the predictiveness of a marker with its performance as a classifier. Working Paper 289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taylor JMG, Yu M, Sandler HM. Individualized predictions of disease progression following radiation therapy for prostate cancer. J Clin Oncol. 2005;23:816–25. doi: 10.1200/JCO.2005.12.156. [DOI] [PubMed] [Google Scholar]
- 39.Altman DG, Lausen B, Sauerbrei W, Schumacher M. Dangers of using “optimal” cutpoints in the evaluation of prognostic factors. J Natl Cancer Inst. 1994;86:829–35. doi: 10.1093/jnci/86.11.829. [DOI] [PubMed] [Google Scholar]
- 40.Brier GW. Verification of weather forecasts expressed in terms of probability. Monthly Weather Review. 1905;78:1–3. [Google Scholar]
- 41.Graf E, Schmoor C, Sauerbrei W, Schumacher M. Assessment and comparison of prognostic classification schemes for survival data. Stat Med. 1999;18:2529–45. doi: 10.1002/(sici)1097-0258(19990915/30)18:17/18<2529::aid-sim274>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 42.Schemper M, Henderson R. Predictive accuracy and explained variation in Cox regression. Biometrics. 2000;56:249–55. doi: 10.1111/j.0006-341x.2000.00249.x. [DOI] [PubMed] [Google Scholar]
- 43.Henderson R, Jones M, Stare J. Accuracy of point predictions in survival analysis. Stat Med. 2001;20:3083–96. doi: 10.1002/sim.913. [DOI] [PubMed] [Google Scholar]