

Abstract
The CRISPR/Cas9 system has attracted significant attention for its potential to transform genome engineering. We and others have recently shown that the RNA-guided Cas9 nuclease can be employed to engineer the Drosophila genome, and that these modifications are efficiently transmitted through the germline. A single targeting RNA can guide Cas9 to a specific genomic sequence where it induces double-strand breaks that, when imperfectly repaired, yield mutations. We have also demonstrated that 2 targeting RNAs can be used to generate large defined deletions and that Cas9 can catalyze gene replacement by homologous recombination. Zinc-finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs) have shown similar promise in Drosophila. However, the ease of producing targeting RNAs over the generation of unique sequence-directed nucleases to guide site-specific modifications makes the CRISPR/Cas9 system an appealingly accessible method for genome editing. From the initial planning stages, engineered flies can be obtained within a month. Here we highlight the variety of genome modifications facilitated by the CRISPR/Cas9 system along with key considerations for starting your own CRISPR genome engineering project.
Keywords: CRISPR, Cas9, genome engineering, homologous recombination, site-directed mutagenesis
Introduction
An efficient, reliable means for precisely modifying the genome in living cells is a long-standing goal of biomedical science. In the clinic, such a tool will enable gene therapies to repair damaged genes, while in the lab it will be used to selectively manipulate genomic elements to study their function. In Drosophila, multiple approaches for precise genome editing have been successfully developed.1-7 However, their significant time and labor requirements have limited the widespread adoption of genome engineering techniques in Drosophila. The CRISPR/Cas9 system is poised to change this. We and others have recently demonstrated that within 1 month CRISPR/Cas9-mediated genome modifications can be efficiently generated in Drosophila and transmitted through the germline.8-10
Endogenous CRISPR RNA/Cas9 systems comprise a single polypeptide nuclease, Cas9, that is guided to target sites by a complex of 2 small RNAs – the CRISPR RNA (crRNA), which contains the targeting sequence, and a common trans-activating CRISPR RNA (tracrRNA).11,12 For use in genome engineering, the S. pyogenes system was simplified to 2 components through the generation of a chimeric RNA (chiRNA or guide RNA [gRNA]) comprising all critical crRNA and tracrRNA sequences12 (Fig. 1). The 2-component system requires only a single gRNA that recognizes a 20-nt target sequence next to a trinucleotide NGG protospacer adjacent motif (PAM) to direct Cas9-dependent cleavage of both DNA strands within the target sequence.12 This elegantly simple system has recently been shown to efficiently generate mutations in mammalian cell lines, human stem cells, yeast, bacteria, mice, zebrafish, worms, and flies.8-10,13-24 The fact that this has all been accomplished over a period of months illustrates the adaptability of the CRISPR/Cas9 system.
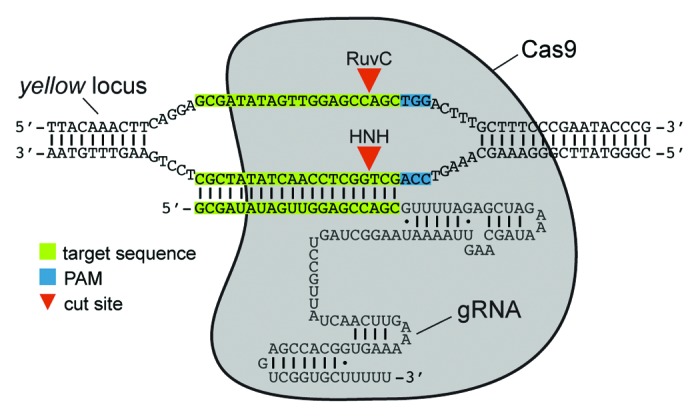
Figure 1. Schematic of the 2-component CRISPR/Cas9 system. A target site in the yellow locus is shown as an example. Cas9 is guided to a cleavage site by a chimeric RNA containing critical crRNA and tracrRNA sequences, including 20-nt of homology to a target site. This RNA has alternately been referred to as a guide RNA (gRNA), a single-guide RNA (sgRNA) or a chimeric RNA (chiRNA). Cas9 (gray) contains 2 distinct endonuclease domains, a HNH domain and a RuvC-like domain, that independently cleave both stands at the target site to generate a DSB (red arrowheads). Cleavage of target sites requires a high degree of homology to the gRNA and a 3-bp PAM (NGG) immediately 3′ of the target sequence.
Engineering Diverse Genome Modifications with CRISPR
In the short time since it was adapted for use in flies, the CRISPR/Cas9 system has already been used to successfully generate a variety of complex genome modifications, and the possibilities for expanding its application are nearly limitless.8-10 Here, we focus on the applications likely to be of broadest interest to the Drosophila community.
Knockouts/deletions
The induction of site-specific double-strand breaks (DSBs) in chromosomal DNA can yield mutations when breaks are inaccurately repaired by non-homologous end joining (NHEJ). This error-prone repair process can generate small insertions and deletions (indels) at the cleavage site that disrupt gene function (Fig. 2A). Separately, we used 4 different gRNAs to target Cas9 to the yellow and rosy loci, and observed germline transmission of frame-shifting indels at each targeted site9 (Table 1). Bassett et al. (2013) and Yu et al. (2013) expanded the number of sites targeted for mutation by Cas9-induced NHEJ, and observed efficient germline transmission of indels at 9 of 12 targeted sites in 8 genes.
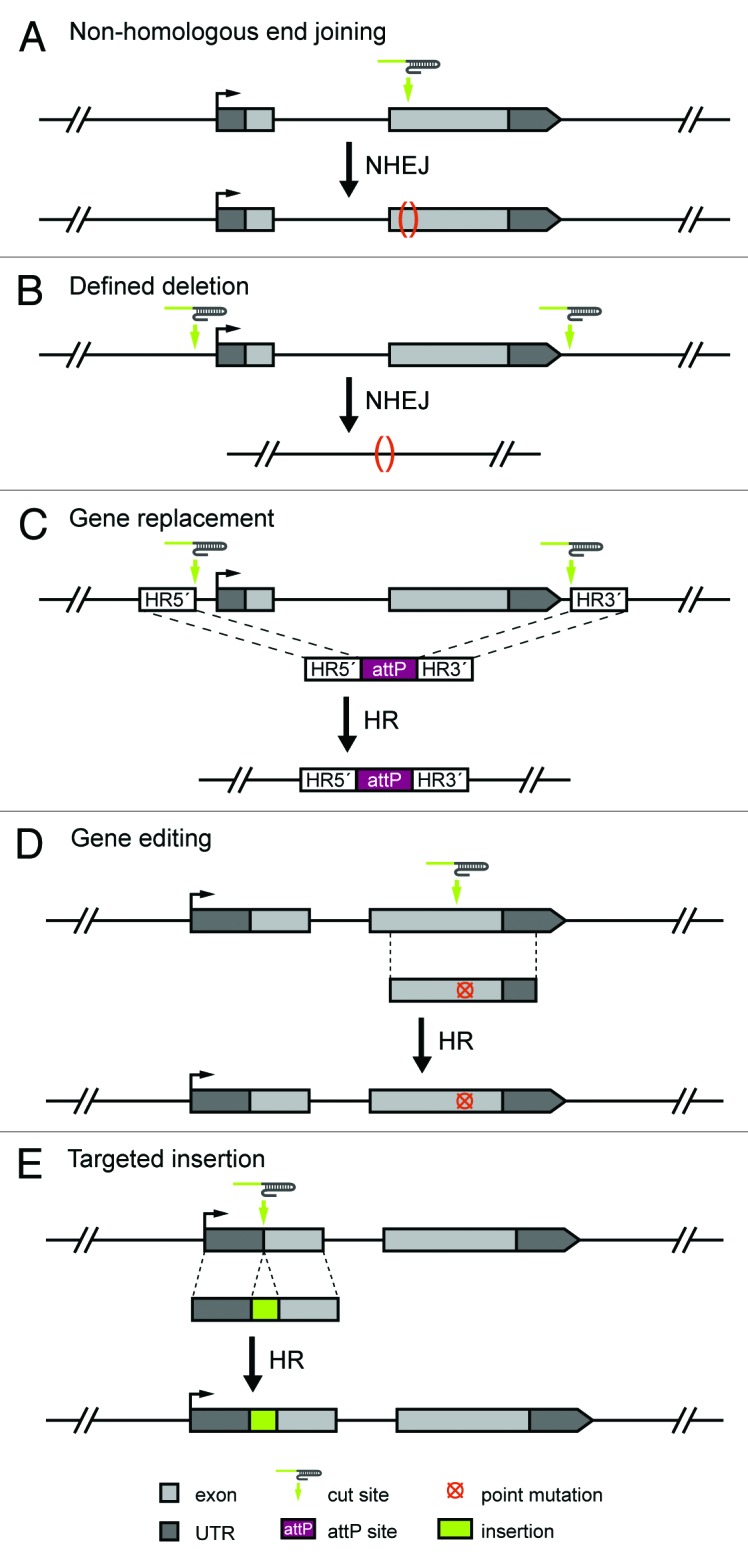
Figure 2. Engineering diverse genome modifications with the CRISPR system. (A) Using a single gRNA, targeted DSBs can be generated and repaired imperfectly by NHEJ resulting in disruptive mutations.8-10 (B) Through a process that is likely mediated by NHEJ, targeted deletions can be generated using 2 gRNAs targeting the limits of the region to be removed. This approach has been used to delete yellow and rosy.9 (C) Employing 2 gRNAs targeting the limits of the region to be replaced and a donor template for HR, targeted genomic regions can be replaced with exogenous sequences. In this example, a gene is replaced with an attP ΦC31 phage recombination site to allow for subsequent manipulation of the locus. In addition to the attP sequence, the donor template contains homology arms corresponding to the sequences immediately adjacent to the predicted cleavage sites. This approach has been used to replace yellow with attP.9 (D) Cas9-mediated HR can be used to engineer point mutations into a target locus. In this example, a target site is chosen near the sequence to be mutated, and novel sequence introduced via a donor template containing the modification flanked by sequences homologous to the target region. This approach has been used to introduce point mutations in mice and zebrafish.23,31 (E) Insertions of exogenous sequence including fluorescent molecules and epitope tags can be incorporated into a target locus via HR. A target site at or near the intended insertion site is chosen and used in conjunction with a donor template comprising two homology regions flanking the sequence to be inserted.
Table 1. Germline transmission rates of targeted mutations in rosy.
| Male crosses | Female crosses | |||||||
| gRNA(s) | ssODN donor | % (#) founders | % (#) progeny | % (#) founders | % (#) progeny | % (#) founders yielding targeted event | % (#) overall germline transmission | % (#) overall progeny |
| R1 | - | 10 (1/10) | 1.2 (6/508) | 20 (2/10) | 2.5 (5/201) | 100 (3/3) | 15 (3/20) | 1.6 (11/709) |
| R2 | - | 27 (3/11) | 22 (88/404) | NA | NA | 100 (3/3) | 27 (3/11) | 22 (88/404) |
| R3 | - | 9.7 (3/31) | 1.7 (27/1621) | 5.3 (1/19) | 0.49 (2/406) | 100 (4/4) | 8 (4/50) | 1.4 (29/2027) |
| R5′, R3′ | + | 33 (4/12) | 5.8 (35/603) | 0 (0/6) | 0 (0/549) | 0 (0/4) | 0 (0/18)* | 3.0 (35/1152) |
Flies injected with plasmids for expression of Cas9 and the indicated gRNAs with or without an ssODN donor template for the HR-mediated replacement of rosy with an attP ΦC31 phage recombination site were outcrossed and progeny screened for rosy eye color. The percentage of injected flies producing 1 or more rosy progeny (founders) is indicated along with the percentage of total progeny exhibiting rosy eyes. At least 1 progeny per founder was sequenced to determine if the targeted event had occurred. The percentage of founders in which the expected event occurred in 1 or more progeny is reported, as is the overall germline transmission rate (% injected flies yielding expected event). *We recovered the precise deletion of the 6.1-kb rosy locus without attP incorporation from 1/18 crosses (5.6%) in 7 progeny (0.6% of total progeny screened).
While the generation of random indels can disrupt function, the precise deletion of genes or other genomic sequences provides a powerful tool for unambiguously elucidating their role (Fig. 2B). To precisely delete the yellow gene, we simultaneously targeted the locus with 2 gRNAs, 1 directed to the 5′ end and the other at the 3′ end. Using this strategy we generated stable transformants harboring precise deletions of the 4.6-kb yellow locus as well as flies with partial deletions of the locus that abolished yellow function.9 We have also successfully deleted the 6.1-kb rosy locus (Table 1). Thus, multiple gRNAs can be simultaneously employed to delete entire open-reading frames.
Knock-ins/insertions
In addition to NHEJ, targeted DSBs in chromosomal DNA can catalyze homologous recombination (HR) using a donor template for repair. The replacement of a gene with an attP ΦC31 phage recombination site has been successfully combined with traditional ends-in and ends-out HR approaches in Drosophila to provide ongoing genetic access to a locus of interest.25,26 Using 2 gRNAs targeting sequences flanking yellow and a single-stranded oligodeoxynucleotide (ssODN) donor template, we were able to replace the endogenous yellow locus with an attP recombination site9 (Fig. 2C). This result demonstrates that the CRISPR/Cas9 system can be employed for homology-based genome engineering in Drosophila.
The finding that targeted insertion of exogenous sequences into the Drosophila genome can be accomplished using the readily programmable CRISPR/Cas9 system opens the door to a wide variety of genome modifications. This approach can potentially be used to introduce specific point mutations, tag endogenous loci, or flank genes with recombination sites for conditional knockout alleles, to cite just a few of the possibilities (Fig. 2D and E).
Starting a CRISPR Genome Engineering Project
The CRISPR/Cas9 system is a straightforward genome engineering technique that can be employed by any laboratory to generate targeted modifications. Here we address 3 key considerations for starting your own CRISPR/Cas9 genome engineering project: (1) options for identifying targeted events, (2) strategies for maximizing targeting specificity, and (3) methods for delivering system components. We also direct you to the flyCRISPR discussion board, accessible through our website (flyCRISPR.molbio.wisc.edu/news), where members of the Drosophila community are sharing their ideas and strategies.
Screening for targeted events
The initial experiments employing the CRISPR/Cas9 system in Drosophila were facilitated by the fact that mutations in yellow, rosy, and white yield easily scored visible phenotypes.8-10 In most cases, however, phenotypic screening will not be an option, necessitating alternative approaches for recognizing targeted events. Here we discuss 3 options to consider as you design your engineering strategy: (1) molecular screening, (2) targeting a marked locus, and (3) supplying a marked donor template.
Molecular screening
PCR-based methods are a universal option, but the associated generation and maintenance of candidate fly stocks can be labor intensive. Thus, to be broadly feasible, molecular screening requires a high rate of targeted events. Using a plasmid-based injection paradigm to generate indels at 4 targets in 2 genes, we observed mutant progeny at rates ranging from 0.25 to 22%9 (Table 1). Injection of CRISPR components as RNAs yielded mutant progeny at a rate of 2 to 35% in experiments targeting 5 genes with 8 different gRNAs.8,10 Although 2 gRNAs yielded no progeny due to lethality or sterility, and there were no mutants among the progeny of flies injected with 1 of 2 gRNAs targeting white, a gRNA targeting the K81 gene generated mutations that were transmitted to 99% of characterized progeny10—indicating that, while highly variable, indels can be generated at very high rates. It is important to note that all the numbers reported here likely include clonal events. These rates allowed Yu and colleagues to screen for mutations in progeny via PCR followed by either restriction enzyme-based analysis or direct sequencing of the products. With continued optimization and greater understanding of the factors that influence targeting efficiency, molecular screening of Cas9-induced mutations is likely to become increasingly feasible.
Negative screening
An alternative to molecular screening is to generate deletions in fly lines that contain visibly marked elements in the target locus (Fig. 3A). Because CRISPR components can be introduced into any genetic background via injection, any line carrying a marked element in a locus of interest can be used. One need only design the deletion or gene replacement experiment to remove the element with 2 flanking gRNAs and screen for loss of the visible marker. A consequence of this approach is an increase in the size of the deletion that must be generated, which might adversely affect efficiency. We previously targeted the 4.6-kb yellow locus for deletion, and found that 21% of injected flies (founders) transmitted deletions resulting in the null yellow phenotype to 1.4% of progeny. One of 18 founders transmitted the full 4.6-kb deletion, while the remainder of recovered events represented partial deletions.9 A 6.1-kb deletion of the rosy locus was fortuitously recovered in experiments designed to replace the locus with an attP site. (Table 1). To date, the CRISPR/Cas9 system has not been used to transmit deletions larger than 6.1 kb through the Drosophila germline.9 Nonetheless, the simplicity of this negative screening approach combined with the presence of marked elements in most Drosophila genes makes it an attractive option for many applications.
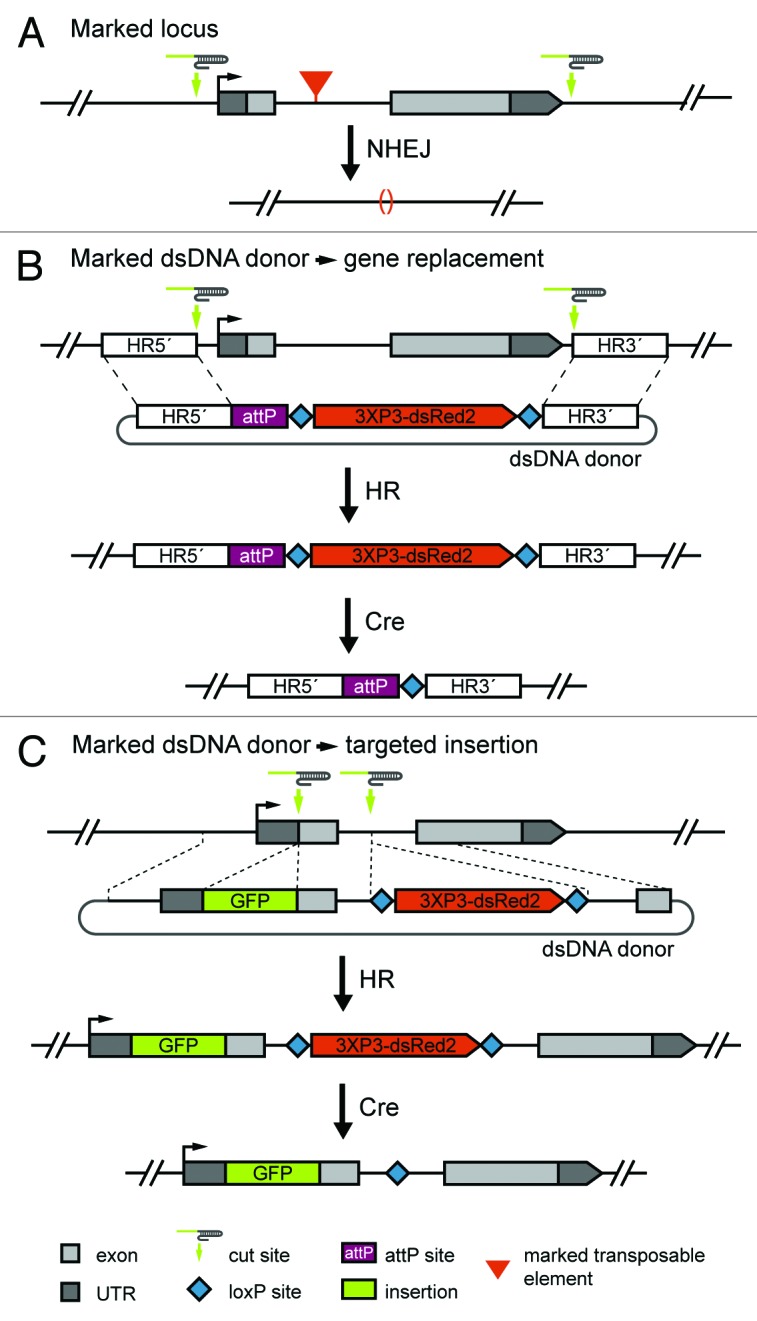
Figure 3. Strategies for detecting targeted events. (A) Targeting a locus with a marked element will allow screening for targeted deletions or gene replacements via loss of the marker. In this example, a locus containing a visibly marked transposable element is targeted for deletion. (B) A dsDNA donor with a removable positive marker can be used to facilitate screening for targeted deletions and gene replacements. In this example, a loxP-flanked fluorescent marker driven in the eye allows for identification of transformants based on eye color. The visible marker can later be removed using Cre recombinase. (C) In this example, a GFP tag is inserted at the translation start site of a gene using a donor template containing a removable positive marker to aid in screening. The donor was designed so the visible marker will be inserted in a non-conserved region of an adjacent intron as its subsequent removal will leave a single loxP site behind. This strategy might be facilitated by the use of 2 target sites—one at the tag insertion site and another at the insertion site for the positive marker. With this approach, the donor vector would contain homology arms corresponding to sequences adjacent to each cleavage site, sequences homologous to the region between the insertion sites, GFP coding sequence, and sequences encoding a visible marker.
Positive screening
A third option is to use a donor template for HR that includes a visible marker (Fig. 3B and C). While ssODN donors such as the one we utilized to integrate an attP docking site are useful for small modifications, larger insertions and the incorporation of visible markers for screening will require double-stranded DNA (dsDNA) donors. Injected dsDNA donor templates have been successfully utilized in Drosophila to incorporate up to 13 kb of exogenous sequence in both P element- and ZFN-induced HR.27,28 Based on a comprehensive analysis of ZFN-induced HR, dsDNA donors containing flanking homology arms of at least 1 kb in length serve as effective donors.27 dsDNA donors have been employed in mammalian cells as templates for CRISPR/Cas9-induced HR and there is every reason to expect they will work as effectively in flies.21 While this strategy requires generating a donor template, it will facilitate simple screening while enabling a broad range of modifications, from gene deletion and replacement (Fig. 3B) to the incorporation of fluorescent tags (Fig. 3C). Flanking the visible marker with FRT or loxP recombination sites will allow for its subsequent removal.
Strategies for maximizing targeting specificity
Once you’ve determined your strategy for recognizing targeted events, you will need to select the sequences to be targeted. Specificity and efficiency of cleavage are critical parameters for genome engineering methods that rely on sequence-specific DSB generation in chromosomal DNA. Although little is known about the factors affecting Cas9 efficiency, significant attention has already been devoted to identifying parameters that maximize specificity.12,15,29,30 Based on these studies, we expect that careful target selection and optimization of injection conditions will enable high specificity when employing the CRISPR/Cas9 system.
Initial CRISPR/Cas9 genome engineering studies identified a 12-nt ‘seed’ region adjacent to the PAM that was necessary for gRNA-guided cleavage by Cas9.12,15 The detrimental effects of seed-region mismatches on cleavage efficiency suggested that targets lacking perfect matches to the 12-nt seed sequence adjacent to an NGG PAM sequence elsewhere in the genome would be highly specific. More recently, 2 studies in mammalian cell lines indicate that somewhat more stringent criteria should be applied when selecting target sequences to minimize off-target cleavage.29,30 Importantly, Hsu et al. (2013) also found that NAG can serve as a PAM in mammalian cell lines, mediating cleavage with approximately 1/5 the efficiency of NGG. Together these findings suggest that targeting specificity can be maximized by selecting targets with the fewest potential off-target cleavage sites as defined by the rules below:
(1) PAM-adjacent sites with ≥ 11/12 matches to the target seed sequence. (2) PAM-adjacent sites with ≥ 18/20 matches to the full target sequence. (3) Sites meeting the above criteria adjacent to a divergent PAM of the form NAG as well as NGG.
An online CRISPR target identification tool is available from Feng Zhang’s laboratory at http://www.genome-engineering.org/crispr. This tool identifies CRISPR target sites lacking perfect matches to the seed sequence elsewhere in the genome; an updated version incorporating the more conservative rules outlined above is currently not available for the Drosophila genome.
In addition to careful selection of target sites, the concentration of CRISPR components can be titrated to maximize specificity by taking advantage of cleavage efficiency differences between on- and off-target sites.30 This has not yet been directly investigated in Drosophila. Finally, to further reduce the potential for generating mutations at off-target cleavage sites, Cas9 can be mutated and supplied as a nickase that will generate targeted single-strand breaks capable of catalyzing HR but unlikely to be repaired by the NHEJ pathway unless targeted as pairs.15,21
Encouragingly, neither we (Gratz et al. 2013) nor Bassett et al. (2013) uncovered evidence of cleavage at potential off-target sites identified by sequence similarity. In both studies, targets were selected to avoid perfect matches to the seed sequence adjacent to a PAM elsewhere in the genome, suggesting that even using looser criteria than above, strategic target selection can limit off-target cleavage in Drosophila. It is also possible that differences in DNA repair between germ cells and transformed cell lines result in lower apparent rates of off-target cleavage in Drosophila since only those cleavage events that are improperly repaired yield mutations. However, comprehensive analyses have not yet been performed, so further work will be required to determine the potential for off-target effects in Drosophila.
Deciding on a delivery system
The final decision to make is how you will introduce CRISPR components into Drosophila embryos. In this section, we discuss 3 alternatives: (1) injection of DNA plasmids encoding Cas9 and gRNA(s) for in vivo transcription/translation, (2) injection of these components as RNAs, or (3) injection of modification-specific components into flies expressing the common system components, Cas9 and/or tracrRNA.
DNA injection
To generate indels in the yellow and rosy loci, we injected DNA plasmids encoding Cas9, and separately, 4 different gRNAs into preblastoderm embryos. 6 to 27% (median = 12) of injected embryos that survived to become fertile adults transmitted targeted mutations to progeny at rates ranging from 0.25 to 22% (median = 2)9 (Table 1). With our expression plasmid, unique gRNA-encoding constructs can be generated in a couple of days using oligonucleotides. This U6-gRNA plasmid is available from Addgene and detailed protocols are available on our website (http://flycrispr.molbio.wisc.edu).
RNA injection
Two Drosophila groups have observed efficient germline transmission when injecting CRISPR components as RNA.8,10 Using single gRNAs to induce indels in 12 targets, mutant progeny were generated at rates ranging from 0–99% (median = 9). The percentage of founders (injected embryos that yielded mutant progeny) ranged from 0–100% (median = 59%) for the 10 targets for which this data was reported. DNA templates for in vitro transcription of gRNAs can be generated as plasmids or PCR products.8,10
Flies expressing CRISPR components
Germline expression of CRISPR/Cas9 components may increase targeting efficiency. To that end, we have generated transgenic flies that express Cas9 under the control of the vasa promoter. In addition, we have generated stable transgenic flies that express tracrRNA under the control of the snRNA:U6:96Ab promoter, and flies that express both Cas9 and tracrRNA. To facilitate rapid and widespread use, we have deposited these flies at the Bloomington Drosophila Stock Center. All lines are homozygous viable, suggesting low toxicity of CRISPR/Cas9 components in the absence of crRNA. Using these fly lines, targeted modifications can in principle be generated by injecting only the targeting component of the system. We do not yet have data on germline transmission using these lines; however, analysis of injected embryos indicates successful generation of Cas9-induced modifications with this approach.
Choosing a delivery system
To date, results have been reported for the generation of indels using DNA and RNA injection-based approaches. Initial reports raise the possibility that RNA injections may yield fewer surviving flies than DNA injections while yielding more affected progeny.8-10 However, no direct comparisons have been made, which will be important for determining how these approaches compare in their cleavage efficiency and toxicity given the significant variability between loci and targets that has been reported. Similarly, future experiments will be required to determine how techniques employing transgenic flies expressing CRISPR components compare with the injection-only techniques reported so far. A drawback of the transgenic approach is decreased flexibility in choosing the genetic background in which to generate modifications. The injection-based systems, on the other hand, place constraints on target selection due to the requirements for 1 or 2 Gs at the start of the gRNA sequence for efficient transcription from the U6 and T7 promoters, respectively. These additional requirements decrease the frequency of target sites in the genome from 1/8 to 1/32 for the plasmid-based system and 1/128 for an RNA injection approach. However, a new report suggests that these constraints may be easily overcome. In zebrafish, the G nucleotides can simply be added to the end of the gRNA construct without undue effects on efficiency.31 Finally, higher or lower cleavage rates may be desirable depending on the particulars of a given genome modification experiment. For instance, when targeting an essential gene, high cleavage rates might frequently yield bi-allelic breaks that would decrease the likelihood of recovering a targeted event. Suggesting this is a significant concern, Yu et al. (2013) observed 100% male infertility in the targeting of 2 out of 3 sex-linked genes and 100% larval lethality when targeting an essential gene. These factors will likely combine to make different approaches ideal for distinct circumstances. The options already available suggest the CRISPR/Cas9 system will be a versatile technique adaptable for a broad range of applications.
Outlook
The CRISPR/Cas9 system has sparked extraordinary interest as a tool for genome engineering due to its simplicity and adaptability. In the short time since the development of the 2-component system, work in Drosophila has already demonstrated that RNA-guided Cas9 can be used to generate indels, gene deletions, and gene replacements—all of which are efficiently transmitted through the germline.8-10,12 In the coming months, these successes are sure to be expanded upon as targeting efficiency and specificity are optimally balanced and more complex modifications are attempted. With Drosophila researchers worldwide eagerly embracing CRISPR technology, we can look forward to realizing the promise of designer flies on demand.
Author’s Note
While this manuscript was in review, germline transmission of indels and defined deletions generated in transgenic flies expressing both Cas9 and a custom gRNA was reported.32 Fillip Port and Simon Bullock also report the efficient generation and transmission of mutations in yellow and ebony in transgenic flies expressing Cas9 ubiquitously under the control of the act5 promoter, while Hui-Min Chen and Tzumin Lee report their successful targeting of yellow in transgenic flies expressing UAS-Cas9, a germline-specific Gal4 and a gRNA. These data are available at http://www.crisprflydesign.org.
Disclosure of Potential Conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful to Alex Cummings, Laura Donohue, Danielle Hamm, Jenny Nguyen, and Anna Zeidman for their assistance with experiments, and to Dustin Rubinstein and David Loehlin for helpful comments on the manuscript. We thank the Genetics Society of America for permission to use the schematic in Figure 1, which was originally published as Figure S1A of Gratz et al., 2013. This work was funded by startup funds from the University of Wisconsin to Harrison MM, Wildonger J, and O’Connor-Giles KM, and grants from the National Institutes of Health to Wildonger J (R00 NS072252) and O’Connor-Giles KM (R00 NS060985 and R01 NS078179). Plasmids and transgenic fly lines described here are available through the non-profit distributor Addgene and the Bloomington Drosophila Stock Center, respectively. Detailed protocols and reagent information are available at flyCRISPR.molbio.wisc.edu.
Glossary
Abbreviations:
- CRISPR
clustered regularly interspaced short palindromic repeats
- crRNA
CRISPR RNA
- tracrRNA
trans-activating CRISPR RNA
- gRNA
guide RNA
- PAM
protospacer adjacent motif
- DSB
double-strand break
- NHEJ
non-homologous end joining
- Indel
insertion-deletion
- HR
homologous recombination
- ssODN
single-stranded oligodeoxynucleotide
- dsDNA
double-stranded DNA
Footnotes
Previously published online: www.landesbioscience.com/journals/fly/article/26566
References
- 1.Banga SS, Boyd JB. Oligonucleotide-directed site-specific mutagenesis in Drosophila melanogaster. Proc Natl Acad Sci U S A. 1992;89:1735–9. doi: 10.1073/pnas.89.5.1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beumer KJ, Trautman JK, Bozas A, Liu JL, Rutter J, Gall JG, Carroll D. Efficient gene targeting in Drosophila by direct embryo injection with zinc-finger nucleases. Proc Natl Acad Sci U S A. 2008;105:19821–6. doi: 10.1073/pnas.0810475105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bibikova M, Golic M, Golic KG, Carroll D. Targeted chromosomal cleavage and mutagenesis in Drosophila using zinc-finger nucleases. Genetics. 2002;161:1169–75. doi: 10.1093/genetics/161.3.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gloor GB, Nassif NA, Johnson-Schlitz DM, Preston CR, Engels WR. Targeted gene replacement in Drosophila via P element-induced gap repair. Science. 1991;253:1110–7. doi: 10.1126/science.1653452. [DOI] [PubMed] [Google Scholar]
- 5.Gong WJ, Golic KG. Ends-out, or replacement, gene targeting in Drosophila. Proc Natl Acad Sci U S A. 2003;100:2556–61. doi: 10.1073/pnas.0535280100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang J, Zhou W, Watson AM, Jan YN, Hong Y. Efficient ends-out gene targeting in Drosophila. Genetics. 2008;180:703–7. doi: 10.1534/genetics.108.090563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rong YS, Golic KG. Gene targeting by homologous recombination in Drosophila. Science. 2000;288:2013–8. doi: 10.1126/science.288.5473.2013. [DOI] [PubMed] [Google Scholar]
- 8.Bassett AR, Tibbit C, Ponting CP, Liu JL. Highly efficient targeted mutagenesis of Drosophila with the CRISPR/Cas9 system. Cell Rep. 2013;4:220–8. doi: 10.1016/j.celrep.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gratz SJ, Cummings AM, Nguyen JN, Hamm DC, Donohue LK, Harrison MM, Wildonger J, O’Connor-Giles KM. Genome engineering of Drosophila with the CRISPR RNA-guided Cas9 nuclease. Genetics. 2013;194:1029–35. doi: 10.1534/genetics.113.152710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu Z, Ren M, Wang Z, Zhang B, Rong YS, Jiao R, Gao G. Highly Efficient Genome Modifications Mediated by CRISPR/Cas9 in Drosophila. Genetics. 2013;195:289–91. doi: 10.1534/genetics.113.153825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gasiunas G, Barrangou R, Horvath P, Siksnys V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci U S A. 2012;109:E2579–86. doi: 10.1073/pnas.1208507109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–21. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chang N, Sun C, Gao L, Zhu D, Xu X, Zhu X, Xiong JW, Xi JJ. Genome editing with RNA-guided Cas9 nuclease in zebrafish embryos. Cell Res. 2013;23:465–72. doi: 10.1038/cr.2013.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cho SW, Kim S, Kim JM, Kim JS. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol. 2013;31:230–2. doi: 10.1038/nbt.2507. [DOI] [PubMed] [Google Scholar]
- 15.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–23. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DiCarlo JE, Norville JE, Mali P, Rios X, Aach J, Church GM. Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res. 2013;41:4336–43. doi: 10.1093/nar/gkt135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Friedland AE, Tzur YB, Esvelt KM, Colaiácovo MP, Church GM, Calarco JA. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nat Methods. 2013;10:741–3. doi: 10.1038/nmeth.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hwang WY, Fu Y, Reyon D, Maeder ML, Tsai SQ, Sander JD, Peterson RT, Yeh JR, Joung JK. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat Biotechnol. 2013;31:227–9. doi: 10.1038/nbt.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol. 2013;31:233–9. doi: 10.1038/nbt.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J. RNA-programmed genome editing in human cells. Elife. 2013;2:e00471. doi: 10.7554/eLife.00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–6. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shen B, Zhang J, Wu H, Wang J, Ma K, Li Z, Zhang X, Zhang P, Huang X. Generation of gene-modified mice via Cas9/RNA-mediated gene targeting. Cell Res. 2013;23:720–3. doi: 10.1038/cr.2013.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, Jaenisch R. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153:910–8. doi: 10.1016/j.cell.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xiao A, Wang Z, Hu Y, Wu Y, Luo Z, Yang Z, Zu Y, Li W, Huang P, Tong X, et al. Chromosomal deletions and inversions mediated by TALENs and CRISPR/Cas in zebrafish. Nucleic Acids Res. 2013;41:e141. doi: 10.1093/nar/gkt464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao, G., Wesolowska, N. & Rong, Y.S. SIRT combines homologous recombination, site-specific integration, and bacterial recombineering for targeted mutagenesis in Drosophila. Cold Spring Harb Protoc2009, pdb prot5236 (2009). [DOI] [PubMed]
- 26.Huang J, Zhou W, Dong W, Watson AM, Hong Y. From the Cover: Directed, efficient, and versatile modifications of the Drosophila genome by genomic engineering. Proc Natl Acad Sci U S A. 2009;106:8284–9. doi: 10.1073/pnas.0900641106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beumer KJ, Trautman JK, Mukherjee K, Carroll D. Donor DNA Utilization during Gene Targeting with Zinc-finger Nucleases. G3 (Bethesda) (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keeler KJ, Dray T, Penney JE, Gloor GB. Gene targeting of a plasmid-borne sequence to a double-strand DNA break in Drosophila melanogaster. Mol Cell Biol. 1996;16:522–8. doi: 10.1128/mcb.16.2.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, Sander JD. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. 2013;31:822–6. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31:827–32. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hwang WY, Fu Y, Reyon D, Maeder ML, Kaini P, Sander JD, Joung JK, Peterson RT, Yeh JR. Heritable and precise zebrafish genome editing using a CRISPR-Cas system. PLoS One. 2013;8:e68708. doi: 10.1371/journal.pone.0068708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kondo S, Ueda R. Highly Improved Gene Targeting by Germline-Specific Cas9 Expression in Drosophila. Genetics. 2013 doi: 10.1534/genetics.113.156737. [DOI] [PMC free article] [PubMed] [Google Scholar]