

Abstract
The anti-CD20 antibody rituximab (RTX; Rituxan®, MabThera®) was the first anti-cancer antibody approved by the US Food and Drug Administration in 1997 and it is now the most-studied unconjugated therapeutic antibody. The knowledge gained over the past 15 y on the pharmacodynamics (PD) of this antibody has led to the development of a new generation of anti-CD20 antibodies with enhanced efficacy in vitro. Studies on the pharmacokinetics (PK) properties and the effect of factors such as tumor load and localization, antibody concentration in the circulation and gender on both PK and clinical response has allowed the design of optimized schedules and novel routes of RTX administration. Although clinical results using newer anti-CD20 antibodies, such as ofatumumab and obinutuzumab, and novel administration schedules for RTX are still being evaluated, the knowledge gained so far on RTX PK and PD should also be relevant for other unconjugated monoclonal antibody therapeutics, and will be critically reviewed here.
Keywords: B-NHL, CLL, FcRn, FcγRs, pharmacodynamics, pharmacokinetics, rituximab
Introduction
Rituximab (RTX; Rituxan®, MabThera®) is a chimeric monoclonal antibody (mAb) that binds the CD20 antigen, a transmembrane phosphoprotein specifically expressed by B-lymphocytes, from the pre-B to the mature germinal center B cells, and by most B cell neoplasms derived from these cells.1-3 RTX induces target cell death and is used in combination with polychemotherapy in the treatment of all histological types of B non-Hodgkin lymphoma (B-NHL) and in chronic lymphocytic leukemia (CLL), both as first-line and as rescue therapy. Furthermore, it is used for maintenance therapy of B-NHL and for treatment of several autoimmune diseases, in particular rheumatoid arthritis.4,5 In the past 15 y, much has been learned about RTX pharmacodynamics (PD) and pharmacokinetics (PK) and about how these affect the clinical response of patients with B cell neoplasia. This information can be applied to optimize treatments with new generation anti-CD20 as well as other anti-tumor mAbs.
RTX PD
Rituximab is an unconjugated IgG1k antibody, and most studies are consistent with the hypothesis that RTX in vivo acts mostly through immune-mediated mechanisms, including complement-dependent cytotoxicity (CDC), antibody-dependent cell-mediated cytotoxity (ADCC) involving NK cells and phagocytosis by macrophages and neutrophils (Fig. 1A).6-13 These mechanisms depend on the Fc portion of the antibody binding to FcγRs on immune cells. In addition, RTX and other anti-CD20 antibodies can activate signaling pathways after binding of the Fab portion to CD20 on B cells and induce homotypic adhesion (aggregation of target cells) and/or cell death to a variable extent (Fig. 1B).14-17 At least for RTX, direct cell death induction is not generally considered a major mechanism of action of the antibody.6 In addition to the mechanisms already mentioned, some evidence suggests that RTX may induce an anti-tumor immune response by cytotoxic T lymphocytes (CTL).18 Indeed, the antibody may promote tumor antigen uptake and peptide presentation by dendritic cells, leading to maturation and activation of specific effector CTL (Fig. 1C). This mechanism could explain the delayed and prolonged responses sometimes observed in patients with lymphoproliferative disorders, which are projected well beyond the time that effective circulating mAb concentrations are still detected. This mechanism, however, still needs to be confirmed in other models, and a direct demonstration that a vaccine effect occurs in patients is not yet available.

Figure 1. Possible mechanisms of action of RTX. (A) immune mediated. (B) direct mechanisms. (C) vaccine effect
The studies on the mechanisms of action of RTX have been amply summarized in other reviews and will not be described here in detail.6,19 It suffices to say that the extent to which each of these mechanisms of action is involved in tumor control probably depends on a number of factors, including tumor localization and load, CD20 expression levels, and the extent of tumor infiltration by immune effector cells such as NK cells and macrophages.
A new generation of anti-CD20 antibodies that have enhanced immune-mediated activities has now been developed. Obinutuzumab (GA101), a humanized and glycoengineered mAb, shows increased binding to FcγRIIIA and enhanced NK-mediated ADCC, increased direct cell death induction; it is in late-stage clinical trials.16,17,20 Ofatumumab (HuMax-CD20), a fully human mAb, has increased complement activation potential, particularly in the presence of low CD20 expression levels.21 Ofatumumab has been tested in clinical trials in CLL patients who are refractory to both fludarabine and alemtuzumab.22
Anti-CD20 antibodies can be divided into type I or type II according to whether they translocate CD20 into membrane microdomains, known as lipid rafts, and activate complement or not.14,23 RTX is a prototype type I antibody, with high capacity to translocate to rafts and high CDC, whereas obinutuzumab is a type II anti-CD20, with low CDC but higher capacity to induce homotypic adhesion and direct cell death with respect to RTX. Caution in the interpretation of cell death data are, however, warranted in the presence of homotypic adhesion, which can produce significant artifacts.24 Of note is that the type I or II properties are not simply dependent upon the CD20 epitopes recognized by the antibodies.25
RTX, like other unconjugated IgG1 antibodies, presents two types of binding: (1) Specific, through binding of Fab region of antibody to the CD20 target antigen (Fig. 2A) and (2) Non-specific, through binding of the antibody Fc region to Fcγ receptors (FcγR) on the immune effector cells, to the neonatal Fc receptor (FcRn) on different cell types and to C1q, the first complement component (Fig. 2B). Whereas the specific binding to target antigen and non-specific binding to Fcγ receptors can affect both PD and PK, binding to C1q is thought to mostly play a role in determining the mechanism of action of the mAb (Fig. 2). These different types of binding are described in more details in the following paragraphs.
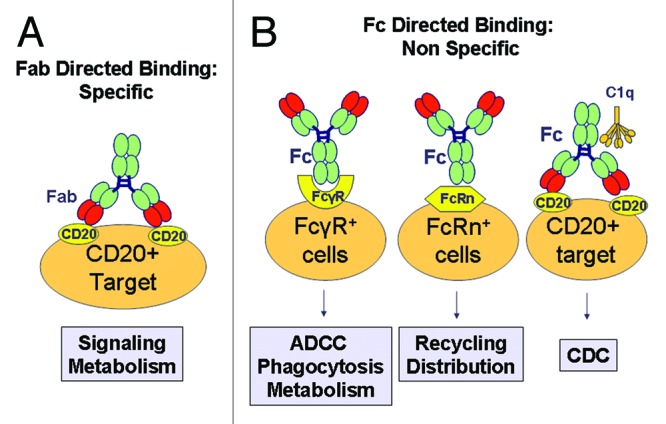
Figure 2. Major types of binding of RTX that affect PK and PD. (A) Specific binding to CD20 target takes place via Fab, leads to intracellular signaling and mAb metabolism. CD20 binding is saturable. (B) Non-specific binding of RTX includes binding to (1) FcγRs which leads to activation of immune cells (ADCC, phagocytosis) and mAb metabolism, (2) FcRn, which is non saturable and plays a major role antibody PK, and (3) C1q which activates CDC.
Specific binding: The anti-CD20 antibodies bind to CD20 with a good affinity (about 5 nM for RTX). The affinity of obinutuzumab seems slightly higher (about 0.4 nM). The rate of separation of the antibody from its target is lower in the case of ofatumumab (lower “off-rate”).21 For all anti-CD20 antibodies, a long residence of the molecule on the cellular membrane is observed, due to the fact that the antigen does not internalize significantly, at least in vitro, after its binding by the different antibodies.
Non-specific binding: Anti-CD20 antibodies can interact with different Fc receptors expressed on effector cells of the human immune system: FcγRI (CD64) with high affinity (10−8-10−9M), FcγRIIA, B or C (CD32A, B, or C) with low affinity (< 10−7M) and FcγRIIIA and B (CD16A and B), with intermediate affinity (1–3x10−7M). All are activating receptors except FcγRIIB (CD32B), which is inhibitory. Binding affinity determines to what extent the receptors may be occupied by free serum IgG or free (not target bound) RTX. Thus, CD64 quite efficiently binds free IgG, whereas lower affinity receptors usually bind mostly target bound RTX. This is because the binding site for FcγR on IgG is a domain on CH2 near the hinge region. Upon binding of mAb to its target, a small conformational change occurs with a slight “opening” of the hinge region, favoring binding of antibody to the FcγRs.26,27
The binding of Fc to activating Fcγ receptors cross-links these molecules, leading generally to the activation of the immune cells expressing them. The activation is more effective the higher the affinity of the Fc for the receptor and the lower the expression of inhibiting FcγRIIB on the same cells.26,27 Nonetheless, the presence of excess IgGs in plasma can dramatically inhibit effector mechanisms that rely on low-intermediate affinity FcγRs, such as ADCC by NK cells or phagocytosis by macrophages.10,26 This is because of competition between free plasma IgGs and RTX. Thus, immune cell activation may not be very strong in the circulation where high IgG levels are present.12,20
Antibodies also interact with the non-specific neonatal FcRn receptor, also known as the Brambell receptor. The binding between IgG and FcRn occurs at the interface between the CH2 and CH3 domains of IgG. FcRn is essential for the stability (half-life) of the antibody in vivo and for the regulation of IgG levels in serum.28,29 Indeed, the FcRn receptor is present on epithelial cells, endothelial cells, monocytes/macrophages and dendritic cells and protects the antibodies from degradation by the lysosomes. Following uptake from the fluid phase by pinocytosis, the IgG binds to FcRn in the acidic environment of endosomes (pH 6–6.5), where the FcRn-IgG complex is protected from proteolytic degradation. IgG can then be recycled to the cell plasma membrane, where it is released into the environment at pH 7.0–7.5 (Fig. 3). If release takes place on the other side of the cell, transcytosis (i.e., transport across a cell layer) takes place. In contrast if endosomes fuse with lysosomes, IgG degradation will occur. The mechanisms that regulate recycling, transcytosis and degradation of IgGs are not well understood. Recirculation of mAbs and transcytosis through FcRn are important mechanisms of mAb stabilization in vivo and distribution across tissues, respectively.28-30 Thus, Fc modifications aimed at improving bioavailability/stability of therapeutic mAbs in vivo are being actively sought by several groups, even though a considerable amount of work is still required to define the best strategies and translate the laboratory findings into the clinic. Indeed, improved binding at acid pH does not necessarily translate into longer half-life and better bioavailability.29,30
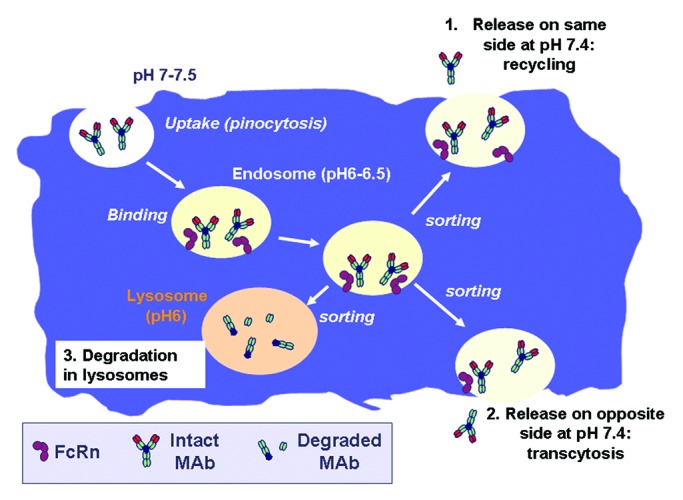
Figure 3. Interaction between IgG and FcRn. The mechanism by which IgG is taken up by cells (pinocytosis), binds inside the cell to FcRn in the acidic endosomes and is either recycled to the cell surface, transcytosed across the cell or degraded in lysosomes is shown.
Finally, RTX can bind to C1q and activate the classical complement cascade. C1q is a protein complex composed of 18 peptides that form 6 identical globular heads on a single stem structure (often compared with a bunch of tulips). Binding of multiple heads of C1q is required to activate complement. This can take place when several antibody molecules bind to the target cell surface and undergo a conformational change that exposes C1q binding sites in the CH2 domain. If the geometry of target-bound mAbs is adequate, C1q may bind through its multiple globular heads, undergo a conformational change and activate the complement cascade. Dissociation constants (KD) between anti-CD20 antibodies and C1q have been reported to be in the 10–100 nM range.31-34
The role of glycosylation
IgG has an important glycosylation site on asparagine (Asn) 297 within the CH2 domain; it is composed of a central heptasaccaride “core,” with the variable addition of residues of galactose, N-acetylglucosamine, sialic acid and fucose. MAbs are always a mixture of different glycosylated forms.35 This is important when considering biosimilar products because different cell lines or culture conditions can modify the glycosylation of the antibodies produced, and, therefore, also their Fc-mediated functionality.36 Indeed, removal of fucose, as is the case with obinutuzumab, results in an increase in its affinity for the FcγRIIIA receptor and increased ADCC.17,20,37 Conversely, glycosylation is not important for binding to FcRn. Therefore, the Asn297 glycosylation does not affect significantly the half-life of the antibody in vivo.35 Finally, C1q binding and CDC may be affected by the glycosylation pattern of IgG1 antibodies, but is not significantly modified by defucosylation38-40
Rituximab PK
Doses and routes of administration
RTX is usually administered by intravenous (i.v.) injection. The first approved schedule for induction therapy of B-NHL was 375 mg/m2 i.v. given for 4 cycles, and this was based on the pivotal trial of the antibody.41 Treatment cycles are generally given once a week for RTX as a single agent, and every 21–28 d when combined with chemotherapy. Modifications were later introduced to the initial schedules in B-NHL and CLL, most prominently additional induction cycles (6–12), increased frequency of administration, and maintenance therapy with the same dose given every 2 or 3 mo.
Limited to the treatment of central nervous system (CNS) lymphomas, an intrathecal or intraventricular route has also been attempted since maximal RTX levels in cerebrospinal fluid are generally not more than 1% of serum levels after i.v. administration.42,43 Doses administered by the intrathecal or intraventricular route are generally 10 or 25 mg antibody every few days.44,45
A more convenient administration would be the oral route, but this is limited by pre-systemic degradation in the gastrointestinal tract, and by inefficient diffusion or convection through the intestinal epithelium. To bypass the low oral bioavailability, and as an alternative to i.v. administration, a number of mAbs are delivered subcutaneously (s.c.). The subcutaneous route has been employed for antibodies used in the treatment of allergy or autoimmune diseases,46-48 but more recently has been extended to trastuzumab, an anti-human epidermal growth factor receptor (HER)-2 antibody approved for treatment of breast cancer. The s.c. formulation of a mAb generally brings substantial benefits for patients and for healthcare workers compared with i.v. administration, in particular shorter infusion times (~5 min vs. 150 min or more for i.v. administration). The proposed s.c. dose for RTX is a fixed dose of 1400 mg.49-51 The recent knowledge gained about RTX PK and PD provide a useful basis to define optimal RTX schedules in different disease contexts, for different recipients and for the different administration routes, as described in the following paragraphs.
General aspects of RTX PK
PK of RTX has been mostly studied after i.v. administration. In this case, RTX disposition is characterized by a 2-exponential decay, with a long elimination half-life of about 3 weeks (Fig. 4). The 2-compartment open PK model with first-order elimination represents the best structural model and seems to provide the best fit of RTX disposition, both during and after treatment, even with different schedules of drug administration.45,52,53 RTX has shown target (CD20)-mediated disposition where antibody-antigen binding influences the rate and extent of antibody distribution and elimination (Fig. 4), as will be further detailed below.

Figure 4. Biphasic PKs of RTX. The model shows high clearance by specific binding /to CD20) which, after saturation, leads to low clearance through non-specific binding via FcγR (RES)
The new generation of antibodies, such as obinutuzumab, ofatumumab, and other therapeutic mAbs, have also shown evidence of nonlinear behavior in humans.54-56 The nonlinear PK of these types of antibodies may in part be explained by binding of the antibodies to their respective targets, with a large component of target-mediated elimination after the first dose that is decreased after subsequent infusions because of a reduction in the available target antigen.54,57
Absorption
After i.v. administration of RTX, all the drug administered reaches the systemic circulation (by definition the bioavailability (F) by this route is 100%), while after s.c. administration only a fraction of RTX dose (F ≅ 60%) is absorbed, because, during the absorption phase, a portion of the drug undergoes proteolytic degradation or phagocytosis.49,58,59 Primary pathways for systemic absorption include convective transport of antibody through lymphatic vessels and into the blood, and diffusion of antibody across blood vessels distributed near the site of injection; however, on the basis of its molecular size, it is considered more likely that the RTX administered via s.c. injection is absorbed mainly via convection through lymphatic vessels.59-61 Indeed the lymphatic capillaries are densely distributed in the subcutaneous tissue and their relatively “open” structure facilitates the absorption of the antibody (a macromolecule) from the interstitial space into the lymph. Generally, after s.c. injection, absorption occurs slowly and the time to reach maximum plasma concentration varies from 2 to 8 d. The bioavailability is determined by the extent to which the drug after s.c. administration undergoes pre-systemic catabolism and systemic absorption. In general, the absolute bioavailability reported varies from 50 to 100%.59 Clearly, RTX will also bind to CD20 on B cells after s.c. administration.
The scheme reported in Figure 5 shows the PK model that seems to better describe RTX absorption after either i.v. or s.c. administration.
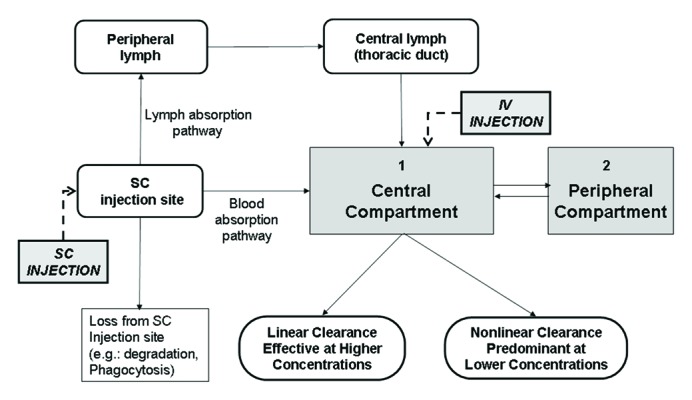
Figure 5. Model of mAb absorption and clearance after i.v. or s.c. administration. Absorption and clearance pathways are shown. Adapted from ref. 56.
Distribution
Antibody distribution kinetics is influenced by rates of convective transport, binding to tissue sites, and rate of catabolism within tissue. After i.v. administration, RTX binds to the CD20 antigen present on the surface of normal or neoplastic B cells in the peripheral blood, bone marrow and lymph nodes.62 After distribution at the level of tissue blood vessels, there are different mechanisms of transport of the antibody from the systemic circulation through the capillary endothelial cells and into tissues. RTX diffusion across vascular endothelial cells is very slow and the movement of RTX through or between the cell membranes mainly occurs via transcellular (endocytosis) (Fig. 3) or paracellular mechanisms, i.e., convective transport of the antibody within the movement of the fluid flow (Fig. 5).63,64 For both i.v. and s.c. administration of antibodies, FcRn plays an important role by reducing mAb catabolism and mediating mAb transport across endothelial cells, thus promoting the distribution of the antibodies across tissues.30,59
The volume of distribution of RTX at steady-state is approximately 9.6 L. Since the plasma volume is only 3‒3.5 L, this suggests that the mAb distributes into the extracellular spaces of tissues, except the CNS.65,66 Indeed, the blood-brain barrier physically impedes entry of macromolecules into the CNS, severely limiting distribution of RTX after i.v. administration. Brain vessels express the FcRn at high levels. Some authors suggest that FcRn promotes antibody egress from the cerebrospinal fluid rather than entry into the CNS, although whether it also plays a role in bidirectional transport has not yet been fully clarified.43
Elimination
The total clearance is the sum of specific target-mediated internalization, which is not linear and saturable, and non-specific clearance, which is linear and mediated by both FcγR-dependent and independent mechanisms (Fig. 4). Binding to FcRn generally reduces clearance because the antibody is recycled through FcRn to the surface and released into the cell environment (Fig. 3). Therefore, FcRn binding protects mAbs from intracellular degradation.
In particular, the mechanisms of antibody eliminations are three: (1) target-mediated elimination; (2) proteolysis by the liver Kupffer cells and by monocytes/macrophages of the reticuloendothelial system (RES); and (3) non-specific, FcγR-independent, endocytosis. In discussing of target-mediated elimination, it is worth recalling that CD20 usually is not rapidly or efficiently internalized by B cells after antibody binding, unlike other antigens like CD19, CD30, HER2 or epidermal growth factor receptor.2,67 Therefore, CD20-mediated elimination may not lead to a high rate of RTX catabolism, but mostly to its clearance from the circulation through absorption by the tumor mass. Proteolysis by Kupffer cells and by monocytes/macrophages of the RES is mediated mostly by binding of the Fc part of the antibody to Fcγ-receptors expressed on phagocytes, followed by receptor-mediated endocytosis and degradation in lysosomes. In this context, it is also worth mentioning that this FcγR mediated endocytosis by phagocytes may lead either to the engulfment of the whole RTX-opsonized target by phagocytes (phagocytosis),12 or, in some cases, to the removal of RTX and bound CD20 from the target membrane, leaving the rest of the target cell intact. This latter mechanism has been called trogocytosis (or shaving) and may lead, in addition to RTX catabolism, to decreased CD20 expression on the target cell. Trogocytosis has been described for mAbs directed against different antigens.67 In non-specific, FcγR-independent, endocytosis, MAbs may enter cells, especially endothelial and dendritic cells, via fluid-phase endocytosis (pinocytosis). Also in this case mAb internalization may be followed by transfer to lysosomes and degradation into peptides or amino acids, unless protection by FcRn binding in endosomes takes place (Fig. 3).
With increasing concentrations of RTX, total clearance (CL) decreases markedly (and the elimination half-life increases), as soon as the target-mediated elimination pathway begins to become saturated, and approaches that of the linear process (CLL). The clearance and half-life reach a constant value (plateau) when contribution from the nonlinear way (CLNL) becomes negligible (Fig. 4).68
Accumulation
RTX accumulation and its extent are the result of the administration frequency (drug half-life relative to the dosing interval). Because RTX distribution and elimination are very long and clearance rate can vary, the extent to which the drug could accumulate after multiple doses is difficult to estimate. A steady-state condition (i.e., the condition in which, during each dosing interval, the intake of a drug is equal to the amount eliminated from the body) is achieved after approximately 3–5 half-lives (Fig. 6). Indeed, immunoglobulins in general take a longer time to reach steady-state compared with small molecules and have a longer elimination half-life;64 in fact, small molecules (e.g., chemotherapy) have an elimination half-life in the magnitude of hours and rapidly achieve steady-state following administration (hours-days), while large molecules (e.g., mAbs) have a very long elimination half-life (in the magnitude of weeks) and may take up to 12 weeks to achieve steady-state.65,66,69,70
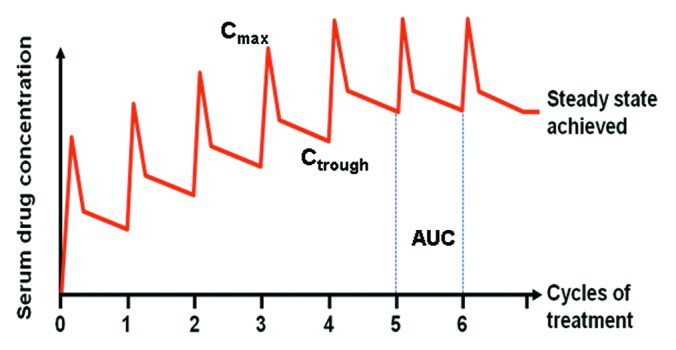
Figure 6. Model of RTX PK after multiple dosing. Cmax, Ctrough, AUC and steady-state, reached after about 5 cycles, are shown.
Factors Affecting RTX PK
The association between tumor burden and RTX levels
Circulating RTX levels have been shown to be affected by the “tumor burden” (tumor volume) in an inversely proportional way. Clinical studies have demonstrated that a high “tumor burden” is associated with low RTX serum levels.71-73 This is because the tumor cells act a sink for the antibody, adsorbing RTX through CD20 binding and inducing target-mediated elimination. A similar phenomenon is associated with the observation that a decrease in serum antibody levels during maintenance treatment can be predictive of relapse.74 Indeed, tumor growth adsorbs antibody, whose levels consequently fall in the circulation, in some cases anticipating clinical relapse.
Association between RTX levels and clinical response
Exposure to RTX (assessed by the area under the serum concentration-time curve (AUC)) and trough/pre-dose concentration (Ctrough) of the drug are the PK parameters most frequently related to the patient's response (Fig. 6). Cmax represents a measure of drug concentration in the blood immediately following i.v. administration, but it should be noted that during the first hours after drug injection, the changes in serum concentration do not always reflect a proportional change in the concentration of RTX in all other tissues, and hence in the amount of drug in the body. The balance between plasma/serum and tissue concentrations is obtained only a few days after drug administration. This is probably the reason why AUC and Ctrough are more directly related to clinical response compared with Cmax.
The therapeutic response to RTX has been correlated with serum drug concentration (Ctrough) in several studies. In the pivotal trial, patients with indolent B-NHL who responded to therapy had higher RTX serum concentrations (median levels of 25.4 µg/ml) compared with patients who did not respond (median 5.9 µg/ml).73 This has suggested that maintaining RTX above 25 µg/ml would be beneficial and has led some groups to attempt to maintain RTX levels > 25 µg/ml with individualized maintenance schedules.75
A further analysis of PK data from the pivotal trial showed how the CD20-mediated pathway of RTX clearance was highly dependent on tumor load pre-infusion, and diminished upon repeated infusions and following target saturation.72 In contrast, the target independent clearance did not change during treatment. This latter study suggested that, in FL patients, Ctrough correlated with clinical response only in patients with RTX serum concentrations in the low range (< 35 µg/ml), i.e., in the presence of a significant tumor burden. This study suggests that sufficient RTX needs to be given to overcome the “sink effect” of high tumor burden.
During maintenance therapy, the NHL9 study by Jager et al. (induction phase with 6 cycles of 375 mg/m2 RTX administered i.v. with fludarabine and mitoxantrone, followed by a maintenance phase with RTX 375 mg/m2 i.v. every 2 mo for 2 y, in patients with follicular lymphoma, FL) demonstrated a correlation of RTX levels (Ctrough) with quality of remission. Patients achieving a complete response at the end of the induction phase had higher median RTX concentrations than those achieving a partial response.74 Furthermore, a correlation between RTX levels and progression-free survival (PFS) was shown with a trend toward a longer PFS in patients with higher Ctrough. This correlation reached statistical significance at maintenance cycle six.74
In patients with CLL, an association between drug exposure and clinical response was found: higher values of AUC and Ctrough were observed in responders than non-responders after 3–6 cycles of drug.68 In CLL, the higher dose of 500 mg/m2 allows an AUC at steady-state similar to that achieved with doses of 375 mg/m2 in patients with B-NHL. Higher doses are required in the initial phase to maintain higher concentrations, which determine a greater clinical benefit for the patients.68 The RTX clearance in CLL patients has been reported to be much higher than in NHL patients. This increased clearance is potentially due to the higher number of malignant cells in circulation for CLL and thus the predominance of the faster receptor (CD20)-mediated clearance component, which overcomes the lower CD20 density in CLL. The kinetics at the end of therapy become equal because the amount of available receptors is reduced.
Gender effects
An interesting aspect that has recently emerged is the effect of gender on RTX PK. Assessment of RTX levels in 17 of 29 previously untreated FL patients in the NHL9 study has shown gender-dependent differences in RTX levels.74 Ctrough and AUC were generally higher in females than males both in the induction phase and in the maintenance phase, resulting in a better quality of response.
Similarly, a PK study of 20 diffuse large B cell lymphoma (DLBCL) patients from the RICOVER-60 and R-CHOP-14-pegfilgrastim trials, treated with 8 doses of RTX + 6 cycles of CHOP-14 (cyclophosphamide, doxorubicin, vincristine, prednisone given every 14 d), revealed gender-dependent differences in RTX PK.66 In the latter study, RTX concentrations were measured 10 min before each infusion, 1 week and 1, 2, 3, 6 and 9 mo after the last infusion. Females were shown to have a longer elimination half-life than males (30.7 vs. 24.7 d, p = 0.003), which resulted in a higher overall exposure to the drug. This gender difference correlated with a better PFS observed in the total female population (n = 287) of the RICOVER-60 trial compared with males (n = 325).66 It was suggested that the higher weight of males contributed to their faster RTX clearance, but sex and weight independently affected RTX clearance and elimination half-life.
These data were also confirmed by the CORAL study performed in relapsed or refractory DLBCL, in which females taking RTX had a better event-free survival (EFS) than males (63% vs. 46%).76 This was evident only in premenopausal and not postmenopausal women, suggesting hormonal factors are involved. A recent retrospective study of 1793 DLBCL patients confirmed the adverse prognostic factor of male gender in response to RTX-containing therapeutic regimen.77
Finally, it is worth mentioning that preliminary evidence shows that gender may affect PK also of other unconjugated therapeutic mAbs, such as antibodies targeting tumor necrosis factor.78 Clearly, further studies are required to define whether the gender effect on PK is applicable to all unconjugated mAbs.
Improving RTX Therapeutic Regimen during Induction and Maintenance
After more than 15 y of RTX use, the dose of the molecule and the best therapeutic schemes in different disease contexts are only starting to be clarified. As described above, we know that Ctrough correlates with response and that a trough concentration of > 25 μg/ml is a reasonable threshold to maintain for efficacy. We also know that the exposure to antibodies depends on a variety of factors, including dose and frequency of administration, distribution, specific and non-specific clearance, amount of tumor.55 An exact quantification of the effect of each factor, which obviously varies from subject to subject, is, however, difficult to define.
Nonetheless, important general rules have started to emerge. In the above mentioned study of 20 DLBCL patients,66 the PK simulation compared both the RTX peak (Cmax) and trough concentrations (Ctrough) following 8 RTX cycles according to the R-CHOP-14 vs. R-CHOP-21 schedules (i.e., cycles CHOP chemotherapy combined with RTX given on day 1, given every 14 vs. 21 d). The model showed that the highest trough serum RTX levels were lower for R-CHOP-21 compared with R-CHOP-14 (Fig. 7); however, the time of drug exposure > 25 µg/ml was longer for R-CHOP-21 due to the longer cycles (3 weeks instead of 2 weeks). Thus, it is possible to understand why female patients in the RICOVER-60 trial benefited more from R-CHOP-14 compared with males (4-y PFS 72% vs. 64%): different RTX dosing schedules affect duration of drug exposure and therefore the R-CHOP-21 schedule provided a longer exposure to the drug than the R-CHOP-14 schedule and maximized exposure to clinically relevant concentrations of RTX (> 25 µg/ml)(Fig. 7).66 Females may be less affected by the shorter schedule because, as was previously noted, the elimination half-life is longer in females.
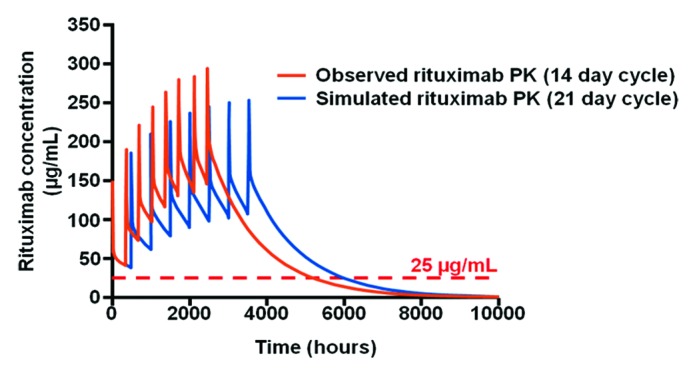
Figure 7. Model of RTX PK with 21 d vs. 14 d cycles. The model shows that RTX levels > 25 µg/ml are maintained for a longer period of time when RTX is administered every 21 d (red) rather than every 14 d (blue). Adapted from ref. 55.
These observations might also explain why the German NHL-B2 study results, which showed higher efficacy of dose-dense chemotherapy CHOP-14 vs. CHOP-21 (without RTX) in DLBCL in first-line therapy,79 were not confirmed in two reports comparing the same schedules combined with RTX (R-CHOP-14 vs. R-CHOP-21).80,81 Indeed, a number of factors should be considered (1) there is no linear dose-effect relation for RTX (in contrast to responses to CHOP chemotherapy), (2) the shorter exposure of RTX in the schedule R-CHOP-14 vs. R-CHOP-21 could not fully exploit the potential provided by the 8 doses of RTX, and (3) on the contrary, the longer exposure to RTX in the R-CHOP-21 regimen could be important from the therapeutic point of view, balancing in the R-CHOP-21 schedule the contribution made by dose-dense chemotherapy in CHOP-14.66 In other words, the optimal schedule for RTX appears to be every 3 weeks, whereas chemotherapy (CHOP) treatment may benefit from the shorter 2 weeks cycles.
To investigate this problem, the SEXIE-R-CHOP-14 study66 was undertaken. This ongoing Phase 2 study in patients with DLBCL in first-line therapy is designed to assess whether (1) by increasing RTX dose in males to 500 mg/m2/cycle, RTX serum levels are achieved similar to those observed in female patients at the standard dose of 375 mg/m2/cycle, and (2) increased RTX dose translates into better results in elderly male patients with DLBCL. This study may also further investigate the interesting possible difference between pre- and post-menopausal women suggested in the study of Gisselbrecht.76
The results of these studies could induce the scientific community to switch from 6 to 8 cycles of RTX in the treatment of DLBCL. The SMARTE-R-CHOP14 study82 will show us, instead, if it is convenient also to change the schedule of RTX administration. The schedule of RTX use in the study is so characterized: 1) RTX administration times are not simultaneous to the administration of chemotherapy, 2) the first 3 RTX doses are used with an intensive approach (days -4, -1, 10 of the whole treatment period) with the aim of achieving in the shortest time the maximum RTX serum level in terms of trough concentration, 3) subsequent RTX doses (doses 4–8) are administered at longer and longer intervals (days 29, 57, 99, 155 and 239 of the entire treatment period), and 4) the last 3 administrations (doses 6–8) are performed after the end of the six cycles of chemotherapy (days 99, 155, and 239 of the whole treatment period), to have a useful RTX serum level for a prolonged period of time (longer period of time compared with the RTX administration simultaneous to the 6th chemotherapy cycle).
PK data supporting drug administration every 2 mo for maintenance
RTX maintenance has shown clear clinical benefit in several clinical trials, but questions remain about what constitutes the best maintenance schedule. Again, PK studies can help define these schedules. As mentioned above, a guiding rule has been obtained from analysis of RTX concentrations in responding and non-responding patients with indolent relapsed/refractory B-NHL 3 mo after induction monotherapy, establishing the > 25 µg/ml level as a target to maintain over time.72,73 Further recent data72 obtained in the RTX maintenance therapy confirmed that, at maintenance cycle 6, patients with relapse had a RTX serum concentration < 25 µg/ml, while patients in continuous remission still had a RTX concentration > 25 µg/ml. These data suggest consideration of RTX serum Ctrough levels as a predictive factor of maintenance of the therapeutic response.
During maintenance therapy, the optimal RTX serum level of 25 µg/ml is achieved when RTX infusions are administered every 2 mo, as indicated by data of Jager,74 Salles (PRIMA study),83 Kahl (RESORT study),84 Berinstein73 and Gordan.75 It is therefore reasonable to assume that a maintenance therapy administered every 2 mo ensures to all patients an optimal level of active drug.
Use of body surface area-dependent dosages vs. fixed dose
The current indication of RTX in the treatment of B-NHL specifies the use of a body surface area (BSA)-dependent dosing (375 mg/m2). The BSA-dependent dosing is associated with possible dosing errors, with a higher PK variability, and is a poor indicator of optimal drug exposure. Generally, the administration of a fixed dose of mAb is always advisable when the drug has a wide therapeutic window; if, instead, the molecule is characterized by a narrow therapeutic window, it is appropriate to assess the effect of body weight on drug distribution (Vd) and elimination (CL) parameters.85
The study by Wang et al.86 relating to a group of mAbs shows that the fixed dose and the BSA-dependent dosing approaches perform similarly, when considering the variability (CV ≅ 30%) observed in the prediction of the RTX PK parameters, in particular the systemic exposure to the drug (AUC).86 These considerations have led some studies to use fixed doses of anti-CD20 antibodies, in particular those using new mAbs like ofatumumab and obinutuzumab.
PK of subcutaneous RTX vs. intravenous RTX
On the basis of the above observations on RTX PK after i.v. administration, it is hypothesized that attaining Ctrough levels of RTX after s.c. administration that are at least as high as those achieved after i.v. administration provides a comparable clinical efficacy. Compared with i.v. administration, the use of a s.c. route requires that RTX dose is increased to compensate for the portion lost during the absorption phase (~40%). The rounding in excess to a s.c. dose of 1400 mg of RTX (fixed dose) would ensure (also on the basis of the inter-individual PK variability) a systemic exposure to the drug “non-inferior” to that obtained after i.v. administration of a dose of 375 mg/m2 .
The available data show that the PK parameters that are usually indicative of the systemic exposure to RTX are comparable to those obtained after i.v. administration:49-51,58 In particular, the values of AUC and Ctrough obtained after s.c. administration of RTX are non-inferior to those obtained after i.v. administration of RTX; this should ensure a similar level of saturation of the target CD20 receptors and therefore a comparable efficacy. Also, there is no difference between s.c. and i.v. administration regarding the time required to reach steady-state; the steady-state after multiple dosing is reached in a time of approximately 4–5 times the elimination half-life, and, therefore, it is not affected by the absorption process. The SPARKTHERA study thus identified a fixed dose of 1400 mg for s.c. RTX that, when compared with the standard i.v. dose of 375 mg/m2, achieved a non-inferior Ctrough and AUC.58 Considering the wide therapeutic window of RTX, the fixed dose is recommended for the above mentioned reasons, and because it is more practical for the dosing, resulting in less drug waste and reduced risks of dosing errors.
RTX treatment of CNS lymphomas
PK analyzes of i.v. RTX has shown that RTX levels in CNS reach less than 1% (or less than 1 µg/ml) that observed in blood.42 Instead, an intrathecal or intraventricular route can significantly increase this value. Mean Cmax of 194 and 580 µg/ml were reported 1 h after 10 mg or 25 mg intraventricular doses given twice a week for 4 weeks in a recent Phase 1 study.45 Ctrough was 5.3 and 12.1 µg/ml before the last dose. PK data after intraventricular administration suggest that the biphasic RTX elimination profile applies here as well. Co-administration of methotrexate seemed to slow RTX elimination, probably by inhibiting RTX egress across the blood brain barrier and to the bloodstream, through an undefined mechanism.45 The good response rate together with the favorable PK results of this study suggests that RTX intraventricular route is a promising treatment option for CNS lymphomas.
The Interaction between RTX PD and PK
The rationale for PK parameters
In vitro studies of the RTX PD can provide a rationale for the use of schedules and doses sufficient to maintain a level of antibody of at least 25 µg/ml in serum for prolonged times. In fact, in whole blood CDC occurs at optimal levels at doses equal to or greater than 10 µg/ml.10,20 These values are thus consistent with the minimum optimal values defined in the in vivo PK studies (25 µg/ml).
Other mechanisms of action, such as ADCC or phagocytosis, require much lower RTX concentrations than CDC, in purified cellular systems. In fact, levels of about 0.1 µg/ml are already optimal under these conditions.12,87 In vivo these mechanisms of action (ADCC, phagocytosis) probably occur in tissues, as they are probably inhibited by complement and excess IgGs in the blood.12,20,88 A level of 25 µg/ml of RTX in the circulation may be the necessary level to achieve lower, but active, levels in tissues and in the tumor.
Factors affecting PD
Experimental evidence obtained using purified cells or whole blood demonstrate that the levels of expression of CD20 have a role in determining the CDC induced by RTX.20,89 Therefore, under conditions in which the neoplastic cells express low levels of CD20 (i.e., relatively few CD20 molecules per cell), like most CLL, a low rate of cell lysis induced by RTX and complement is observed (also in whole blood), while neoplastic cells that express high levels of CD20 show a high rate of lysis induced by the drug. There does seem to be a threshold of CD20 below which complement does not become activated by RTX. Some new mAbs such as ofatumumab appear to reduce this threshold value and induce higher CDC with targets expressing low levels of CD20.10,21 The importance of the mechanisms of complement-mediated cell cytotoxicity is further confirmed by data indicating a possible role of the complement inhibitors CD55 and CD59, which are expressed on the surface of target cells and may inhibit CDC in vitro and in vivo.89,90 Furthermore CD59 has been reported to be increased in RTX-resistant cells.91
There are various factors that can affect the efficacy of RTX in vivo. One is the polymorphisms of FcγRIIIA expressed on NK cells and monocytes/macrophages (V/F at position 158) and implicated in the response of FL patients to RTX as monotherapy.9,92,93 RTX efficacy in this case correlates with higher binding to the receptor bearing a Val residue at position 158. These data have demonstrated a role for immune-mediated mechanisms, most prominently ADCC and phagocytosis in the mechanism of action of RTX. Furthermore, the fact that NK cells from F/F donors require higher concentrations of RTX for ADCC in vitro compared with V/V carriers87 suggests that increasing RTX doses in vivo may overcome the effect of FcγRIIIA polymorphisms.94 An effect of FcγRIIIA polymorphism has, however, not been observed in CLL95 or in FL patients treated with RTX and chemotherapy, probably due to dilution of the effect by standard drugs.96 In large studies of DLBCL treated with R-CHOP, there are reports of a trend for better response of V/V and V/F patients compared with F/F.97 Also, FcγRIIA, which is expressed by macrophages and PMN, has been implicated in fewer studies,92 but this may be due to linkage disequilibrium between FcγRIIIA and FcγRIIA.98
In some cases, the immune effectors can be defective (for example in patients heavily treated with chemotherapeutics or advanced stage disease), decreasing the efficacy of RTX.99 Furthermore effectors, in particular macrophages mediating phagocytosis of targets, may infiltrate the tumors to a variable extent, in turn modifying the response to RTX.12,100
The immune effectors may also become partly or temporarily depleted after RTX administration due to their consumption in vivo.101 For example, after i.v. administration of RTX, complement is activated very quickly and some limiting components of the system, such as the C2 fragment, are exhausted after repeated administrations.102,103 In this case, the patient is not able to reconstitute all the elements of the complement cascade at optimal levels before another dose of RTX is administered. This phenomenon can lead to decreased efficacy of the antibody. Likewise, activated NK cells, which are involved in ADCC, could be exhausted after repeated dosing and thus no longer effective in lysing tumor targets. These mechanisms may explain in part the requirement for long exposure to RTX to achieve maximal response, with enough time being required to continuously replenish immune effector mechanisms to achieve the best response.
A large tumor mass will be even more difficult to eliminate because of the factors described above. In fact, the tumor mass and the number of CD20 molecules per cell also affect PK because they absorb a greater or lesser amount of circulating RTX. These phenomena may explain why greater amounts of RTX could be necessary in the case of large tumor masses, as are often present in CLL, although in these cases the levels of CD20 per cell are rather modest.
There is also evidence that the achievement of an excessive level of antibody in a rather short time can have a negative effect on efficacy because this can lead to a rapid depletion of immune mechanisms, which would then be followed by removal of CD20 from the cell membrane by exhausted macrophages, a phenomenon called shaving.67 The decrease in the expression of CD20 after shaving would lead to a partial resistance to the drug. On the contrary, a more gradual increase in the levels of antibody would entail a lower exhaustion of effector mechanisms and decreased shaving.104,105
Conclusions and Future Prospects
RTX doses were initially established on a very empirical basis, without a thorough knowledge of the mechanisms of action, the factors that determine the efficacy or the resistance to this new biologic drug, or PK. Over the past 15 y, however, many data have been obtained on PK and PD and different prognostic factors have been identified, allowing better prediction of what could be the best treatment regimens. At least in the context of B cell malignancies, the maintenance of a minimum level of drug (currently defined as > 25 µg/ml) for a prolonged time (at least 200 d for induction therapy and up to 2 y for maintenance), seems to be more important rather than the rapid achievement of a very high dose (200‒300 µg/ml) for a shorter time. Better timing, simplified administration (fixed doses, s.c.) and different schedules with respect to chemotherapy are being introduced, on the basis of the present knowledge and of the widely different characteristics of therapeutic mAbs compared with standard drugs in terms of both PK and PD. Structural modifications of anti-CD20 and other therapeutic mAbs are at different stages of pre-clinical and clinical development, and these may lead not only to improved mechanisms of action in vivo, but also to more favorable PK and biodistribution of these drugs, including in difficult organs such as the CNS. Several of the lessons learned from RTX studies should be valuable for other anti-cancer mAbs. Open questions remain whether and which structural modifications of unconjugated mAbs will allow better efficacy or PK properties in vivo, as well as improved clinical response.
Acknowledgments
This work was supported in part by the “Associazione Italiana Ricerca sul Cancro (AIRC)(to JG and GS), by the Italian Ministry Instruction, University and Research (MIUR, PRIN grant to FP).
Glossary
Abbreviations:
- ADCC
antibody dependent cellular cytotoxicity
- Asn
asparagine
- AUC
area under the curve
- CLL
chronic lymphocytic leukemia
- B-NHL
B-cell non-Hodgkin lymphoma
- BSA
body surface area
- CDC
complement dependent cytotoxicity
- CHOP
cyclophosphamide, doxorubicin, vincristine, prednisone
- CL
clearance
- Cmax
maximum concentration
- CNS
central nervous system
- CTL
cytotoxic T lymphocytes
- Ctrough
trough concentration
- DLBCL
diffuse large B-cell lymphoma
- EFS
event-free survival
- FcγRs
Fcγ receptors
- FcRn
Fc neonatal receptor
- FDA
Food and Drug Administration
- FL
follicular lymphoma
- IgG
immunoglobulin G
- i.v.
intravenous
- MAb
monoclonal antibody
- NK
natural killer
- PD
pharmacodynamics
- PFS
progression-free survival
- PK
pharmacokinetics
- PMN
polymorphonuclear neutrophil
- R-CHOP
rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone
- RES
reticuloendothelial system
- RTX
rituximab
- s.c.
subcutaneous
- Val
valine
- Vd
volume of distribution
- Vs.
versus
Disclosure of Potential Conflicts of Interest
JG, GS, AR, RF, GG, FP, AP, GS, FZ and MR have received honoraries from Roche Italia, the firm distributing rituximab and developing subcutaneous rituximab. JG has received a research grant from Roche Glycart AG, the firm developing new anti-CD20 antibodies. EG is an employee of Roche Italia.
Footnotes
Previously published online: www.landesbioscience.com/journals/mabs/article/26008
References
- 1.Riley JK, Sliwkowski MX. CD20: a gene in search of a function. Semin Oncol. 2000;27(Suppl 12):17–24. [PubMed] [Google Scholar]
- 2.Tedder TF, Engel P. CD20: a regulator of cell-cycle progression of B lymphocytes. Immunol Today. 1994;15:450–4. doi: 10.1016/0167-5699(94)90276-3. [DOI] [PubMed] [Google Scholar]
- 3.Kehrl JH, Riva A, Wilson GL, Thévenin C. Molecular mechanisms regulating CD19, CD20 and CD22 gene expression. Immunol Today. 1994;15:432–6. doi: 10.1016/0167-5699(94)90273-9. [DOI] [PubMed] [Google Scholar]
- 4.Martin P, Furman RR, Coleman M, Leonard JP. Phase I to III trials of anti-B cell therapy in non-Hodgkin’s lymphoma. Clin Cancer Res. 2007;13:5636s–42s. doi: 10.1158/1078-0432.CCR-07-1085. [DOI] [PubMed] [Google Scholar]
- 5.St Clair EW. Novel targeted therapies for autoimmunity. Curr Opin Immunol. 2009;21:648–57. doi: 10.1016/j.coi.2009.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weiner GJ. Rituximab: mechanism of action. Semin Hematol. 2010;47:115–23. doi: 10.1053/j.seminhematol.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Golay J, Zaffaroni L, Vaccari T, Lazzari M, Borleri GM, Bernasconi S, Tedesco F, Rambaldi A, Introna M. Biologic response of B lymphoma cells to anti-CD20 monoclonal antibody rituximab in vitro: CD55 and CD59 regulate complement-mediated cell lysis. Blood. 2000;95:3900–8. [PubMed] [Google Scholar]
- 8.Di Gaetano N, Cittera E, Nota R, Vecchi A, Grieco V, Scanziani E, Botto M, Introna M, Golay J. Complement activation determines the therapeutic activity of rituximab in vivo. J Immunol. 2003;171:1581–7. doi: 10.4049/jimmunol.171.3.1581. [DOI] [PubMed] [Google Scholar]
- 9.Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, Watier H. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood. 2002;99:754–8. doi: 10.1182/blood.V99.3.754. [DOI] [PubMed] [Google Scholar]
- 10.Bologna L, Gotti E, Da Roit F, Intermesoli T, Rambaldi A, Introna M, Golay J. Ofatumumab is more efficient than rituximab in lysing B chronic lymphocytic leukemia cells in whole blood and in combination with chemotherapy. J Immunol. 2013;190:231–9. doi: 10.4049/jimmunol.1202645. [DOI] [PubMed] [Google Scholar]
- 11.Cartron G, Watier H, Golay J, Solal-Celigny P. From the bench to the bedside: ways to improve rituximab efficacy. Blood. 2004;104:2635–42. doi: 10.1182/blood-2004-03-1110. [DOI] [PubMed] [Google Scholar]
- 12.Leidi M, Gotti E, Bologna L, Miranda E, Rimoldi M, Sica A, Roncalli M, Palumbo GA, Introna M, Golay J. M2 macrophages phagocytose rituximab-opsonized leukemic targets more efficiently than m1 cells in vitro. J Immunol. 2009;182:4415–22. doi: 10.4049/jimmunol.0713732. [DOI] [PubMed] [Google Scholar]
- 13.Uchida J, Hamaguchi Y, Oliver JA, Ravetch JV, Poe JC, Haas KM, Tedder TF. The innate mononuclear phagocyte network depletes B lymphocytes through Fc receptor-dependent mechanisms during anti-CD20 antibody immunotherapy. J Exp Med. 2004;199:1659–69. doi: 10.1084/jem.20040119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deans JP, Li H, Polyak MJ. CD20-mediated apoptosis: signalling through lipid rafts. Immunology. 2002;107:176–82. doi: 10.1046/j.1365-2567.2002.01495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ivanov A, Beers SA, Walshe CA, Honeychurch J, Alduaij W, Cox KL, Potter KN, Murray S, Chan CH, Klymenko T, et al. Monoclonal antibodies directed to CD20 and HLA-DR can elicit homotypic adhesion followed by lysosome-mediated cell death in human lymphoma and leukemia cells. J Clin Invest. 2009;119:2143–59. doi: 10.1172/JCI37884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alduaij W, Ivanov A, Honeychurch J, Cheadle EJ, Potluri S, Lim SH, Shimada K, Chan CH, Tutt A, Beers SA, et al. Novel type II anti-CD20 monoclonal antibody (GA101) evokes homotypic adhesion and actin-dependent, lysosome-mediated cell death in B-cell malignancies. Blood. 2011;117:4519–29. doi: 10.1182/blood-2010-07-296913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mössner E, Brünker P, Moser S, Püntener U, Schmidt C, Herter S, Grau R, Gerdes C, Nopora A, van Puijenbroek E, et al. Increasing the efficacy of CD20 antibody therapy through the engineering of a new type II anti-CD20 antibody with enhanced direct and immune effector cell-mediated B-cell cytotoxicity. Blood. 2010;115:4393–402. doi: 10.1182/blood-2009-06-225979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abès R, Gélizé E, Fridman WH, Teillaud JL. Long-lasting antitumor protection by anti-CD20 antibody through cellular immune response. Blood. 2010;116:926–34. doi: 10.1182/blood-2009-10-248609. [DOI] [PubMed] [Google Scholar]
- 19.Boross P, Leusen JH. Mechanisms of action of CD20 antibodies. Am J Cancer Res. 2012;2:676–90. [PMC free article] [PubMed] [Google Scholar]
- 20.Bologna L, Gotti E, Manganini M, Rambaldi A, Intermesoli T, Introna M, Golay J. Mechanism of action of type II, glycoengineered, anti-CD20 monoclonal antibody GA101 in B-chronic lymphocytic leukemia whole blood assays in comparison with rituximab and alemtuzumab. J Immunol. 2011;186:3762–9. doi: 10.4049/jimmunol.1000303. [DOI] [PubMed] [Google Scholar]
- 21.Teeling JL, Mackus WJ, Wiegman LJ, van den Brakel JH, Beers SA, French RR, van Meerten T, Ebeling S, Vink T, Slootstra JW, et al. The biological activity of human CD20 monoclonal antibodies is linked to unique epitopes on CD20. J Immunol. 2006;177:362–71. doi: 10.4049/jimmunol.177.1.362. [DOI] [PubMed] [Google Scholar]
- 22.Cang S, Mukhi N, Wang K, Liu D. Novel CD20 monoclonal antibodies for lymphoma therapy. J Hematol Oncol. 2012;5:64. doi: 10.1186/1756-8722-5-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beers SA, Chan CH, French RR, Cragg MS, Glennie MJ. CD20 as a target for therapeutic type I and II monoclonal antibodies. Semin Hematol. 2010;47:107–14. doi: 10.1053/j.seminhematol.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 24.Golay J, Bologna L, André PA, Buchegger F, Mach JP, Boumsell L, Introna M. Possible misinterpretation of the mode of action of therapeutic antibodies in vitro: homotypic adhesion and flow cytometry result in artefactual direct cell death. Blood. 2010;116:3372–3, author reply 3373-4. doi: 10.1182/blood-2010-06-289736. [DOI] [PubMed] [Google Scholar]
- 25.Klein C, Lammens A, Schäfer W, Georges G, Schwaiger M, Mössner E, Hopfner KP, Umaña P, Niederfellner G. Epitope interactions of monoclonal antibodies targeting CD20 and their relationship to functional properties. MAbs. 2013;5:22–33. doi: 10.4161/mabs.22771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bruhns P. Properties of mouse and human IgG receptors and their contribution to disease models. Blood. 2012;119:5640–9. doi: 10.1182/blood-2012-01-380121. [DOI] [PubMed] [Google Scholar]
- 27.Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. 2008;8:34–47. doi: 10.1038/nri2206. [DOI] [PubMed] [Google Scholar]
- 28.Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol. 2007;7:715–25. doi: 10.1038/nri2155. [DOI] [PubMed] [Google Scholar]
- 29.Kuo TT, Aveson VG. Neonatal Fc receptor and IgG-based therapeutics. MAbs. 2011;3:422–30. doi: 10.4161/mabs.3.5.16983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deng R, Meng YG, Hoyte K, Lutman J, Lu Y, Iyer S, DeForge LE, Theil FP, Fielder PJ, Prabhu S. Subcutaneous bioavailability of therapeutic antibodies as a function of FcRn binding affinity in mice. MAbs. 2012;4:101–9. doi: 10.4161/mabs.4.1.18543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Idusogie EE, Presta LG, Gazzano-Santoro H, Totpal K, Wong PY, Ultsch M, Meng YG, Mulkerrin MG. Mapping of the C1q binding site on rituxan, a chimeric antibody with a human IgG1 Fc. J Immunol. 2000;164:4178–84. doi: 10.4049/jimmunol.164.8.4178. [DOI] [PubMed] [Google Scholar]
- 32.Idusogie EE, Wong PY, Presta LG, Gazzano-Santoro H, Totpal K, Ultsch M, Mulkerrin MG. Engineered antibodies with increased activity to recruit complement. J Immunol. 2001;166:2571–5. doi: 10.4049/jimmunol.166.4.2571. [DOI] [PubMed] [Google Scholar]
- 33.Michaelsen TE, Sandlie I, Bratlie DB, Sandin RH, Ihle O. Structural difference in the complement activation site of human IgG1 and IgG3. Scand J Immunol. 2009;70:553–64. doi: 10.1111/j.1365-3083.2009.02338.x. [DOI] [PubMed] [Google Scholar]
- 34.Pawluczkowycz AW, Beurskens FJ, Beum PV, Lindorfer MA, van de Winkel JG, Parren PW, Taylor RP. Binding of submaximal C1q promotes complement-dependent cytotoxicity (CDC) of B cells opsonized with anti-CD20 mAbs ofatumumab (OFA) or rituximab (RTX): considerably higher levels of CDC are induced by OFA than by RTX. J Immunol. 2009;183:749–58. doi: 10.4049/jimmunol.0900632. [DOI] [PubMed] [Google Scholar]
- 35.Jefferis R. Isotype and glycoform selection for antibody therapeutics. Arch Biochem Biophys. 2012;526:159–66. doi: 10.1016/j.abb.2012.03.021. [DOI] [PubMed] [Google Scholar]
- 36.Schneider CK, Vleminckx C, Gravanis I, Ehmann F, Trouvin JH, Weise M, Thirstrup S. Setting the stage for biosimilar monoclonal antibodies. Nat Biotechnol. 2012;30:1179–85. doi: 10.1038/nbt.2447. [DOI] [PubMed] [Google Scholar]
- 37.Ferrara C, Brünker P, Suter T, Moser S, Püntener U, Umaña P. Modulation of therapeutic antibody effector functions by glycosylation engineering: influence of Golgi enzyme localization domain and co-expression of heterologous beta1, 4-N-acetylglucosaminyltransferase III and Golgi alpha-mannosidase II. Biotechnol Bioeng. 2006;93:851–61. doi: 10.1002/bit.20777. [DOI] [PubMed] [Google Scholar]
- 38.Jefferis R. Recombinant antibody therapeutics: the impact of glycosylation on mechanisms of action. Trends Pharmacol Sci. 2009;30:356–62. doi: 10.1016/j.tips.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 39.Kanda Y, Yamada T, Mori K, Okazaki A, Inoue M, Kitajima-Miyama K, Kuni-Kamochi R, Nakano R, Yano K, Kakita S, et al. Comparison of biological activity among nonfucosylated therapeutic IgG1 antibodies with three different N-linked Fc oligosaccharides: the high-mannose, hybrid, and complex types. Glycobiology. 2007;17:104–18. doi: 10.1093/glycob/cwl057. [DOI] [PubMed] [Google Scholar]
- 40.Cérutti M, Golay J. Lepidopteran cells, an alternative for the production of recombinant antibodies? MAbs. 2012;4:294–309. doi: 10.4161/mabs.19942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McLaughlin P, Grillo-López AJ, Link BK, Levy R, Czuczman MS, Williams ME, Heyman MR, Bence-Bruckler I, White CA, Cabanillas F, et al. Rituximab chimeric anti-CD20 monoclonal antibody therapy for relapsed indolent lymphoma: half of patients respond to a four-dose treatment program. J Clin Oncol. 1998;16:2825–33. doi: 10.1200/JCO.1998.16.8.2825. [DOI] [PubMed] [Google Scholar]
- 42.Harjunpää A, Wiklund T, Collan J, Janes R, Rosenberg J, Lee D, Grillo-López A, Meri S. Complement activation in circulation and central nervous system after rituximab (anti-CD20) treatment of B-cell lymphoma. Leuk Lymphoma. 2001;42:731–8. doi: 10.3109/10428190109099335. [DOI] [PubMed] [Google Scholar]
- 43.Lampson LA. Monoclonal antibodies in neuro-oncology: Getting past the blood-brain barrier. MAbs. 2011;3:153–60. doi: 10.4161/mabs.3.2.14239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pels H, Schulz H, Manzke O, Hom E, Thall A, Engert A. Intraventricular and intravenous treatment of a patient with refractory primary CNS lymphoma using rituximab. J Neurooncol. 2002;59:213–6. doi: 10.1023/A:1019999830455. [DOI] [PubMed] [Google Scholar]
- 45.Rubenstein JL, Li J, Chen L, Advani R, Drappatz J, Gerstner E, Batchelor T, Krouwer H, Hwang J, Auerback G, et al. Multicenter phase 1 trial of intraventricular immunochemotherapy in recurrent CNS lymphoma. Blood. 2013;121:745–51. doi: 10.1182/blood-2012-07-440974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Milgrom H, Berger W, Nayak A, Gupta N, Pollard S, McAlary M, Taylor AF, Rohane P. Treatment of childhood asthma with anti-immunoglobulin E antibody (omalizumab) Pediatrics. 2001;108:E36. doi: 10.1542/peds.108.2.e36. [DOI] [PubMed] [Google Scholar]
- 47.Wenzel S, Ford L, Pearlman D, Spector S, Sher L, Skobieranda F, Wang L, Kirkesseli S, Rocklin R, Bock B, et al. Dupilumab in persistent asthma with elevated eosinophil levels. N Engl J Med. 2013;368:2455–66. doi: 10.1056/NEJMoa1304048. [DOI] [PubMed] [Google Scholar]
- 48.Keystone EC, Genovese MC, Klareskog L, Hsia EC, Hall ST, Miranda PC, Pazdur J, Bae SC, Palmer W, Zrubek J, et al. GO-FORWARD Study Golimumab, a human antibody to tumour necrosis factor alpha given by monthly subcutaneous injections, in active rheumatoid arthritis despite methotrexate therapy: the GO-FORWARD Study. Ann Rheum Dis. 2009;68:789–96. doi: 10.1136/ard.2008.099010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Salar A, Avivi I, Larouche JF, Janikova A, Pereira J, Brewster M, Catalani O, McIntyre C, Sayyed P, Haynes A. Final results of the BP22333 Study demonstrate Non-inferior pharmacokinetics (PK) and safety of subcutaneous (SC) administration of rituximab compared to intravenous (IV) administration as maintenance therapy in patients with follicular lymphoma (FL) Blood. 2012;120:1641a. [Google Scholar]
- 50.Assouline S, Buccheri V, Delmer A, Doelken G, Gaidano G, McIntyre C, Brewster M, Hourcade-Potelleret F, Sayyed P, Badoux X. Subcutaneous rituximab in combination with fludarabine and cyclophosphamide for patients with CLL: Initial results of a phase Ib study (SAWYER- BO25341) show non-inferior pharmacokinetics and comparable safety to that of intravenous rituximab. Blood. 2012;120:1637a. [Google Scholar]
- 51.Davies A, Merli F, Mihaljevik B, Siritanaratkul N, Solal-Celigny P, Boehnke A, Berge C, McIntyre C, Barrett M, Macdonald D. Pharmacokinetics (PK), safety and overall response rate (ORR) achieved with subcutaneous /SC) administration of rituximab in combination with chemotherapy were comparable to those achieved with intravenous (IV) administration in patients (pts) with follicular lymphoma (FL) in the first-line setting: Stage 1 results of the Phase III SABRINA study (BO22334) Blood. 2012;120:1629a. [Google Scholar]
- 52.Regazzi MB, Iacona I, Avanzini MA, Arcaini L, Merlini G, Perfetti V, Zaja F, Montagna M, Morra E, Lazzarino M. Pharmacokinetic behavior of rituximab: a study of different schedules of administration for heterogeneous clinical settings. Ther Drug Monit. 2005;27:785–92. doi: 10.1097/01.ftd.0000184162.60197.c1. [DOI] [PubMed] [Google Scholar]
- 53.Dostalek M, Gardner I, Gurbaxani BM, Rose RH, Chetty M. Pharmacokinetics, pharmacodynamics and physiologically-based pharmacokinetic modelling of monoclonal antibodies. Clin Pharmacokinet. 2013;52:83–124. doi: 10.1007/s40262-012-0027-4. [DOI] [PubMed] [Google Scholar]
- 54.Salles G, Morschhauser F, Lamy T, Milpied N, Thieblemont C, Tilly H, Bieska G, Asikanius E, Carlile D, Birkett J, et al. Phase 1 study results of the type II glycoengineered humanized anti-CD20 monoclonal antibody obinutuzumab (GA101) in B-cell lymphoma patients. Blood. 2012;119:5126–32. doi: 10.1182/blood-2012-01-404368. [DOI] [PubMed] [Google Scholar]
- 55.Sehn LH, Assouline SE, Stewart DA, Mangel J, Gascoyne RD, Fine G, Frances-Lasserre S, Carlile DJ, Crump M. A phase 1 study of obinutuzumab induction followed by 2 years of maintenance in patients with relapsed CD20-positive B-cell malignancies. Blood. 2012;119:5118–25. doi: 10.1182/blood-2012-02-408773. [DOI] [PubMed] [Google Scholar]
- 56.Czuczman MS, Fayad L, Delwail V, Cartron G, Jacobsen E, Kuliczkowski K, Link BK, Pinter-Brown L, Radford J, Hellmann A, et al. 405 Study Investigators Ofatumumab monotherapy in rituximab-refractory follicular lymphoma: results from a multicenter study. Blood. 2012;119:3698–704. doi: 10.1182/blood-2011-09-378323. [DOI] [PubMed] [Google Scholar]
- 57.Mould DR, Sweeney KR. The pharmacokinetics and pharmacodynamics of monoclonal antibodies--mechanistic modeling applied to drug development. Curr Opin Drug Discov Devel. 2007;10:84–96. [PubMed] [Google Scholar]
- 58.Salar A, Bouabdallah R. MaIntyre C, Sauyyed P, Bittner B. A two stage phase Ib study to investigate the pharmacokinetics, safety and tolerability of subcutaneous rituximab in patients with follicular lymphoma as part of maintenance treatment. Blood. 2010;116:2858a. [Google Scholar]
- 59.Wang W, Wang EQ, Balthasar JP. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther. 2008;84:548–58. doi: 10.1038/clpt.2008.170. [DOI] [PubMed] [Google Scholar]
- 60.Kagan L, Turner MR, Balu-Iyer SV, Mager DE. Subcutaneous absorption of monoclonal antibodies: role of dose, site of injection, and injection volume on rituximab pharmacokinetics in rats. Pharm Res. 2012;29:490–9. doi: 10.1007/s11095-011-0578-3. [DOI] [PubMed] [Google Scholar]
- 61.McLennan DN, Porter CJ, Edwards GA, Martin SW, Heatherington AC, Charman SA. Lymphatic absorption is the primary contributor to the systemic availability of epoetin Alfa following subcutaneous administration to sheep. J Pharmacol Exp Ther. 2005;313:345–51. doi: 10.1124/jpet.104.078790. [DOI] [PubMed] [Google Scholar]
- 62.Keating GM. Rituximab: a review of its use in chronic lymphocytic leukaemia, low-grade or follicular lymphoma and diffuse large B-cell lymphoma. Drugs. 2010;70:1445–76. doi: 10.2165/11201110-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 63.Muller PY, Brennan FR. Safety assessment and dose selection for first-in-human clinical trials with immunomodulatory monoclonal antibodies. Clin Pharmacol Ther. 2009;85:247–58. doi: 10.1038/clpt.2008.273. [DOI] [PubMed] [Google Scholar]
- 64.Lobo ED, Hansen RJ, Balthasar JP. Antibody pharmacokinetics and pharmacodynamics. J Pharm Sci. 2004;93:2645–68. doi: 10.1002/jps.20178. [DOI] [PubMed] [Google Scholar]
- 65.de Jonge ME, Huitema AD, Rodenhuis S, Beijnen JH. Clinical pharmacokinetics of cyclophosphamide. Clin Pharmacokinet. 2005;44:1135–64. doi: 10.2165/00003088-200544110-00003. [DOI] [PubMed] [Google Scholar]
- 66.Müller C, Murawski N, Wiesen MH, Held G, Poeschel V, Zeynalova S, Wenger M, Nickenig C, Peter N, Lengfelder E, et al. The role of sex and weight on rituximab clearance and serum elimination half-life in elderly patients with DLBCL. Blood. 2012;119:3276–84. doi: 10.1182/blood-2011-09-380949. [DOI] [PubMed] [Google Scholar]
- 67.Beum PV, Peek EM, Lindorfer MA, Beurskens FJ, Engelberts PJ, Parren PW, van de Winkel JG, Taylor RP. Loss of CD20 and bound CD20 antibody from opsonized B cells occurs more rapidly because of trogocytosis mediated by Fc receptor-expressing effector cells than direct internalization by the B cells. J Immunol. 2011;187:3438–47. doi: 10.4049/jimmunol.1101189. [DOI] [PubMed] [Google Scholar]
- 68.Li J, Zhi J, Wenger M, Valente N, Dmoszynska A, Robak T, Mangat R, Joshi A, Visich J. Population pharmacokinetics of rituximab in patients with chronic lymphocytic leukemia. J Clin Pharmacol. 2012;52:1918–26. doi: 10.1177/0091270011430506. [DOI] [PubMed] [Google Scholar]
- 69.Rahman A, Carmichael D, Harris M, Roh JK. Comparative pharmacokinetics of free doxorubicin and doxorubicin entrapped in cardiolipin liposomes. Cancer Res. 1986;46:2295–9. [PubMed] [Google Scholar]
- 70.Nelson RL, Dyke RW, Root MA. Comparative pharmacokinetics of vindesine, vincristine and vinblastine in patients with cancer. Cancer Treat Rev. 1980;7(Suppl 1):17–24. doi: 10.1016/S0305-7372(80)80003-X. [DOI] [PubMed] [Google Scholar]
- 71.Harrold JM, Straubinger RM, Mager DE. Combinatorial chemotherapeutic efficacy in non-Hodgkin lymphoma can be predicted by a signaling model of CD20 pharmacodynamics. Cancer Res. 2012;72:1632–41. doi: 10.1158/0008-5472.CAN-11-2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yin A, Li J, Hurst D, Visich J. Population pharmacokinetics (PK) and association of PK and clinical outcomes of rituximab in patients with non-Hodgkin's lymphoma. J Clin Oncol. 2010;28(suppl 15):e13108. [Google Scholar]
- 73.Berinstein NL, Grillo-López AJ, White CA, Bence-Bruckler I, Maloney D, Czuczman M, Green D, Rosenberg J, McLaughlin P, Shen D. Association of serum Rituximab (IDEC-C2B8) concentration and anti-tumor response in the treatment of recurrent low-grade or follicular non-Hodgkin’s lymphoma. Ann Oncol. 1998;9:995–1001. doi: 10.1023/A:1008416911099. [DOI] [PubMed] [Google Scholar]
- 74.Jäger U, Fridrik M, Zeitlinger M, Heintel D, Hopfinger G, Burgstaller S, Mannhalter C, Oberaigner W, Porpaczy E, Skrabs C, et al. Arbeitsgemeinschaft Medikamentöse Tumortherapie (AGMT) Investigators Rituximab serum concentrations during immuno-chemotherapy of follicular lymphoma correlate with patient gender, bone marrow infiltration and clinical response. Haematologica. 2012;97:1431–8. doi: 10.3324/haematol.2011.059246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gordan LN, Grow WB, Pusateri A, Douglas V, Mendenhall NP, Lynch JW. Phase II trial of individualized rituximab dosing for patients with CD20-positive lymphoproliferative disorders. J Clin Oncol. 2005;23:1096–102. doi: 10.1200/JCO.2005.12.171. [DOI] [PubMed] [Google Scholar]
- 76.Gisselbrecht C, Schmitz N, Mounier N, Singh Gill D, Linch DC, Trneny M, Bosly A, Milpied NJ, Radford J, Ketterer N, et al. Rituximab maintenance therapy after autologous stem-cell transplantation in patients with relapsed CD20(+) diffuse large B-cell lymphoma: final analysis of the collaborative trial in relapsed aggressive lymphoma. J Clin Oncol. 2012;30:4462–9. doi: 10.1200/JCO.2012.41.9416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Carella AM, de Souza CA, Luminari S, Marcheselli L, Chiappella A, di Rocco A, Cesaretti M, Rossi A, Rigacci L, Gaidano G, et al. Prognostic role of gender in diffuse large B-cell lymphoma treated with rituximab containing regimens: a Fondazione Italiana Linfomi/Grupo de Estudos em Moléstias Onco-Hematológicas retrospective study. Leuk Lymphoma. 2013;54:53–7. doi: 10.3109/10428194.2012.691482. [DOI] [PubMed] [Google Scholar]
- 78.Ordás I, Mould DR, Feagan BG, Sandborn WJ. Anti-TNF monoclonal antibodies in inflammatory bowel disease: pharmacokinetics-based dosing paradigms. Clin Pharmacol Ther. 2012;91:635–46. doi: 10.1038/clpt.2011.328. [DOI] [PubMed] [Google Scholar]
- 79.Pfreundschuh M, Trümper L, Kloess M, Schmits R, Feller AC, Rübe C, Rudolph C, Reiser M, Hossfeld DK, Eimermacher H, et al. German High-Grade Non-Hodgkin’s Lymphoma Study Group Two-weekly or 3-weekly CHOP chemotherapy with or without etoposide for the treatment of elderly patients with aggressive lymphomas: results of the NHL-B2 trial of the DSHNHL. Blood. 2004;104:634–41. doi: 10.1182/blood-2003-06-2095. [DOI] [PubMed] [Google Scholar]
- 80.Cunningham D, Hawkes EA, Jack A, Qian W, Smith P, Mouncey P, Pocock C, Ardeshna KM, Radford JA, McMillan A, et al. Rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisolone in patients with newly diagnosed diffuse large B-cell non-Hodgkin lymphoma: a phase 3 comparison of dose intensification with 14-day versus 21-day cycles. Lancet. 2013;381:1817–26. doi: 10.1016/S0140-6736(13)60313-X. [DOI] [PubMed] [Google Scholar]
- 81.Delarue R, Tilly H, Mounier N, Petrella T, Salles G, Thieblemont C, Bologna S, Ghesquières H, Hacini M, Fruchart C, et al. Dose-dense rituximab-CHOP compared with standard rituximab-CHOP in elderly patients with diffuse large B-cell lymphoma (the LNH03-6B study): a randomised phase 3 trial. Lancet Oncol. 2013;14:525–33. doi: 10.1016/S1470-2045(13)70122-0. [DOI] [PubMed] [Google Scholar]
- 82.Pfreundschuh M, Zeynalova S, Poeschel V, Haenel M, Scmitz N, Ho D, Reiser M, Loeffler M, Schubert J. Improved outcome of elderly patients with poor prognosis diffuse large B-cell lymphoma (DLBCL) after dose-dense rituximab: Results of the DENSE-R-CHOP-14 trial of the german High Grade Non Hodgkin's Lymphoma Study Group (DSHNHL) J Clin Oncol. 2008;26:a8508. [Google Scholar]
- 83.Salles G, Seymour JF, Offner F, López-Guillermo A, Belada D, Xerri L, Feugier P, Bouabdallah R, Catalano JV, Brice P, et al. Rituximab maintenance for 2 years in patients with high tumour burden follicular lymphoma responding to rituximab plus chemotherapy (PRIMA): a phase 3, randomised controlled trial. Lancet. 2011;377:42–51. doi: 10.1016/S0140-6736(10)62175-7. [DOI] [PubMed] [Google Scholar]
- 84.Kahl BD, Williams ME, Hong F, Gascoyne RD, Horning SJ. Preliminary pharmacokinetic (PK) analysis of eastern cooperative oncology group protocol E4402: rituximab extended schedule or re-treatment trial (RESORT) Blood. 2007;110:3420a. [Google Scholar]
- 85.Bai S, Jorga K, Xin Y, Jin D, Zheng Y, Damico-Beyer LA, Gupta M, Tang M, Allison DE, Lu D, et al. A guide to rational dosing of monoclonal antibodies. Clin Pharmacokinet. 2012;51:119–35. doi: 10.2165/11596370-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 86.Wang DD, Zhang S, Zhao H, Men AY, Parivar K. Fixed dosing versus body size-based dosing of monoclonal antibodies in adult clinical trials. J Clin Pharmacol. 2009;49:1012–24. doi: 10.1177/0091270009337512. [DOI] [PubMed] [Google Scholar]
- 87.Dall’Ozzo S, Tartas S, Paintaud G, Cartron G, Colombat P, Bardos P, Watier H, Thibault G. Rituximab-dependent cytotoxicity by natural killer cells: influence of FCGR3A polymorphism on the concentration-effect relationship. Cancer Res. 2004;64:4664–9. doi: 10.1158/0008-5472.CAN-03-2862. [DOI] [PubMed] [Google Scholar]
- 88.Wang SY, Racila E, Taylor RP, Weiner GJ. NK-cell activation and antibody-dependent cellular cytotoxicity induced by rituximab-coated target cells is inhibited by the C3b component of complement. Blood. 2008;111:1456–63. doi: 10.1182/blood-2007-02-074716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Golay J, Lazzari M, Facchinetti V, Bernasconi S, Borleri G, Barbui T, Rambaldi A, Introna M. CD20 levels determine the in vitro susceptibility to rituximab and complement of B-cell chronic lymphocytic leukemia: further regulation by CD55 and CD59. Blood. 2001;98:3383–9. doi: 10.1182/blood.V98.12.3383. [DOI] [PubMed] [Google Scholar]
- 90.Macor P, Tripodo C, Zorzet S, Piovan E, Bossi F, Marzari R, Amadori A, Tedesco F. In vivo targeting of human neutralizing antibodies against CD55 and CD59 to lymphoma cells increases the antitumor activity of rituximab. Cancer Res. 2007;67:10556–63. doi: 10.1158/0008-5472.CAN-07-1811. [DOI] [PubMed] [Google Scholar]
- 91.Dalle S, Dupire S, Brunet-Manquat S, Reslan L, Plesa A, Dumontet C. In vivo model of follicular lymphoma resistant to rituximab. Clin Cancer Res. 2009;15:851–7. doi: 10.1158/1078-0432.CCR-08-1685. [DOI] [PubMed] [Google Scholar]
- 92.Weng WK, Levy R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J Clin Oncol. 2003;21:3940–7. doi: 10.1200/JCO.2003.05.013. [DOI] [PubMed] [Google Scholar]
- 93.Cartron G, Trappe RU, Solal-Céligny P, Hallek M. Interindividual variability of response to rituximab: from biological origins to individualized therapies. Clin Cancer Res. 2011;17:19–30. doi: 10.1158/1078-0432.CCR-10-1292. [DOI] [PubMed] [Google Scholar]
- 94.Ternant D, Cartron G, Hénin E, Tod M, Girard P, Paintaud G. Model-based design of rituximab dosage optimization in follicular non-Hodgkin’s lymphoma. Br J Clin Pharmacol. 2012;73:597–605. doi: 10.1111/j.1365-2125.2011.04125.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Farag SS, Flinn IW, Modali R, Lehman TA, Young D, Byrd JC. Fc gamma RIIIa and Fc gamma RIIa polymorphisms do not predict response to rituximab in B-cell chronic lymphocytic leukemia. Blood. 2004;103:1472–4. doi: 10.1182/blood-2003-07-2548. [DOI] [PubMed] [Google Scholar]
- 96.Carlotti E, Palumbo GA, Oldani E, Tibullo D, Salmoiraghi S, Rossi A, Golay J, Pulsoni A, Foà R, Rambaldi A. FcgammaRIIIA and FcgammaRIIA polymorphisms do not predict clinical outcome of follicular non-Hodgkin’s lymphoma patients treated with sequential CHOP and rituximab. Haematologica. 2007;92:1127–30. doi: 10.3324/haematol.11288. [DOI] [PubMed] [Google Scholar]
- 97.Ahlgrimm M, Pfreundschuh M, Kreuz M, Regitz E, Preuss KD, Bittenbring J. The impact of Fc-γ receptor polymorphisms in elderly patients with diffuse large B-cell lymphoma treated with CHOP with or without rituximab. Blood. 2011;118:4657–62. doi: 10.1182/blood-2011-04-346411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lejeune J, Thibault G, Ternant D, Cartron G, Watier H, Ohresser M. Evidence for linkage disequilibrium between Fcgamma RIIIa-V158F and Fcgamma RIIa-H131R polymorphisms in white patients, and for an Fcgamma RIIIa-restricted influence on the response to therapeutic antibodies. J Clin Oncol. 2008;26:5489–91, author reply 5491-2. doi: 10.1200/JCO.2008.19.4118. [DOI] [PubMed] [Google Scholar]
- 99.Danielou-Lazareth A, Henry G, Geromin D, Khaznadar Z, Briere J, Tamouza R, Cayuela JM, Thieblemont C, Toubert A, Dulphy N. At diagnosis, diffuse large B-cell lymphoma patients show impaired rituximab-mediated NK-cell cytotoxicity. Eur J Immunol. 2013;43:1383–8. doi: 10.1002/eji.201242733. [DOI] [PubMed] [Google Scholar]
- 100.Taskinen M, Karjalainen-Lindsberg ML, Nyman H, Eerola LM, Leppä S. A high tumor-associated macrophage content predicts favorable outcome in follicular lymphoma patients treated with rituximab and cyclophosphamide-doxorubicin-vincristine-prednisone. Clin Cancer Res. 2007;13:5784–9. doi: 10.1158/1078-0432.CCR-07-0778. [DOI] [PubMed] [Google Scholar]
- 101.Beurskens FJ, Lindorfer MA, Farooqui M, Beum PV, Engelberts P, Mackus WJ, Parren PW, Wiestner A, Taylor RP. Exhaustion of cytotoxic effector systems may limit monoclonal antibody-based immunotherapy in cancer patients. J Immunol. 2012;188:3532–41. doi: 10.4049/jimmunol.1103693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.van der Kolk LE, Grillo-López AJ, Baars JW, Hack CE, van Oers MH. Complement activation plays a key role in the side-effects of rituximab treatment. Br J Haematol. 2001;115:807–11. doi: 10.1046/j.1365-2141.2001.03166.x. [DOI] [PubMed] [Google Scholar]
- 103.Kennedy AD, Beum PV, Solga MD, DiLillo DJ, Lindorfer MA, Hess CE, Densmore JJ, Williams ME, Taylor RP. Rituximab infusion promotes rapid complement depletion and acute CD20 loss in chronic lymphocytic leukemia. J Immunol. 2004;172:3280–8. doi: 10.4049/jimmunol.172.5.3280. [DOI] [PubMed] [Google Scholar]
- 104.Aue G, Lindorfer MA, Beum PV, Pawluczkowycz AW, Vire B, Hughes T, Taylor RP, Wiestner A. Fractionated subcutaneous rituximab is well-tolerated and preserves CD20 expression on tumor cells in patients with chronic lymphocytic leukemia. Haematologica. 2010;95:329–32. doi: 10.3324/haematol.2009.012484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lindorfer MA, Wiestner A, Zent CS, Taylor RP. Monoclonal antibody (mAb)-based cancer therapy: Is it time to reevaluate dosing strategies? Oncoimmunology. 2012;1:959–61. doi: 10.4161/onci.20368. [DOI] [PMC free article] [PubMed] [Google Scholar]