

Abstract
Here, we describe a fast, easy-to-use, and sensitive method to profile in-depth structural micro-heterogeneity, including intricate N-glycosylation profiles, of monoclonal antibodies at the native intact protein level by means of mass spectrometry using a recently introduced modified Orbitrap Exactive Plus mass spectrometer. We demonstrate the versatility of our method to probe structural micro-heterogeneity by describing the analysis of three types of molecules: (1) a non-covalently bound IgG4 hinge deleted full-antibody in equilibrium with its half-antibody, (2) IgG4 mutants exhibiting highly complex glycosylation profiles, and (3) antibody-drug conjugates. Using the modified instrument, we obtain baseline separation and accurate mass determination of all different proteoforms that may be induced, for example, by glycosylation, drug loading and partial peptide backbone-truncation. We show that our method can handle highly complex glycosylation profiles, identifying more than 20 different glycoforms per monoclonal antibody preparation and more than 30 proteoforms on a single highly purified antibody. In analyzing antibody-drug conjugates, our method also easily identifies and quantifies more than 15 structurally different proteoforms that may result from the collective differences in drug loading and glycosylation. The method presented here will aid in the comprehensive analytical and functional characterization of protein micro-heterogeneity, which is crucial for successful development and manufacturing of therapeutic antibodies
Keywords: native mass spectrometry, monoclonal antibodies, glycosylation, antibody-drug conjugates, biopharmaceuticals, biosimilars, protein micro-heterogeneity, proteoforms
Introduction
Controlling and understanding the protein micro-heterogeneity of monoclonal antibodies (mAbs) both in a qualitative and quantitative manner represents one of the main focuses in the development and manufacturing of this class of therapeutics. Post-translational modification (PTM) on mAbs needs to be minutely characterized because it may affect antibody structure, efficacy, and potency, and its potential antigenicity or immunogenicity. The most widespread PTM occurring on mAbs is N-glycosylation. The nature of the glycan chains influences Fc-effector function and serum half-life.1,2 In particular, the lack of core fucosylation enhances antibody dependent cellular cytotoxicity (ADCC),3,4 while the presence of (α2–6)-linked sialic acids (N-acetylneuraminic acids) may be beneficial for anti-inflammatory activity.5-7 mAb glycosylation can be very diverse in nature, leading to an extensive molecular heterogeneity of the glycoprotein. Wild-type mAbs typically exist as a mixture of 3–5 different glycoforms, with their nature and abundance highly dependent on the cell line and expression system used. Human antibodies expressed in non-human cell lines can bear non-human carbohydrate chains (e.g., containing N-glycolylneuraminic acid, the galactose(α1–3)galactose epitope or xylose) that may trigger undesired immunogenic responses. Therefore, regulatory authorities require a thorough qualitative and quantitative analysis of mAb glycosylation for the preparation of medicinal products.
Other modifications frequently occurring on mAbs are disulfide pairings, N- and C-terminal modifications such as pyroGlu, Lys and Gly clipping,8 which also require qualitative and quantitative analysis. At present, it is not possible to analyze all this micro-heterogeneity using a single method. For the most part, efforts so far have been directed toward the analysis of mAbs glycosylation. Several analytical approaches to characterize antibody N-glycosylation targeted at different molecular levels, i.e., the glycan, the glycopeptide and the glycoprotein level, have been developed. One of the dominant approaches currently used is analysis at the glycan level, whereby the glycan chains are first enzymatically released from the protein. Subsequently, these glycans, eventually derivatized with a fluorescent label at the reducing side (reductive amination), are analyzed using chromatographic methods, such as normal-phase liquid chromatography (NP-LC), hydrophilic interaction liquid chromatography (HILIC), and high-performance anion-exchange chromatography (HPAEC), regularly combined with intermediate exo-glycosidase digestions.9-12 Alternatively, mass spectrometry (MS) can also be used for the analysis at the glycan level, mostly by using matrix-assisted laser desorption ionization (MALDI) in combination with time of flight (TOF) analyzers. A general drawback of analyzing only at the glycan level is that the connectivity to the protein of origin is lost, which becomes a problem if the protein to be analyzed is not very pure. An additional drawback of using MALDI-MS for the analysis of glycans is represented by the relatively poor ionization efficiency, especially of sialylated glycans, that severely hampers their detection, and thus quantitative analysis.13-15
Analysis by MS at the glycopeptide level has recently become more perceptible, partly to overcome some of the aforementioned shortcomings.16 In this sort of analysis, the connectivity issue is evidently solved; however, analysis on the glycopeptide level is still immature, needing more advanced and dedicated chromatographic approaches. Moreover, in this case proteolysis (e.g., by trypsin) needs to be complete and reproducibly controlled, which typically becomes more difficult when a protein is highly glycosylated. Additionally, glycopeptide fragmentation in MS has not always been sufficiently informative. This last issue may be (partly) solved as it has recently been shown that glycopeptides can be more efficiently fragmented and characterized using in series collision-induced dissociation (CID) and electron transfer dissociation (ETD), which target the structures of the glycans and peptide backbone, respectively, and can also provide information on their structural branching.17-19
Antibody glycosylation can also be investigated at the intact protein level through a combination of chromatography and MS. Investigations at the intact protein level have the advantage of requiring less sample handling, but glycan analyses of intact proteins by chromatography and MS are still in their infancy compared with measurements at the peptide and glycan level. Most commonly, these mass spectrometric analyses are performed under denaturing conditions, with the protein eluted in an acidified organic water mixture using LC-MS,20-22 prior to ionization by electrospray. Current TOF mass analyzers provide sufficient resolving power to resolve (less complex) glycan profiles on the intact proteins in a qualitative and relative quantitative manner. Alternatively, mAbs can be mass analyzed by native MS, wherein the protein is ionized in an aqueous ammonium acetate buffer.23 Native MS seems to retain the protein in a more folded structure, whereby the proteins also become substantially less charged in the ionization process. This implies, however, that they need to be detected at high m/z values, which so far has been little explored on ion traps or Orbitraps.
In this work, we aim for the detailed characterization of highly complex micro-heterogeneity, including glycosylation profiles, on intact native mAbs. We use the recently described Orbitrap Exactive Plus (ThermoFisher Scientific) that has been modified to perform native MS.24 We show that this instrument is capable, through its high sensitivity, mass accuracy and resolving power, of providing baseline separation of the different proteoforms on intact half- (~75 kDa) and full-mAbs (~150 kDa). The analysis by native MS on the Orbitrap at the intact protein level provides a number of advantages. Most importantly, a single highly resolved profile of all protein micro-heterogeneity could be obtained within a few minutes using a few femto-mole of sample, making it a time- and cost-efficient tool for routine analysis. Very little sample preparation is required, as the direct injection into the mass spectrometer excludes the need for a chromatographic step prior to MS analysis. Additionally, differences in the chemical nature of the glycan chains do not substantially affect the ionization efficiency of the intact protein, allowing the relative quantification of all proteoforms/glycoforms, including highly sialylated glycans. The detailed qualitative and quantitative profiles we observe reveal, in some cases, more than 30 different proteoforms of a single mAb,25 extending the depth of structural characterization usually obtained by current technologies.
Results
Benchmarking the performance of native MS using an Orbitrap mass analyzer in the characterization of protein micro-heterogeneity of intact full-length mAbs
To test the performance and demonstrate the versatility of the new analysis workflow, we selected three different samples: (1) a full-length (150 kDa), hinge deleted, IgG4 that exists in equilibrium with its half-antibody (75 kDa); (2) IgG4 mutants exhibiting highly complex glycosylation profiles; and (3) an IgG1 antibody-drug conjugate (ADC).
We benchmarked our approach evaluating the glycosylation profile on a wild-type, hinge-deleted IgG4 antibody (ΔhingeIgG4). The deletion of the hinge region excludes intermolecular disulfide bonds between the two heavy chains, making the dimerization of the two half-antibodies occur solely through non-covalent interactions.26 The full native mass spectrum of the ΔhingeIgG4WT antibody is shown in Figure 1A. Notably, as described earlier27 this spectrum can be generated in a matter of a few minutes, consuming just a few femtomoles of sample. The native MS spectrum provides a glimpse of the equilibrium, caused by the deletion of the hinge region, that exists between the half- and full-antibody in solution at the particular concentration used, from which dimerization constants can be determined.26 This feature enables the dedicated analysis of the glycosylation profile at both the half- and full-antibody level in a single spectrum. The mAb protein micro-heterogeneity caused by the diverse glycosylation becomes apparent when zooming-in on a single charge state (Fig. 1A in-sets). Multiple peaks corresponding to the different glycoforms are easily baseline-resolved at high S/N levels, allowing very accurate mass measurement and, therefore, reliable proteoform assignment.

Figure 1. Antibody glycosylation analysis at the intact protein level by native Orbitrap MS. In (A) the full native mass spectrum of an ΔhingeIgG4WT antibody is shown, revealing two charge-states envelopes originating from the half- (~m/z 4000) and full-antibody (~m/z 6000) being in equilibrium. The in-sets show enlarged single charge state spectra for both the half- and full-antibody, highlighting the micro-heterogeneity caused by glycosylation. The convoluted zero-charge mass spectra and glycan assignments, of the major components, are shown for the half-antibody in (B) and the full-antibody in (C). In case of more than one possible schematic structure for a (Neu5Aca)(Galb)MancGlcNAcdFuce composition, only one isoform is included; additional isomeric structures are displayed in Figure S4
First, we evaluated the micro-heterogeneity profile on the half-antibody (~75 kDa). Therefore, the spectra were convoluted to zero-charge, taking the signals at all detected charge states into account, which facilitated mass assignment (Fig. 1B). The masses found experimentally were in very good agreement with the theoretical masses of the protein sequence with the addition of glycan structures typically observed when expressing mAbs in a human (HEK-293F) cell line (Table S1). After identification and assignment of the most naive glycan (G0F in this case), the subsequent peaks in the spectrum can be easily assigned by examination of the mass shifts, which correspond to the addition of single successive monosaccharide residues.
Additionally, convolution to a zero charge mass spectrum provides a means to measure peak intensities, which are based on the sum of the intensities over all detected charge states peaks. Therefore, we also have quantitative information on the abundance of the different proteoforms largely caused by the micro-heterogeneity in the glycosylation of the protein backbone. The likely confidence of this quantitative data are very high, as it may be hypothesized that different glycan structures have negligible influence on the ionization efficiency of the much larger protein backbones. We used the quantitative experimental information of the different glycovariants of the half-antibody to predict the identity and the abundance of the glycovariants present in the full-antibody (~150 kDa) (Fig. 1C). Because the full-antibody consists of a pair of two half-antibodies, all glycan combinations are possible, including asymmetric species, i.e., full-antibodies carrying two different glycans on the two halves. We used a statistical model to predict the abundances of all possible pair combinations based on the abundances of the glycans on the half-antibody. A comparison between the predicted and experimental glycan profiles is given in Figure S1. The quantitative similarity between the predicted and measured spectrum is excellent, providing evidence that, at the ensemble level, the glycan profiles are alike on both halves of the antibody.
Glycosylation profiles and further micro-heterogeneity on ΔhingeIgG4 antibody mutants
We next analyzed several ΔhingeIgG4 single-point mutants that are known to display more complex glycosylation profiles than the wild-type ΔhingeIgG4.28 Recently, Rose et al. revealed that specific point mutations in the CH3 domain of ΔhingeIgG4 antibodies affect both the CH3-CH3 interaction strength, and therefore their dimerization constant, and their glycosylation profiles.28 The change in glycosylation is a surprising finding because the glycosylation site is quite distant from the CH3 domain. Among all samples previously analyzed with more conventional methods such as HPAEC-PAD and MALDI-TOF, the Y407E, Y407A, Y407Q and Y407K mutants showed high predominance of the half-antibody form, as well as the most diverse glycosylation profiles.
The zero-charge convoluted spectra of Y407E, Y407A, Y407Q and Y407K are displayed in Figure 2. The high resolving power (see also Figs. S2 and S3) allows the discrimination of a plethora of peaks that are quite close in mass. The mass and structural assignment is rather straightforward, even for the lower abundant species in the very congested spectra. Based on the MS compositional analysis in terms of hexose, N-acetylhexosamine, deoxyhexose and sialic acid (Table S1), more than 20 different complex-type N-glycan compositions could be assigned within a single spectrum. Our data reveals a considerable increase in the number of N-glycan species compared with previous studies.28 Most notably, next to the glycan structures most frequently observed on mAbs expressed in human cell lines, we could identify lower abundant glycans that occur less frequently, but that are still allowed considering the N-glycan biosynthetic pathways typical of the human (HEK) cells. Notably, we could even detect tetra-antennary and trisialylated glycan structures, these being most prominent in the Y407K, Y407E and Y407Q mutants, and a structure with two Fuc residues that seemed to hint at the presence of an H- or Lewis-type antigen, which is not frequently detected in mAbs.

Figure 2. Overview of glycosylation profiles at the intact protein level of four IgG4 half-antibody mutants. As shown previously,26 mutants at Y407 in ΔhingeIgG4 induce a dramatic change in the half- and full-antibody equilibrium and substantially alter the glycosylation pattern. From top left to bottom right, the spectra obtained for Y407A, Y407K, Y407E and Y407Q are shown. Compared with the WT, these mutants display a relatively high sialic acid content. The schematic structures of the most abundant assigned N-glycans, taking into account the N-glycan biosynthetic pathways in human and the known N-glycosylation patterns of human mAbs, are displayed. In case of more than one possible schematic structure for a (Neu5Aca)(Galb)MancGlcNAcdFuce composition, only one isoform is included; additional isomeric structures are displayed in Figure S4. *Antibody species bearing the glycine truncation.
A glimpse of some of the most extended glycan structures assigned is provided in Figure 3, while a summary of all assigned structures is given in Figure S4. The glycan mass data were used to determine the stoichiometry of the Man, Gal, GlcNAc, Fuc, and Neu5Ac building blocks (Table S1), translated into the most likely N-glycan structures, based on literature data. Our analysis reveals even more clearly than previously reported that there is an intricate interplay between the mutations of Y407 in the CH3 domain and the glycosylation at N297 distantly located in the CH2 domain.28 A more detailed investigation of the data disclosed re-occurring lower abundant satellite peaks that appeared at the lower mass-side of each peak and had a mass consistently 57 Da lighter, most likely corresponding to the truncation of the C-terminal glycine. The amount of truncated antibody was consistently 13 ± 5%. If we also take these species into account, some of the recorded spectra displayed more than 30 proteoforms, due to the combination of glycosylation and Gly-truncation. Generally, the glycosylation profile was alike for the intact and the Gly-truncated antibody. This further demonstrates the advantage of the new method of analyzing intact antibodies by native MS on the modified Orbitrap, as all co-occurring micro-heterogeneities are observed in a single analysis.
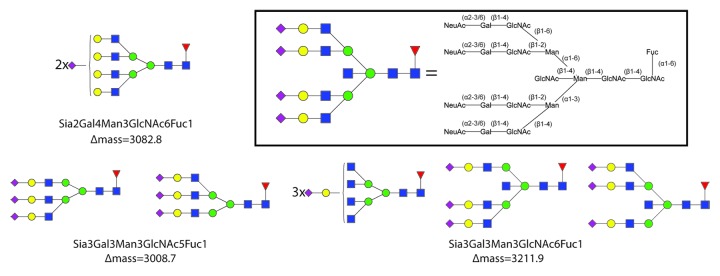
Figure 3. Overview of some of the most extended glycan structures identified on the IgG4 mutant antibodies. Typical examples of (sialylated) tri- and tetra-antennary N-glycans are presented. The in-set shows the relationship between the structure of a tetra-sialylated tetra-antennary N-glycan and the used symbolic notation for such a structure, clarifying also the symbolic notation of diantennary and triantennary structures. The Δmass, i.e., the mass shift of the glycosylated form compared with the deglycosylated form, is also indicated. See Figure S4 for an overview of all glycan structures identified and structurally assigned.
Next, we subjected the ΔhingeIgG4 Y407E mutant, which we found to be particularly rich in sialic acids, to enzymatic desialylation using the enzyme neuraminidase. Removal of the (α2–3)/(α2–6)-linked sialic acid residues could be achieved, resulting in a much more simplified native MS spectrum (Fig. S5). As a consequence of the loss of sialylated glycoforms, the relative abundances of the non-sialylated species do change as each sialylated species turns into its corresponding non-sialylated version. The increase of peak intensities in the desialylated sample was found to exactly match the intensities of their corresponding sialylated versions, confirming that both glycan identification and quantitation were correct.
Mass resolution and mass accuracy
Mass accuracy and resolution often decrease with increasing mass of the analyte, impeding the analysis of intact proteins, especially when compared with lower molecular weight species such as glycopeptides and released glycans. This may be due to the general lower instrumental resolution at high m/z and the incomplete desolvation of the analyte in ESI caused by salt or solvent adducts. To probe the accuracy of our method, we assessed the mass accuracy for all samples analyzed. The theoretical masses are compared with the experimental ones in Table S1. Theoretical masses were obtained from the protein and glycan sequences; experimental masses were assessed from well-calibrated native MS spectra. A systematic mass error of around +3 Da was observed for the half-antibodies, while this error doubles to 6 Da for the full-antibody (Fig. 4). From our experiments on the calibrant, i.e., CsI clusters generated by ESI,24 we know that the instrumental mass accuracy is two orders of magnitude better at 6000 m/z range (± 0.02 Da). Therefore, the shift in the peak maximum cannot be correlated with instrumental mass accuracy, but it might be attributed to an incomplete desolvation or substitution of one or more protons acquired by the mAb in the ESI process by Na+ or K+. In its current settings, our Orbitrap cannot fully resolve such very small mass differences. To explore this hypothesis, we simulated spectra of the wild-type ΔhingeIgG4 antibody: completely desolvated, or with one Na+, K+ or H2O adduct, taking also into account the natural isotopic peak widths. These simulations, as displayed in Figure 4, clearly reveal that the majority of mAb ions are completely “naked,” i.e., completely desolvated, while likely a very small fraction bears a single small molecule or cation adduct, causing a slight shift of the peak maximum (the 6 Da mentioned above) and a peak shoulder.
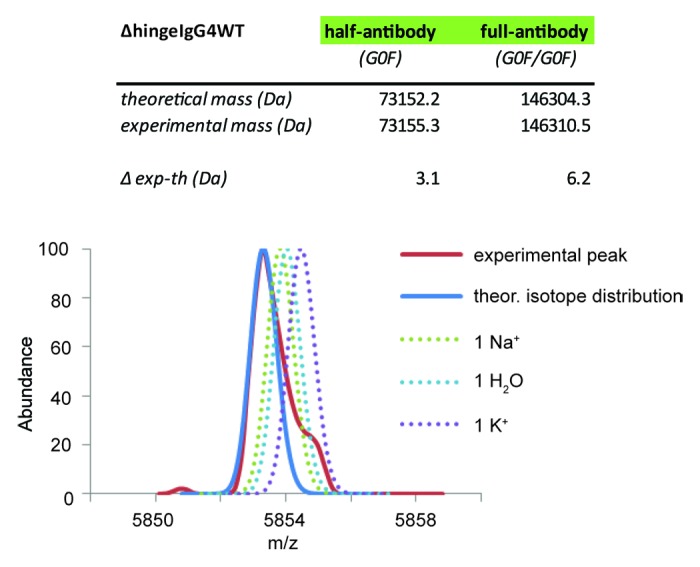
Figure 4. Limitations in accurate mass measurements in native Orbitrap MS of intact proteins. Comparison of the theoretical (blue line) and the experimental (red line) peak signal at a single charge state peak. The simulated peak of the ΔhingeIgG4WT antibody is displayed in solid blue when the ions are entirely desolvated (theoretical isotope distribution). Modeling into this peak the partial presence of a single water molecule, sodium (Na+) or potassium (K+) ion broadens the signal, leading to an increase in the experimentally measured mass.
Analyzing micro-heterogeneity in antibody-drug conjugates
The method described here may easily be extended to study induced protein micro-heterogeneity caused, for instance, by chemically-induced modifications. To illustrate this, we analyzed brentuximab vedotin (ADCETRIS®). This ADC is of particular interest because it carries a potent cytotoxic drug, monomethyl auristatin E, covalently attached via a maleimidecaproyl linker to cysteine residues of the IgG1 mAb that are usually involved in intermolecular disulfide bridges. To allow drug conjugation, disulfide bridges in the IgG1 are first (partially) reduced, where after the drug can be coupled. Because each reduced disulfide bridge exposes two free cysteine residues, it is expected that two drug molecules are conjugated per reduced disulfide bridge. Figure 5A depicts the native MS spectrum of the brentuximab vedotin ADC following deglycosylation by PNGase-F. The peaks arising originate from the intact mAb (150 kDa), but clearly show a diverse conjugation profile of the drug molecules. Species originating from the different drug-loads are clearly baseline resolved. Binding of the drug induces a mass shift of 2636 Da, corresponding to the binding of two drug molecules (one to each available cysteine residue). These spectra clearly indicate that the ADC product is not homogeneous in terms of drug-load, and antibodies carrying 0, 2, 4, 6, and 8 drug molecules co-exist. The average load could be semi-quantified as ~4.4, which is in agreement with previous findings.29,30 To examine whether deglycosylation by PGNase-F was really indispensable to characterization of the ADC, we also recorded the native MS spectrum of the unprocessed ADC (Fig. 5B). These spectra clearly demonstrate that the glycosylation-induced protein micro-heterogeneity profile of this ADC can be monitored in parallel with the drug-load in a single analysis.
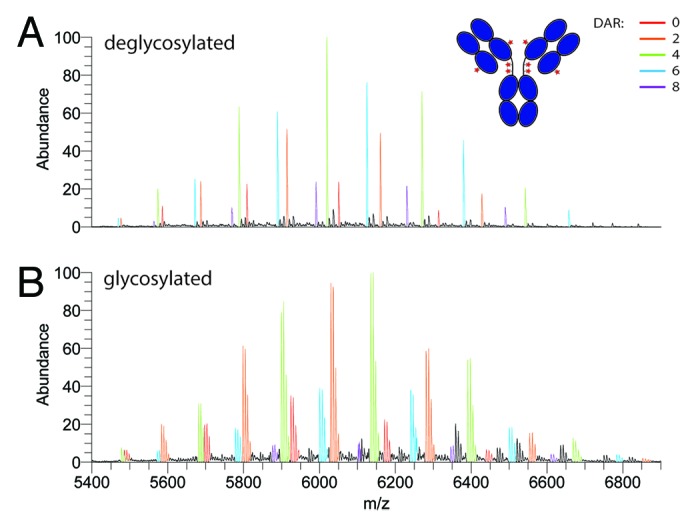
Figure 5. Analysis of the antibody-drug conjugate brentuximab vedotin (ADCETRIS®) at the intact protein level by native Orbitrap MS. Native spectra of the (A) deglycosylated and (B) glycosylated ADC. The differentially colored charge-state envelopes correspond to the different amount of drug molecules loaded onto the antibody. The drug loading clearly increases in steps of two, linked to the two accessible cysteine amino acids when a disulfide bridge is reduced.
Discussion
Here, we describe a method to minutely characterize at the intact protein level micro-heterogeneity originating from complex N-glycosylation, truncations and chemically- or biologically-induced modifications on mAbs. Performing native MS using an Orbitrap Exactive Plus instrument (ThermoFisher Scientific), we were able to identify, assign and quantify up to 25 different glycan structures present on IgG4 antibodies produced in human (HEK-293F) cells. We recently described the modifications made on the Orbitrap Exactive Plus instrument making it amendable for native MS.24,27 The high sensitivity, resolving power and mass accuracy make this instrument particularly interesting for the characterization of protein micro-heterogeneities. Hence, we focus here primarily on protein glycosylation. Our investigation on mass accuracy revealed that a large majority of the ions generated in the electrospray source reach the detector completely “naked” and, thus provide clear evidence that complete desolvation is feasible in the native state for 150 kDa protein assemblies. As a result, experimental mass measurements were found to be in agreement with the theoretical masses obtained from the mAb primary sequence and glycan structures. Incomplete desolvation and salt adducts (e.g., Na+ and K+) have always been a limitation in the analysis of intact proteins, especially in native MS where aqueous buffers are used. Water molecules and salt adducts cause broadening of the peaks and the shift of the peak maximum, both of which typically hamper exact mass measurements on larger proteins.31 As argued before, we hypothesize that the slightly harsher conditions in the source region of the Exactive Plus instrument ensure more efficient desolvation,24 thereby still retaining non-covalent protein-protein interactions.
The improved instrumentation performances led to an increase of the number of identified species, with special regard to the low abundant ones. In particular, referring to the analysis performed by Rose et al., we were now able to detect, besides other glycoforms, low abundant trisialylated glycoforms in mutant ΔhingeIgG4-Y407A and ΔhingeIgG4-Y407K, which were not observed previously. Glycosylation studies of highly sialylated antibodies using MS can be rather challenging, especially when the analysis is performed on released glycans, because the presence of a negatively charged carboxyl group severely affects their ionization efficiency. This issue is solved when the analysis is performed at the intact protein level. Efficient ionization is ensured by the protonation of basic residues of the protein, while the glycan chains remain neutral. This is confirmed by the fact that a native mass spectrum of a glycosylated antibody and a deglycosylated antibody show alike charge-state distributions. More importantly, because the electrospray-induced charging process primarily involves the protein backbone, which is identical in all components (or very similar in case of sequence variants), the ionization efficiency is not substantially affected by the differences in the glycan chains. This aspect is crucial to obtain (semi-) quantitative data.
A thorough characterization of glycosylation, both qualitatively and quantitatively, is imperative, especially for therapeutic antibodies. Such characterization may help in the analysis of batch-to-batch variability, and in biosimilar / reference product comparability exercises.32 The data acquired enable relative quantitation,33 directly from the native MS spectra.
As previously demonstrated by Rose et al.28 and confirmed by our analysis, the IgG4 glycosylation profile is highly influenced by a single point mutation in the CH3 domain, even though the glycosylation site is located in the CH2 domain. We previously showed using HD exchange MS that single mutations can significantly alter the tertiary structure, which may be a cause for the observed change in glycosylation.28
Some obvious advantages of performing protein micro-heterogeneity analysis by using native MS are the ease of sample preparation and the overall analysis speed. After antibody purification, just a buffer exchange step is required prior to mass spectrometric analysis. This normally requires only a couple of minutes per sample.31,34 Moreover, by using native MS, non-covalently assembled proteins, such as ΔhingeIgG4 antibodies and cysteine-conjugated ADCs (brentuximab vedotin in our case), can be still be maintained and analyzed in their native quaternary state. This allows the assessment of the drug-antibody ratio (DAR) for cysteine-linked ADCs.29,35
An obvious remaining limitation is represented by the inability to deduce monosaccharide stereoisomers, linkages, anomeric configurations, and glycan branching, which would still require dedicated glycan analysis by MS/MS or, for instance, NMR spectroscopy or LC combined with exo-glycosidases and methylation analysis.9,17,36-38
In conclusion, the results described here show the amount of information that can be obtained from a single native mass spectrum on the modified Orbitrap mass analyzer. A detailed picture of the most complex antibody glycosylation profiles can be drawn, but the presence of sequence variants can also be detected. In addition, quantitative data of all species identified can be easily obtained from peak intensities. In our opinion, biotechnology and biopharmaceutical companies working on therapeutic antibodies or other types of biotherapeutics can benefit substantially from the accuracy and the speed of this method. This kind of analysis can also be seen as a high-resolution fingerprint that can be used, for instance, for batch-to-batch comparisons or for comparability studies between biosimilar antibodies and their reference products.
Materials and Methods
Sample preparation
A ΔhingeIgG4WT antibody and related mutants ΔhingeIgG4Y407A, ΔhingeIgG4Y407E, ΔhingeIgG4Y407Q and ΔhingeIgG4Y407K were expressed in HEK-293F cells and purified as previously described.28 After purification, 25 µg of each sample were buffer-exchanged into 150 mM ammonium acetate pH 7.5, using 10 kDa MWCO centrifugal filter units (Amicon® Ultra, Millipore). Enzymatic desialylation of the ΔhingeIgG4Y407E sample was performed using the neuraminidase (sialidase) enzyme (Roche). After digestion, the desialylated sample was buffer-exchanged prior to mass spectrometric analysis. Brentuximab vedotin (Seattle Genetics) was dissolved in sterile water. Twenty-five micrograms were buffer-exchanged and analyzed as described.
Native MS
After buffer exchange, approximately 1 µL of 3 µM sample (antibody tetramer equivalent) was directly injected into the mass spectrometer using a gold-coated borosilicate capillary made in-house using a Sutter P-97 puller (Sutter Instrument Co) and an Edwards Scancoat six sputter-coater (Edwards Laboratories). All samples were analyzed using a slightly modified Exactive Plus instrument (ThermoFisher Scientific). Modifications and main settings were previously described.24 In particular, ions were trapped in the HCD cell filled with nitrogen at a pressure of 5 e−10 mbar. Resolution was set at 35 000, scans were acquired for a few minutes, combining 10 microscans.
Data analysis
Protein Deconvolution V2.0 (ThermoFisher Scientific) was used to convolute raw spectra and for mass assignment and relative quantitation.
Supplementary Material
Acknowledgments
This work was supported in part by STW (project 10805), the PRIME-XS project, Grant Agreement Number 262067, funded by the European Union Seventh Framework Program. The Netherlands Proteomics Centre, embedded in The Netherlands Genomics Initiative is acknowledged for funding. ETJvdB, JS and PWHIP are Genmab employees and have stock and/or warrants.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/mabs/article/26282
Footnotes
Previously published online: www.landesbioscience.com/journals/mabs/article/26282
References
- 1.Jefferis R. Glycosylation of recombinant antibody therapeutics. Biotechnol Prog. 2005;21:11–6. doi: 10.1021/bp040016j. [DOI] [PubMed] [Google Scholar]
- 2.Raju TS. Terminal sugars of Fc glycans influence antibody effector functions of IgGs. Curr Opin Immunol. 2008;20:471–8. doi: 10.1016/j.coi.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 3.Shinkawa T, Nakamura K, Yamane N, Shoji-Hosaka E, Kanda Y, Sakurada M, Uchida K, Anazawa H, Satoh M, Yamasaki M, et al. The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J Biol Chem. 2003;278:3466–73. doi: 10.1074/jbc.M210665200. [DOI] [PubMed] [Google Scholar]
- 4.Ferrara C, Grau S, Jäger C, Sondermann P, Brünker P, Waldhauer I, Hennig M, Ruf A, Rufer AC, Stihle M, et al. Unique carbohydrate-carbohydrate interactions are required for high affinity binding between FcgammaRIII and antibodies lacking core fucose. Proc Natl Acad Sci U S A. 2011;108:12669–74. doi: 10.1073/pnas.1108455108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaneko Y, Nimmerjahn F, Ravetch JV. Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science. 2006;313:670–3. doi: 10.1126/science.1129594. [DOI] [PubMed] [Google Scholar]
- 6.Anthony RM, Nimmerjahn F, Ashline DJ, Reinhold VN, Paulson JC, Ravetch JV. Recapitulation of IVIG anti-inflammatory activity with a recombinant IgG Fc. Science. 2008;320:373–6. doi: 10.1126/science.1154315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anthony RM, Wermeling F, Karlsson MC, Ravetch JV. Identification of a receptor required for the anti-inflammatory activity of IVIG. Proc Natl Acad Sci U S A. 2008;105:19571–8. doi: 10.1073/pnas.0810163105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beck A, Wagner-Rousset E, Ayoub D, Van Dorsselaer A, Sanglier-Cianférani S. Characterization of therapeutic antibodies and related products. Anal Chem. 2013;85:715–36. doi: 10.1021/ac3032355. [DOI] [PubMed] [Google Scholar]
- 9.Tharmalingam T, Adamczyk B, Doherty MA, Royle L, Rudd PM. Strategies for the profiling, characterisation and detailed structural analysis of N-linked oligosaccharides. Glycoconj J. 2013;30:137–46. doi: 10.1007/s10719-012-9443-9. [DOI] [PubMed] [Google Scholar]
- 10.Zauner G, Deelder AM, Wuhrer M. Recent advances in hydrophilic interaction liquid chromatography (HILIC) for structural glycomics. Electrophoresis. 2011;32:3456–66. doi: 10.1002/elps.201100247. [DOI] [PubMed] [Google Scholar]
- 11.Zaia J. On-line separations combined with MS for analysis of glycosaminoglycans. Mass Spectrom Rev. 2009;28:254–72. doi: 10.1002/mas.20200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Leeuwen SS, Schoemaker RJW, Timmer CJAM, Kamerling JP, Dijkhuizen L. N- and o-glycosylation of a commercial bovine whey protein product. J Agric Food Chem. 2012;60:12553–64. doi: 10.1021/jf304000b. [DOI] [PubMed] [Google Scholar]
- 13.Harvey DJ. Structural determination of N-linked glycans by matrix-assisted laser desorption/ionization and electrospray ionization mass spectrometry. Proteomics. 2005;5:1774–86. doi: 10.1002/pmic.200401248. [DOI] [PubMed] [Google Scholar]
- 14.Gil GC, Iliff B, Cerny R, Velander WH, Van Cott KE. High throughput quantification of N-glycans using one-pot sialic acid modification and matrix assisted laser desorption ionization time-of-flight mass spectrometry. Anal Chem. 2010;82:6613–20. doi: 10.1021/ac1011377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tep S, Hincapie M, Hancock WS. A MALDI-TOF MS method for the simultaneous and quantitative analysis of neutral and sialylated glycans of CHO-expressed glycoproteins. Carbohydr Res. 2012;347:121–9. doi: 10.1016/j.carres.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 16.Dell A, Morris HR. Glycoprotein structure determination by mass spectrometry. Science. 2001;291:2351–6. doi: 10.1126/science.1058890. [DOI] [PubMed] [Google Scholar]
- 17.Scott NE, Parker BL, Connolly AM, Paulech J, Edwards AV, Crossett B, Falconer L, Kolarich D, Djordjevic SP, Højrup P, et al. Simultaneous glycan-peptide characterization using hydrophilic interaction chromatography and parallel fragmentation by CID, higher energy collisional dissociation, and electron transfer dissociation MS applied to the N-linked glycoproteome of Campylobacter jejuni. Mol Cell Proteomics. 2011;10:M000031–MCP201. doi: 10.1074/mcp.M000031-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singh C, Zampronio CG, Creese AJ, Cooper HJ. Higher energy collision dissociation (HCD) product ion-triggered electron transfer dissociation (ETD) mass spectrometry for the analysis of N-linked glycoproteins. J Proteome Res. 2012;11:4517–25. doi: 10.1021/pr300257c. [DOI] [PubMed] [Google Scholar]
- 19.Ye H, Boyne MT, 2nd, Buhse LF, Hill J. Direct approach for qualitative and quantitative characterization of glycoproteins using tandem mass tags and an LTQ Orbitrap XL electron transfer dissociation hybrid mass spectrometer. Anal Chem. 2013;85:1531–9. doi: 10.1021/ac3026465. [DOI] [PubMed] [Google Scholar]
- 20.Srebalus Barnes CA, Lim A. Applications of mass spectrometry for the structural characterization of recombinant protein pharmaceuticals. Mass Spectrom Rev. 2007;26:370–88. doi: 10.1002/mas.20129. [DOI] [PubMed] [Google Scholar]
- 21.Olivova P, Chen W, Chakraborty AB, Gebler JC. Determination of N-glycosylation sites and site heterogeneity in a monoclonal antibody by electrospray quadrupole ion-mobility time-of-flight mass spectrometry. Rapid Commun Mass Spectrom. 2008;22:29–40. doi: 10.1002/rcm.3330. [DOI] [PubMed] [Google Scholar]
- 22.Kuribayashi R, Hashii N, Harazono A, Kawasaki N. Rapid evaluation for heterogeneities in monoclonal antibodies by liquid chromatography/mass spectrometry with a column-switching system. J Pharm Biomed Anal. 2012;67-68:1–9. doi: 10.1016/j.jpba.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 23.Heck AJ. Native mass spectrometry: a bridge between interactomics and structural biology. Nat Methods. 2008;5:927–33. doi: 10.1038/nmeth.1265. [DOI] [PubMed] [Google Scholar]
- 24.Rose RJ, Damoc E, Denisov E, Makarov A, Heck AJ. High-sensitivity Orbitrap mass analysis of intact macromolecular assemblies. Nat Methods. 2012;9:1084–6. doi: 10.1038/nmeth.2208. [DOI] [PubMed] [Google Scholar]
- 25.Smith LM, Kelleher NL, Consortium for Top Down Proteomics Proteoform: a single term describing protein complexity. Nat Methods. 2013;10:186–7. doi: 10.1038/nmeth.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rose RJ, Labrijn AF, van den Bremer ET, Loverix S, Lasters I, van Berkel PH, van de Winkel JG, Schuurman J, Parren PW, Heck AJ. Quantitative analysis of the interaction strength and dynamics of human IgG4 half molecules by native mass spectrometry. Structure. 2011;19:1274–82. doi: 10.1016/j.str.2011.06.016. [DOI] [PubMed] [Google Scholar]
- 27.Rosati S, Rose RJ, Thompson NJ, van Duijn E, Damoc E, Denisov E, Makarov A, Heck AJ. Exploring an orbitrap analyzer for the characterization of intact antibodies by native mass spectrometry. Angew Chem Int Ed Engl. 2012;51:12992–6. doi: 10.1002/anie.201206745. [DOI] [PubMed] [Google Scholar]
- 28.Rose RJ, van Berkel PH, van den Bremer ET, Labrijn AF, Vink T, Schuurman J, Heck AJ, Parren PW. Mutation of Y407 in the CH3 domain dramatically alters glycosylation and structure of human IgG. MAbs. 2013;5:219–28. doi: 10.4161/mabs.23532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Valliere-Douglass JF, McFee WA, Salas-Solano O. Native intact mass determination of antibodies conjugated with monomethyl Auristatin E and F at interchain cysteine residues. Anal Chem. 2012;84:2843–9. doi: 10.1021/ac203346c. [DOI] [PubMed] [Google Scholar]
- 30.Sievers EL, Senter PD. Antibody-drug conjugates in cancer therapy. Annu Rev Med. 2013;64:15–29. doi: 10.1146/annurev-med-050311-201823. [DOI] [PubMed] [Google Scholar]
- 31.Lorenzen K, van Duijn E. Native mass spectrometry as a tool in structural biology. Curr Protoc Protein Sci 2010; Chapter 17:Unit17 2. [DOI] [PubMed] [Google Scholar]
- 32.Beck A, Sanglier-Cianférani S, Van Dorsselaer A. Biosimilar, biobetter, and next generation antibody characterization by mass spectrometry. Anal Chem. 2012;84:4637–46. doi: 10.1021/ac3002885. [DOI] [PubMed] [Google Scholar]
- 33.Rosati S, Thompson NJ, Barendregt A, Hendriks LJ, Bakker AB, de Kruif J, Throsby M, van Duijn E, Heck AJ. Qualitative and semiquantitative analysis of composite mixtures of antibodies by native mass spectrometry. Anal Chem. 2012;84:7227–32. doi: 10.1021/ac301611d. [DOI] [PubMed] [Google Scholar]
- 34.Thompson NJ, Rosati S, Heck AJ. Performing native mass spectrometry analysis on therapeutic antibodies. Methods. 2013 doi: 10.1016/j.ymeth.2013.05.003. [DOI] [PubMed] [Google Scholar]
- 35.Chen J, Yin S, Wu Y, Ouyang J. Development of a native nanoelectrospray mass spectrometry method for determination of the drug-to-antibody ratio of antibody-drug conjugates. Anal Chem. 2013;85:1699–704. doi: 10.1021/ac302959p. [DOI] [PubMed] [Google Scholar]
- 36.Hokke CH, Bergwerff AA, Van Dedem GWK, Kamerling JP, Vliegenthart JFG. Structural analysis of the sialylated N- and O-linked carbohydrate chains of recombinant human erythropoietin expressed in Chinese hamster ovary cells. Sialylation patterns and branch location of dimeric N-acetyllactosamine units. Eur J Biochem. 1995;228:981–1008. doi: 10.1111/j.1432-1033.1995.tb20350.x. [DOI] [PubMed] [Google Scholar]
- 37.Llop E, Gallego RG, Belalcazar V, Gerwig GJ, Kamerling JP, Segura J, Pascual JA. Evaluation of protein N-glycosylation in 2-DE: Erythropoietin as a study case. Proteomics. 2007;7:4278–91. doi: 10.1002/pmic.200700572. [DOI] [PubMed] [Google Scholar]
- 38.Hocking HG, Gerwig GJ, Dutertre S, Violette A, Favreau P, Stöcklin R, Kamerling JP, Boelens R. Structure of the O-glycosylated conopeptide CcTx from Conus consors venom. Chemistry. 2013;19:870–9. doi: 10.1002/chem.201202713. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.