

Abstract
Background and Purpose
Given the diverse phenotypes including combined non-dyskinetic symptoms in patients harboring mutations of the gene encoding proline-rich transmembrane protein 2 (PRRT2), the clinical significance of these mutations in paroxysmal kinesigenic dyskinesia (PKD) is questionable. In this study, we investigated the clinical characteristics of PKD patients with PRRT2 mutations.
Methods
Familial and sporadic PKD patients were enrolled and PRRT2 gene sequencing was performed. Demographic and clinical data were compared between PKD patients with and without a PRRT2 mutation.
Results
Among the enrolled PKD patients (8 patients from 5 PKD families and 19 sporadic patients), PRRT2 mutations were detected in 3 PKD families (60%) and 2 sporadic cases (10.5%). All familial patients with a PRRT2 gene mutation had the c.649dupC mutation, which is the most commonly reported mutation. Two uncommon mutations (c.649delC and c.629dupC) were detected only in the sporadic cases. PKD patients with PRRT2 mutation were younger at symptom onset and had more non-dyskinetic symptoms than those without PRRT2 mutation. However, the characteristics of dyskinetic movement did not differ between the two groups.
Conclusions
This is the first study of PRRT2 mutations in Korea. The presence of a PRRT2 mutation was more strongly related to familial PKD, and was clinically related with earlier age of onset and common non-dyskinetic symptoms in PKD patients.
Keywords: paroxysmal dyskinesia, paroxysmal, dyskinesia, chorea, dystonia, PRRT2
Introduction
Paroxysmal kinesigenic dyskinesia (PKD) is the most common paroxysmal movement disorder and is characterized by brief, unilateral or bilateral involuntary movements that are precipitated by sudden movements.1-3 Recently, the gene encoding proline-rich transmembrane protein 2 (PRRT2) mutations are identified as the causative gene for PKD.4,5 In familial PKD patients, PRRT2 gene mutations, most commonly c.649dupC, were present in the majority of patients.4,6-8 However, in sporadic cases, the prevalence of PRRT2 gene mutations have been variously reported as 20-45%,7,9,10 and diverse PRRT2 gene mutations have been identified.5-7,9,11 In addition, many non-dyskinetic symptoms, such as migraine, dystonia, and seizure, can be combined in PKD patients.12 Even in PKD families, PRRT2 gene mutations can be inherited with only non-dyskinetic symptoms.6-9
Interestingly, PRRT2 gene mutations can manifest as a broad spectrum of phenotypes, and have been reported in patients with infantile convulsions with paroxysmal choreoathetosis or benign familial infantile epilepsy,8,10,13 and even in patients with migraine and episodic ataxia.14,15 Based on the diverse prevalence of PRRT2 mutations and the various phenotypes associated with these mutations, the meaning of the PRRT2 gene mutation in PKD remains a matter of debate.
This study investigated the clinical significance and genetic characteristics of PRRT2 gene mutations in PKD by comparing the clinical manifestations of Korean PKD patients with and without PRRT2 gene mutations.
Methods
Subjects
Patients with PKD at a movement disorders and pediatric outpatient clinic in Samsung Medical Center, and Soonchunhyang University Hospital, Seoul, Korea, were enrolled between July 2011 and November 2012. All subjects met the diagnostic criteria for PKD.12 The genealogical, clinical, and genetic data of the enrolled patients were investigated and analyzed. We compared the data of PKD patients with and without PRRT2 gene mutations. Seizure, writer's cramp, and migraine were assessed as non-dyskinetic symptoms.6-9,12 All subjects were briefed in detail concerning the study and provided written informed consent to participate. The study was approved by the Institutional Review Board of Samsung Medical Center.
Genetic analysis
Genomic DNA was isolated from peripheral blood leukocytes using the Wizard Genomic DNA Purification kit according to the manufacturer's instructions (Promega, Madison, WI, USA). We performed polymerase chain reaction (PCR) and direct sequencing of all coding exons and their flanking sequences for the PRRT2 gene using primer pairs designed by the authors (available upon request). PCR was performed in a Verti® thermal cycler (Applied Biosystems, Foster City, CA, USA) and cycle sequencing was performed using an ABI Prism 3100×l Genetic Analyzer with the BigDye Terminator Cycle Sequencing Ready Reaction kit (Applied Biosystems). Sequence variations were analyzed with reference to the wild-type sequence (RefSeq No. NM_145239.2) using the Sequencher program (Gene Codes, Ann Arbor, MI, USA).
Statistical analyses
All demographic and clinical data are presented as mean, SD, and median values. Differences between the PKD patients with and without PRRT2 gene mutations were evaluated using unpaired Student's t-test or Mann-Whitney U test for continuous and ordinary variables, while Pearson's chi-square test or Fisher's exact test was used for categorical variables. The level of statistical significance was set at p<0.05. All statistical analyses were conducted using PASW for Windows, version 18.0 (SPSS, Chicago, IL, USA).
Results
Clinical characteristics of the enrolled PKD patients
Eight familial PKD patients from 5 families and 19 sporadic PKD patients were enrolled (Fig. 1). In all enrolled subjects the paroxysmal movement was typically induced by sudden movements, and lasted for less than 1 min. Premonitory sensation or aura was reported in 15 patients (55.6%) and facial involvement was evident in 6 (22.2%). Dyskinetic movement usually involved the limbs (92.6%). In 55.6% of enrolled patients, the dyskinesia was shown unilaterally. The results of a physical examination, electroencephalography, and magnetic resonance imaging of the brain were normal in all subjects. Of the 27 patients, 5 did not need medication either because of remission or because they experienced only mild disturbance in their daily life; 18 of the 22 medically treated patients (18.8%) were in complete remission after medication.
Fig. 1.

The pedigrees of five Korean PKD families. Filled and open symbols indicate relatives with and without PKD, respectively. *Subjects who underwent PRRT2 gene mutation analysis, #Subjects with PRRT2 gene mutation. PKD: Paroxysmal kinesigenic dyskinesia, PRRT2: proline-rich transmembrane protein 2.
Non-dyskinetic symptoms were reported in three of the eight familial PKD patients (37.5%) and one (5.3%) sporadic patient. Among the four PKD patients with non-dyskinetic symptoms, non-febrile benign infantile seizure was noted in two subjects (family A II:3 and one sporadic patient). Migraine without aura was also seen in one subject (family C II:1). The remaining PKD patient had typical writer's cramp (family C III:1).
Identification of the PRRT2 gene mutation
Genetic analysis identified 3 PRRT2 gene mutations in 3 of the 5 PKD families (60%) and in 2 of the 19 sporadic cases (10.5%). The c.649dupC mutation, which is a known as hotspot mutation, was detected in three PKD families, but not in the sporadic cases. Two uncommon mutations (c.649delC and c.629dupC) that have mostly been reported in sporadic cases7,10,11 were also identified in two of the sporadic subjects in this study.
Clinical characteristics: PKD patients with and without PRRT2 mutations
Comparison of demographic data revealed that PKD patients with PRRT2 gene mutations reported being younger at symptom onset than those without gene mutations (Table 1). The clinical manifestation of paroxysmal dyskinesia did not differ between PKD patients with and without PRRT2 gene mutations. Although premonitory sensation was appeared to be more common in patients with PRRT2 gene mutations, the difference was not statistically significant. Comparison of non-dyskinetic symptoms revealed that PKD patients with PRRT2 gene mutations had more non-dyskinetic symptoms than those without. Assessment of the medications for treatment and the responses to medication revealed no significant difference between the PKD patients with and without PRRT2 gene mutations.
Table 1.
Comparison of demographic and clinical data between the PKD patients with and without PRRT2 gene mutations
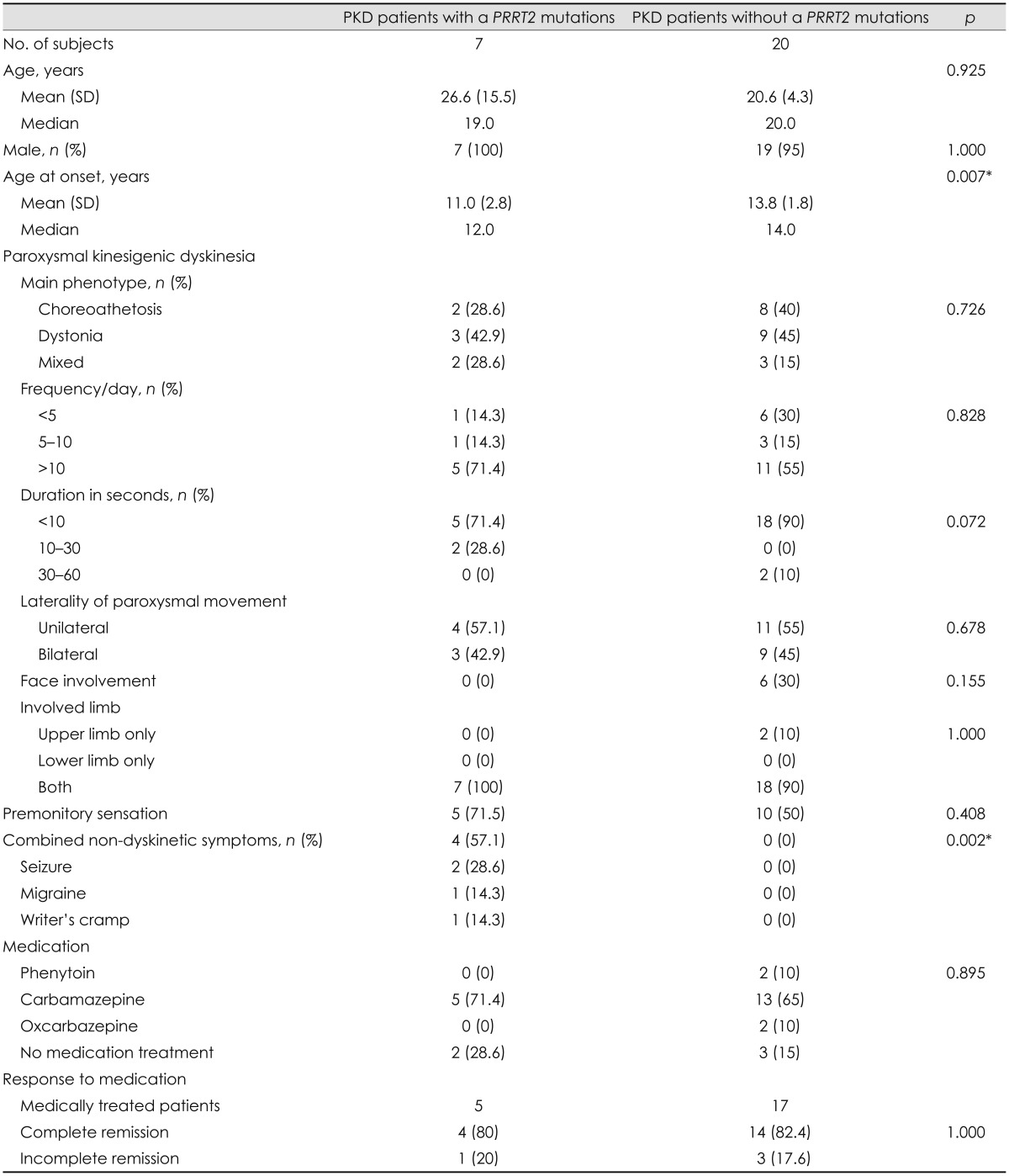
*p<0.05.
PKD: paroxysmal kinesigenic dyskinesia, PRRT2: proline-rich transmembrane protein 2.
Discussion
We investigated the clinical meaning of PRRT2 gene mutations in Korean PKD patients. This is the first study on the PRRT2 mutation in Korean population. Despite many reports of PRRT2 mutations in patients with PKD and other diseases, their clinical role in PKD remains unclear and debatable. Only a few studies have compared the clinical characteristics of PKD patients with and without PRRT2 gene mutations, and their results have been equivocal.16,17 PKD patients with PRRT2 gene mutations typically report an earlier age of symptom onset and are more likely to have non-dyskinetic symptoms than those without gene mutations.
Echoing previous studies,16,17 PKD patients with PRRT2 gene mutations were younger at disease onset in the present study. Assessment of the clinical data revealed that the clinical features of paroxysmal movement were highly typical and homogeneous, similar to previous studies.12,18 Although premonitory sensation appeared to be more commonly observed in the PKD patients with PRRT2 gene mutations than those without, as reported previously,16 the difference was not statistically significant. These findings correspond well with a previous study that found no difference between these two groups, with the exception of premonitory sensation.16 However, another study found a higher prevalence of the choreoathetotic phenotype and longer duration of paroxysmal dyskinesia in PKD patients with PRRT2 gene mutations.17 In the present study, there was no significant difference between the two groups, even when the duration of episodes was dichotomized into <5 s and >5 s (p=0.580) just like the previous study.
Of great interest, PKD patients with PRRT2 mutations had more non-dyskinetic symptoms than those without gene mutations. This contrasts with a previous study that found no evidence of a relationship between the presence of a gene mutation and non-dyskinetic symptoms.16 This discrepancy may be attributable to the particular non-dyskinetic symptoms assessed in each study; while in the present study, seizure, migraine, and writer's cramp were assessed, that previous study assessed only seizure as non-dyskinetic symptoms.16 Comparison of the percentage of patients with seizure revealed no statistically significant difference between patients with and without PPRT2 gene mutations (p=0.060), in accordance with previous observations.16 The evaluation of various non-dyskinetic symptoms in PKD patients could be important because many previous studies already have found various non-dyskinetic symptoms in PKD patients6-9,12 and PRRT2 gene mutations in other diseases.14,15 Furthermore, PRRT2 gene mutations can be inherited with only non-dyskinetic symptoms.6,8,9
Presently, PRRT2 gene mutations were found in three of the five PKD families (60%); all of the detected PRRT2 gene mutation in the familial cases was c.649dupC. This finding corresponds well with previous studies identifying PRRT2 gene mutations as the cause in familial PKD.4,6-11 Although PRRT2 gene mutations have been detected in up to 90% of Chinese familial PKD patients,6 in other studies, the prevalence rates were 58-75% of familial PKD patients in Asian countries.4,10,16
In addition, among the PRRT2 gene mutations, c.649dupC is the most commonly reported in familial PKD patients.4-8,10 Interestingly, in the present study the prevalence of PRRT2 mutations in sporadic PKD patients was lower than those in previous studies,5-7,9,11 and the c.649dupC mutation was not found in sporadic patients. Similarly various mutations other than c.649dupC have been detected in sporadic PKD patients.8,9,11
The present study was subject to some limitations. Since only a small number of patients was enrolled, there is a possibility of selection bias. The differences between the results of the our study and those of previous studies could be attributable to this limitation. However, despite the small sample it was still possible to elucidate statistically the clinical features of PKD patients with PRRT2 gene mutation. In addition, since exon analysis of the PRRT2 was conducted using Sanger sequencing, large deletions, duplications or intron mutations may have been overlooked.
In conclusion, the prevalence of PRRT2 gene mutations was strongly related with familial PKD, and partially with sporadic PKD. Clinically, PKD patients with PRRT2 gene mutations may be associated with earlier onset and non-dyskinetic symptoms. Further studies involving larger patient populations and evaluations of diverse non-dyskinetic symptoms are needed.
Footnotes
The authors have no financial conflicts of interest.
References
- 1.Goodenough DJ, Fariello RG, Annis BL, Chun RW. Familial and acquired paroxysmal dyskinesias. A proposed classification with delineation of clinical features. Arch Neurol. 1978;35:827–831. doi: 10.1001/archneur.1978.00500360051010. [DOI] [PubMed] [Google Scholar]
- 2.Katschnig P, Schwingenschuh P, Chaudhary UJ, Edwards MJ, Lemieux L, Walker MC, et al. Paroxysmal limb dyskinesia induced by weight: an unusual case of cortical reflex seizures. Mov Disord. 2011;26:2438–2439. doi: 10.1002/mds.23874. [DOI] [PubMed] [Google Scholar]
- 3.Kertesz A. Paroxysmal kinesigenic choreoathetosis An entity within the paroxysmal choreoathetosis syndrome. Description of 10 cases, including 1 autopsied. Neurology. 1967;17:680–690. doi: 10.1212/wnl.17.7.680. [DOI] [PubMed] [Google Scholar]
- 4.Chen WJ, Lin Y, Xiong ZQ, Wei W, Ni W, Tan GH, et al. Exome sequencing identifies truncating mutations in PRRT2 that cause paroxysmal kinesigenic dyskinesia. Nat Genet. 2011;43:1252–1255. doi: 10.1038/ng.1008. [DOI] [PubMed] [Google Scholar]
- 5.Li J, Zhu X, Wang X, Sun W, Feng B, Du T, et al. Targeted genomic sequencing identifies PRRT2 mutations as a cause of paroxysmal kinesigenic choreoathetosis. J Med Genet. 2012;49:76–78. doi: 10.1136/jmedgenet-2011-100635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu XR, Wu M, He N, Meng H, Wen L, Wang JL, et al. Novel PRRT2 mutations in paroxysmal dyskinesia patients with variant inheritance and phenotypes. Genes Brain Behav. 2013;12:234–240. doi: 10.1111/gbb.12008. [DOI] [PubMed] [Google Scholar]
- 7.Méneret A, Grabli D, Depienne C, Gaudebout C, Picard F, Dürr A, et al. PRRT2 mutations: a major cause of paroxysmal kinesigenic dyskinesia in the European population. Neurology. 2012;79:170–174. doi: 10.1212/WNL.0b013e31825f06c3. [DOI] [PubMed] [Google Scholar]
- 8.van Vliet R, Breedveld G, de Rijk-van Andel J, Brilstra E, Verbeek N, Verschuuren-Bemelmans C, et al. PRRT2 phenotypes and penetrance of paroxysmal kinesigenic dyskinesia and infantile convulsions. Neurology. 2012;79:777–784. doi: 10.1212/WNL.0b013e3182661fe3. [DOI] [PubMed] [Google Scholar]
- 9.Liu Q, Qi Z, Wan XH, Li JY, Shi L, Lu Q, et al. Mutations in PRRT2 result in paroxysmal dyskinesias with marked variability in clinical expression. J Med Genet. 2012;49:79–82. doi: 10.1136/jmedgenet-2011-100653. [DOI] [PubMed] [Google Scholar]
- 10.Lee YC, Lee MJ, Yu HY, Chen C, Hsu CH, Lin KP, et al. PRRT2 mutations in paroxysmal kinesigenic dyskinesia with infantile convulsions in a Taiwanese cohort. PLoS One. 2012;7:e38543. doi: 10.1371/journal.pone.0038543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Groffen AJ, Klapwijk T, van Rootselaar AF, Groen JL, Tijssen MA. Genetic and phenotypic heterogeneity in sporadic and familial forms of paroxysmal dyskinesia. J Neurol. 2013;260:93–99. doi: 10.1007/s00415-012-6592-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bruno MK, Hallett M, Gwinn-Hardy K, Sorensen B, Considine E, Tucker S, et al. Clinical evaluation of idiopathic paroxysmal kinesigenic dyskinesia: new diagnostic criteria. Neurology. 2004;63:2280–2287. doi: 10.1212/01.wnl.0000147298.05983.50. [DOI] [PubMed] [Google Scholar]
- 13.Heron SE, Grinton BE, Kivity S, Afawi Z, Zuberi SM, Hughes JN, et al. PRRT2 mutations cause benign familial infantile epilepsy and infantile convulsions with choreoathetosis syndrome. Am J Hum Genet. 2012;90:152–160. doi: 10.1016/j.ajhg.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gardiner AR, Bhatia KP, Stamelou M, Dale RC, Kurian MA, Schneider SA, et al. PRRT2 gene mutations: from paroxysmal dyskinesia to episodic ataxia and hemiplegic migraine. Neurology. 2012;79:2115–2121. doi: 10.1212/WNL.0b013e3182752c5a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sheerin UM, Stamelou M, Charlesworth G, Shiner T, Spacey S, Valente EM, et al. Migraine with aura as the predominant phenotype in a family with a PRRT2 mutation. J Neurol. 2013;260:656–660. doi: 10.1007/s00415-012-6747-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tan LC, Methawasin K, Teng EW, Ng AR, Seah SH, Au WL, et al. Clinico-genetic comparisons of paroxysmal kinesigenic dyskinesia patients with and without PRRT2 mutations. Eur J Neurol. 2013 doi: 10.1111/ene.12142. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 17.Li HF, Chen WJ, Ni W, Wang KY, Liu GL, Wang N, et al. PRRT2 mutation correlated with phenotype of paroxysmal kinesigenic dyskinesia and drug response. Neurology. 2013;80:1534–1535. doi: 10.1212/WNL.0b013e31828cf7e1. [DOI] [PubMed] [Google Scholar]
- 18.Houser MK, Soland VL, Bhatia KP, Quinn NP, Marsden CD. Paroxysmal kinesigenic choreoathetosis: a report of 26 patients. J Neurol. 1999;246:120–126. doi: 10.1007/s004150050318. [DOI] [PubMed] [Google Scholar]