

NOD CD8+ DCs express increased CD40; targeting of autoantigen to these cells induces Th1 responses, not tolerance, unless CD40/CD40L interactions are blocked.
Keywords: type 1 diabetes, DEC-205, CD40, peripheral tolerance
Abstract
DCs are important mediators of peripheral tolerance for the prevention of autoimmunity. Chimeric αDEC-205 antibodies with attached antigens allow in vivo antigen-specific stimulation of T cells by CD8+ DCs, resulting in tolerance in nonautoimmune mice. However, it is not clear whether DC-mediated tolerance induction occurs in the context of ongoing autoimmunity. We assessed the role of CD8+ DCs in stimulation of autoreactive CD4+ T cells in the NOD mouse model of type 1 diabetes. Targeting of antigen to CD8+ DCs via αDEC-205 led to proliferation and expansion of β-cell specific BDC2.5 T cells. These T cells also produced IL-2 and IFN-γ and did not up-regulate FoxP3, consistent with an activated rather than tolerant phenotype. Similarly, endogenous BDC peptide-reactive T cells, identified with I-Ag7 tetramers, did not become tolerant after antigen delivery via αDEC-205: no deletion or Treg induction was observed. We observed that CD8+ DCs from NOD mice expressed higher surface levels of CD40 than CD8+ DCs from C57BL/6 mice. Blockade of CD40–CD40L interactions reduced the number of BDC2.5 T cells remaining in mice, 10 days after antigen targeting to CD8 DCs, and blocked IFN-γ production by BDC2.5 T cells. These data indicate that the ability of autoreactive CD4+ T cells to undergo tolerance mediated by CD8+ DCs is defective in NOD mice and that blocking CD40–CD40L interactions can restore tolerance induction.
Introduction
Defects in peripheral T cell tolerance can result in development of autoimmune diseases, such as type 1 diabetes. DCs are critical for induction and maintenance of peripheral tolerance of self-specific T cells, but conversely, DCs also play a significant role in autoimmune pathogenesis by aberrantly activating autoreactive T cells [1, 2]. During diabetes pathogenesis, DCs stimulate self-specific T cells that infiltrate the pancreas, causing the death of the insulin-producing β cells [3, 4]. NOD mice develop spontaneous autoimmune diabetes and model many aspects of type 1 diabetes pathogenesis in humans [5, 6]. Lymphocytes and APCs (macrophages and DCs) infiltrate the pancreatic islets of NOD mice early in life, and this progresses to destructive insulitis and development of hyperglycemia beginning ∼12 weeks of age [5, 7].
Genes that affect T cells and APCs have been linked closely with autoimmune diabetes susceptibility in NOD mice and type 1 diabetes patients, including MHCII, as well as IL-2 and IL-2Rα, CTLA-4, and IL-1R [8, 9]. Some of these genes that are known to affect T cell populations can also affect DCs: IL-2 inhibits Flt3L-dependent DC development, and NOD mice, with lower IL-2 levels, have more plasmacytoid DCs compared with C57BL/6 mice [10]. These genetic susceptibilities interact with environmental influences to allow for autoreactive T cell stimulation and β cell destruction. For example, both NOD mice and type 1 diabetes patients have IL-1 signatures associated with disease [11–13]. Inflammation plays a role in disease initiation and progression, possibly by altering DC number or phenotype that then increases autoreactive T cell stimulation [14]. DCs in NOD mice have been reported to have altered phenotypes, but current data do not present a clear picture [15]. For example, some studies report increased levels of NF-κB and costimulatory markers on NOD bone marrow-derived DCs, but other studies find decreased maturation markers on DCs [16, 17]. Experiments using Flt3L to boost DC numbers in NOD mice demonstrate that the timing of DC stimulation may determine whether DCs are therapeutic or pathogenic for diabetes [18].
Two main populations of conventional DCs reside in peripheral lymphoid tissues: CD11b+ DCs that are strong stimulators of CD4+ T cell effector responses and CD8+ DCs that can efficiently cross-present extracellular antigens. CD8+ DCs can induce tolerance in CD8+ and CD4+ T cells to antigens derived from apoptotic cells [19, 20]. These DCs can also induce the generation of CD4+ Tregs upon stimulation via surface expression of TGF-β [21]. Thus, CD8+ DCs are important for maintaining peripheral immune homeostasis in steady-state mice.
Endocytic lectins on DCs can internalize proteins for loading antigenic peptides on MHCI and MHCII [22, 23]. DEC-205 is one such lectin that is expressed specifically by CD8+ DCs [24]. Antigens can be delivered to CD8+ DCs by linking antigenic peptides to antibodies against DEC-205, leading to efficient MHCII-antigen complex expression on the DC surface [23]. If no secondary immune activation is given, such delivery of antigen to CD8+ DCs in nonautoimmune mice leads to tolerance of CD8+ and CD4+ T cells through deletion, anergy, or Treg induction [21, 25, 26]. Tolerance can be broken by addition of DC-maturing stimuli, such as TLRLs or CD40 stimulation [25, 26]. Under these conditions, stimulation of CD4+ T cells by antigen targeted to matured CD8+ DCs drives Th1 differentiation [27].
Little is known about DC-mediated tolerance induction in the context of autoimmunity. CD8+ T cells responsive to β cell autoantigens can undergo deletion in NOD mice after stimulation by CD8+ DCs after targeting of concomitant antigen to DEC-205 by chimeric antibodies [28]. However, despite the ability of DCs in nonautoimmune mice to tolerize CD4+ T cells, it is not yet clear whether CD4+ T cells, which are consistently responding to endogenous antigen during an ongoing autoimmune response, can be tolerized in a similar manner. Although targeting self-antigens to CD8+ DCs can inhibit disease in autoimmune models, such as experimental autoimmune encephalomyelitis and diabetes, induced against a pseudo-self antigen, in these systems the antigen was delivered prior to autoimmunity, induced by immunization, when the environment is in steady-state [29, 30].
In this study, we examined responses of naturally autoreactive BDC2.5 CD4+ T cells after stimulation by CD8+ DC in NOD mice. BDC2.5 T cells were cloned originally from spleen and LNs of a diabetic NOD mouse and respond to a peptide from chromagranin A, a self-antigen expressed in the granules of pancreatic β cells [31–33]. Here, we demonstrate that in NOD mice, in vivo stimulation of T cells from BDC2.5 TCR Tg mice with antigen targeted to CD8+ DCs leads to effector responses rather than T cell tolerance. These aberrant CD4+ T cell effector responses in NOD mice point to a loss of peripheral tolerance mechanisms and increased immune responses rather than the tolerance observed in nonautoimmune mice. Likewise, endogenous BDC-reactive CD4+ T cells expand rather than become tolerant after stimulation by CD8+ DCs. The observed BDC2.5 effector responses in NOD mice are inhibited by blocking CD40–CD40L interactions between the T cells and CD8+ DCs, indicating that this loss of tolerance can be corrected by inhibition of a proinflammatory pathway that may be aberrantly activated in the autoimmune microenvironment.
MATERIALS AND METHODS
Mice
All mice were bred and housed under specific pathogen-free conditions in facilities at the NIH, according to protocols approved by the Institutional Animal Care and Use Committee. Mice used in these studies were 7–10 weeks of age, with the exception of NOD.BDC2.5.Rag1+/− and Rag1−/− mice that were used at 2 weeks of age. Original breeding pairs of NOD, NOD.BDC2.5 TCR Tg, NOD.Thy1.1, NOD.scid, NOR, and C57BL/6 mice were obtained from The Jackson Laboratory (Bar Harbor, ME, USA), NOD.Idd3/5 mice from Taconic (Derwood, MD, USA), and NOD.MyD88−/− mice from Diane Mathis (Harvard University, Cambridge, MA, USA). Experiments were done with one to two mice/group in all experiments unless indicated otherwise.
Antibodies and flow cytometry
αCD40(1C10) and FoxP3(FJK-16s)-allophycocyanin antibodies were purchased from eBioscience (San Diego, CA, USA). I-Ag7(Ox-6)-PE, CD40(3/23)-FITC, IFN-γ(XMG1.2)-allophycocyanin, and IL-17(TC11-18H10)-PE antibodies were purchased from Becton Dickinson (San Jose, CA, USA). CD40L(MR1) and hamster IgG control antibodies were purchased from Bio X Cell (West Lebanon, NH, USA). CD4(GK1.5)-Pacific blue, Thy1.1(OX7)-allophycocyanin, Thy1.1-PerCP, Thy1.2(30-H12)-allophycocyanin, Thy1.2-PerCP, IL-2(JES6-5H4)-Alexa 488, IL-4(11B11)-PE, TNF-α(MP6-XT22)-PE, IL-12p40(C15.6)-allophycocyanin, CD11c(N418)-PerCP, CD8(53-6.7)-Pacific blue, CD80(16-10A1)-allophycocyanin, and CD86(GL-1)-FITC were purchased from BioLegend (San Diego, CA, USA). Viability was assessed by Aqua LIVE/DEAD fixable staining (Life Technologies, Grand Island, NY, USA). FMO controls were used to set gates. FMO controls are samples that include all fluorophores, except the color being analyzed, and are a good control for MFI measurements [34]. MFI from isotype controls were similar to FMO controls (data not shown).
Production of chimeric antibodies
αDEC-205 antibodies were generated from the NLDC-145 hybridoma and isotype controls from the III/10 hybridoma [35, 36]. Antibody heavy chains contained an altered IgG1 heavy-chain constant region that ablates binding by FcRs, followed by a linker sequence and the antigenic peptides [25, 37]. Iso-BDC and αDEC-BDC (1040-55, sequence RVRPLWVRME) were produced in a similar method, as reported previously [25]. Chimeric antibodies were purified using endotoxin-free protein G-sepharose (BioVision, Milpitas, CA, USA), and quantity and purity were checked by spectrophotometry and SDS-PAGE analysis. They were also tested for binding to CHO cells stably transfected with DEC-205 or DCIR2 (from Juliana Idoyaga, Steinman lab, The Rockefeller University, New York, NY, USA) and spleen DCs. αDEC-BDC binding to CHO lines was analyzed using a polyclonal goat anti-mouse IgG-PE (Jackson ImmunoResearch, West Grove, PA, USA), and binding to spleen DCs was analyzed by an anti-mouse IgG1-Alexa 488 (Life Technologies). All antibody batches were also tested for functional activity by BDC2.5 T cell proliferation in vivo. Chimeric antibody stocks were also assessed for endotoxin levels by Limulus amoebocyte lysate assay (Lonza, Basel, Switzerland). Only preparations with <0.2 EU/ml endotoxin were used in these studies.
Purification and stimulation of T cells
CD4+ T cells were recovered from spleens of BDC2.5 TCR Tg mice by negative selection of CD8-, CD11b-, DX5-, Gr1-, B220-, and Ter119-expressing cells over MACS columns (Miltenyi Biotec, Auburn, CA, USA) using biotin-labeled antibodies (BioLegend) and anti-biotin beads (Miltenyi Biotec). T cells were labeled with 5 μM CFSE (Life Technologies), and 1–2 × 106 cells were injected i.v. One day after T cell injection, 100 ng chimeric antibodies or controls were injected i.p., unless indicated otherwise. During CD40L-blocking experiments, 250 μg MR1 or hamster IgG was injected i.p., 1 day prior to T cell transfer and every 3 days thereafter to the conclusion of the experiment.
Induction of diabetes
CD4+CD25− BDC2.5 T cells were isolated from spleen by negative selection as above, with the addition of biotinylated αCD25 (BioLegend). T cells (5×104) were injected i.v. to NOD.scid mice. These scid mice were treated 1 day prior and concurrently with T cells with PBS or 500 ng αDEC-BDC. Mice were tested for increased glycemia by urine 3–4 days after T cell administration and by blood glucose measurement with a OneTouch Ultra glucose meter, daily, beginning 7 days after T cell administration. Mice were considered diabetic on the 1st day of 2 consecutive days with blood glucose levels >250 mg/dl. In indicated experiments, mice were treated i.p. with 250 μg anti-CD40L or control IgG (MR1 or Armenian hamster IgG; Bio X Cell), concurrently with T cell transfer and 3 days later.
Cytokine production
Splenocytes or LN cells (5×106) were cultured in 48-well cell-culture plates, with or without 4 ng/ml PMA (Sigma, St. Louis, MO, USA) and 1 μM ionomycin (Sigma) for 4 h in the presence of 5 μg/ml brefeldin A (Sigma). In indicated experiments, cells were cultured with or without 2.5 μg/ml BDC mimetope peptide (1040-55) overnight, followed by 4 h in brefeldin A. Cells were then washed and stained for surface markers, followed by fixation with Fixation/Permeabilization buffer (eBioscience). After permeabilization with Perm/Wash buffer (BD Biosciences, San Jose, CA, USA), cells were stained intracellularly for cytokines. All flow cytometry was performed on a CyAn (Beckman Coulter, Brea, CA, USA) or an LSRII (Becton Dickinson). Total cell numbers in lymphoid tissues were counted by gathering the total number of BDC2.5 T cells within the flow sample and the percentage of the total cell count in the tissue was assessed by trypan blue exclusion. The number of BDC2.5 T cells was divided by the fraction of the tissue examined by flow cytometry to determine the total number of BDC2.5 T cells in each tissue.
Endogenous BDC T cell responses
Female NOD mice between 8 and 10 weeks of age were given PBS or 100 ng αDEC-BDC i.p. One week later, mice were challenged with PBS or 500 ng αDEC-BDC plus 20 μg αCD40 (eBioscience) and 50 μg poly(I:C) (Invivogen, San Diego, CA, USA). Three or 4 days following the challenge, the pancreas, pLN, LN, and spleens were harvested, and CD4+ T cells were stained for BDC/I-Ag7 (NIH core facility) and the Treg markers CD25 and FoxP3. The pancreas was inflated with collagenase 3, followed by tissue disruption and filtration to obtain the lymphoid cells.
DC stimulation
poly(I:C) RNA (50 μg; Invivogen), 5 μg LPS (Sigma), and 20 μg αCD40 (eBioscience) were injected i.p., and spleens were taken 12 h later. DCs were obtained through collagenase D digest of the spleen as in ref. [10]. Cells were stained immediately for surface markers (CD11c, CD11b, CD8, B220) and costimulatory proteins or incubated for 4 h in the presence of 1 μg/ml LPS and 5 μg/ml poly(I:C) with 5 μg/ml brefeldin A, followed by surface staining and fixation/permeabilization and intracellular staining as with T cells. All DC stains were done in the presence of TruStain FcX (BioLegend).
Statistics
Experiments were compared by Student's t-test or Mann-Whitney test in GraphPad Prism (GraphPad Software, La Jolla, CA, USA).
RESULTS
Stimulation of BDC2.5 T cells by CD8+ DCs causes T cell proliferation and accumulation
Antigens targeted to and presented by CD8+ DCs via antibodies specific for the endocytic lectin DEC-205 induce T cell tolerance in nonautoimmune-prone mice. Here, we address the use of DC-targeted antigen for tolerance of autoreactive BDC2.5 CD4+ T cells in autoimmune-prone mice using chimeric αDEC-205. Antibodies against DEC-205, with a constant region of the Ig heavy chain that was mutated to inhibit FcR binding [25, 37], were linked to the 1040-55 peptide mimetope that has a high T cell stimulatory capacity for BDC2.5 T cells in vitro (hence, termed αDEC-BDC; Fig. 1A) [38]. These chimeric antibodies bind specifically to DEC-205 and in the spleen target antigen only to CD8+ DCs (Fig. 1B and C). Injection of αDEC-BDC induced proliferation of BDC2.5 T cells in vivo, whereas the nontargeted BDC2.5 mimetope peptide conjugated to an isotype control antibody did not induce proliferation of the BDC2.5 T cells in the spleen or LN, and the number of T cells observed was similar to T cell numbers from PBS-treated mice (Fig. 1D and E, and data not shown).
Figure 1. Targeting of a BDC-mimetope peptide to DEC-205+ DCs induces T cell expansion not tolerance.

(A) GelCode Blue staining of SDS-PAGE of αDEC-BDC and control IgG in a reduced gel, showing the increase in size of the chimeric antibody heavy chain (near 50 kDa) as a result of peptide addition. (B) αDEC-BDC staining on CD11c+CD8+ cells (black solid) and CD11c+CD11b+ cells (gray solid) or secondary-only staining on CD11c+CD8+ cells (black dashed) from spleen. (C) αDEC-BDC binding to CHO cells that stably express DEC-205 (left) or DCIR2 (right). (D) CFSE-labeled CD4+ T cells from BDC2.5 Thy1.1+ mice were transferred to NOD mice followed by treatment with PBS or Iso-BDC, and BDC2.5 T cells from lymphoid tissues were analyzed at Day 3. (E) CFSE-labeled CD4+ T cells from BDC2.5 Thy1.1+ mice were transferred to NOD mice followed by treatment with PBS or αDEC-BDC. Spleen, pLNs, and peripheral LNs were recovered at Day 3 or Day 10 after stimulation, and BDC2.5 T cells were assessed for proliferation by CFSE dilution. Histograms are gated on lymphocytes/live cell marker/CD4+/Thy1.1+ cells. Number above the bar indicates the percentage of CFSElo BDC2.5 T cells. (F) Total number of BDC2.5 T cells in spleen, pLN, and LN after PBS or αDEC-BDC at the indicated time-points. (G) Fold expansion of BDC2.5 T cells stimulated by αDEC-BDC compared with unstimulated (PBS-treated) BDC2.5 T cells at the indicated time-points. Proliferation was assessed by the number of CFSElo cells. Data are representative of five (E–G), three (B and C), or two (D) experiments. Error bars represent + sem (F and G). *P < 0.05 compared with PBS control; **P < 0.005 compared with PBS control.
To study the effects of targeting antigen to CD8+ DCs on CD4+ T cell responses from hosts with ongoing autoimmune responses, we adoptively transferred CFSE-labeled BDC2.5 T cells from Thy1.1+ mice to NOD (Thy1.2+) mice and then treated those mice i.p. with PBS or αDEC-BDC, 1 day later. Three days after stimulation, we examined the BDC2.5 T cells in the spleen, pLN, and peripheral LNs. αDEC-BDC induced several rounds of division in BDC2.5 T cells in all lymphoid tissues (Fig. 1E) and increased the total number of BDC2.5 T cells recovered (Fig. 1F and G). This early proliferative response of BDC2.5 T cells in NOD mice is similar to nonautoreactive CD4+ T cell responses observed at the same time-point in nonautoimmune-prone strains [23, 25]. BDC2.5 T cells from control mice not given the chimeric antibody remain CFSEhi in the spleen and LN (Fig. 1E). As expected, T cells found in the pLN respond to presentation of endogenous pancreatic antigen even in control mice. The level of proliferation varied between animals in all tissues but especially in the pLN; whereas αDEC-BDC treatment caused >85% of the cells to be CFSElo, proliferation of BDC2.5 T cells in PBS-treated NOD mice was generally between 15% and 40% (data not shown). This variable proliferation is likely the result of differing levels of immune activation in prediabetic NOD mice, with changes in subsequent autoantigen presentation to BDC2.5 T cells [4, 39]. This proliferation to endogenous antigen models the disease state in individuals with type 1 diabetes, where diabetogenic T cells will be in the presence of autoantigen presented by DCs.
We then examined the BDC2.5 T cells in lymphoid tissues 10 days after stimulation, a time-point at which DC-mediated tolerance induction would be expected [25, 26]. In NOD mice given αDEC-BDC, however, BDC2.5 T cells continue to proliferate and persist through Day 10 in spleen, pLN, and LN (Fig. 1E and F). Significantly increased numbers of BDC2.5 T cells were observed in spleen and LNs after αDEC-BDC treatment compared with cells from PBS-treated mice (Fig. 1F and G). BDC2.5 T cells expanded approximately sixfold compared with PBS-treated BDC2.5 T cells in LNs by Day 3 and maintain approximately fivefold-greater numbers than unstimulated cells at Day 10 (Fig. 1G). Proliferation of BDC2.5 T cells to endogenous antigen in the pLN of control mice meant that T cells proliferated in mice with or without targeted antigen, and thus there was not as strong an increase in cell numbers after treatment with αDEC-BDC (Fig. 1F and G). Expansion of BDC2.5 T cells at Day 10 was observed consistently over several log changes in dose of αDEC-BDC (from 2 ng to 8 μg chimeric antibody; data not shown). Therefore, T cells exhibit expansion, not deletion, after antigen targeting to DEC-205+ DCs.
BDC2.5 T cells stimulated by CD8+ DCs secrete Th1 effector cytokines and do not increase Foxp3 expression or block diabetes development
To determine if the T cell population stimulated by αDEC-BDC were anergic or displayed effector functions, cytokine secretion was measured. Even at 10 days when T cells would normally become tolerant, stimulated BDC2.5 T cells produced significant amounts of IL-2 and IFN-γ (Fig. 2A and B). T cells from control, PBS-treated mice produced much lower levels of both cytokines. This combination of Th1 effector cytokines is pathogenic during diabetes onset [40]. Little IL-4 or IL-17 was detected in BDC2.5 T cells after stimulation by CD8+ DCs, indicating that a conversion of the T cells to Th2 or Th17 subsets is not occurring (Supplemental Fig. 1). To determine if PMA and ionomycin stimulation is bypassing partial tolerance induced in BDC2.5 T cells stimulated by CD8+ DCs [41], we also stimulated the T cells in vitro with the BDC2.5 mimetope peptide. BDC2.5 T cells stimulated in vivo by CD8+ DCs secrete significantly more IFN-γ in response to in vitro peptide restimulation than BDC2.5 T cells from PBS-treated mice (Fig. 2C), indicating that targeting of antigen to DEC-205+ DCs is boosting the T cell response and is not tolerogenic.
Figure 2. BDC2.5 T cells stimulated by CD8+ DCs produce a Th1 effector phenotype and do not convert to Tregs.

(A) Ten days after in vivo stimulation with αDEC-BDC or PBS, BDC2.5 T cells from spleen and LNs were stained intracellularly for IL-2 and IFN-γ, with or without PMA and ionomycin (Iono.) stimulation. Cells are gated on size, a live marker, and CD4+Thy1.1+ cells. (B) Percentage of IFN-γ+ or IL-2+ BDC2.5 T cells after PMA/ionomycin stimulation at the indicated time-points. Average of four independent experiments + sem. (C) Percentage of BDC2.5 T cells in the indicated tissues producing IFN-γ in response to 16 h of in vitro stimulation, with and without antigenic peptide, after 10 days of in vivo stimulation with PBS (white) or αDEC-BDC (black). Average of two independent experiments + sem. (D) Percentage of BDC2.5 T cells that are positive for intracellular FoxP3 in the spleen of PBS-treated (open) or αDEC-BDC-treated (filled) mice after 10 days. Average of three independent experiments + sem. *P < 0.05 compared with PBS control.
We also examined if Treg numbers increased from conversion of BDC2.5 T cells to Tregs or proliferation of existing Tregs. Although previous studies have observed Treg induction after stimulation of CD4+ T cells by CD8+ DCs [21], we observed no significant increase in Treg numbers after stimulation by αDEC-BDC (Fig. 2D). Therefore, autoreactive CD4+ T cells stimulated by CD8+ DCs in NOD mice are not deleted or anergic and do not become Tregs, suggesting that they bypass tolerogenic signals that normally inhibit T cell responses in steady-state mice.
Another measure of tolerance induction is a decrease in the ability of T cells to cause disease. Thus, we isolated CD4+CD25− BDC2.5 T cells and injected them into NOD.scid mice in the presence or absence of αDEC-BDC. Targeting of antigen to CD8+ DCs did not block diabetes development in the NOD.scid mice, further confirming that this stimulation does not induce tolerance (Supplemental Fig. 2A).
Stimulation by CD8+ DCs elicits similar responses in naive, monospecific BDC2.5 T cells as total CD4+ T cells
As BDC2.5 T cells are not tolerized by CD8+ DC stimulation, we assessed whether differences in the BDC2.5 T cells related to previous antigen experience could be responsible for the lack of tolerance. BDC2.5 T cells proliferate as they traffic through the pLN, starting ∼18 days of age, as a result of developmental pancreatic antigen release [2, 4]. Therefore, many T cells in adult BDC2.5 Tg mice are antigen-experienced. Autoreactive T cells in NOD mice can also exhibit increased expression of secondary TCRs through endogenous TCRα rearrangement, and these secondary TCRs can alter T cell stimulation [42]. BDC2.5 TCR Tg mice were therefore bred onto a NOD.Rag1−/− background to prevent endogenous TCRα chain rearrangement and ensure a monospecific population. BDC2.5.Rag1−/− mice develop diabetes before 4 weeks of age, soon after T cells begin responding to endogenous antigen, as they lack Tregs [43]. We obtained T cells from 2-week-old BDC2.5.Rag1−/− mice or BDC2.5.Rag1+/− littermates to transfer T cells prior to stimulation by endogenous antigens [4, 44] and measured CFSE dilution and fold expansion 10 days after stimulation with αDEC-BDC. BDC2.5 T cells from BDC2.5.Rag1−/− and BDC.2.5.Rag1+/− mice proliferated even in PBS mice in response to endogenous antigen, likely because the T cells were antigen-naive [4, 44]. However, these T cells proliferated and expanded more in response to αDEC-BDC, indicating the lack of tolerance induction (Fig. 3A and B). Therefore, monospecific BDC2.5 T cells that have not been previously exposed to antigen also respond to antigenic stimulation by CD8+ DCs with immunogenic effector responses rather than tolerance induction.
Figure 3. Monospecific BDC2.5 T cells respond similarly to total BDC2.5 T cells.
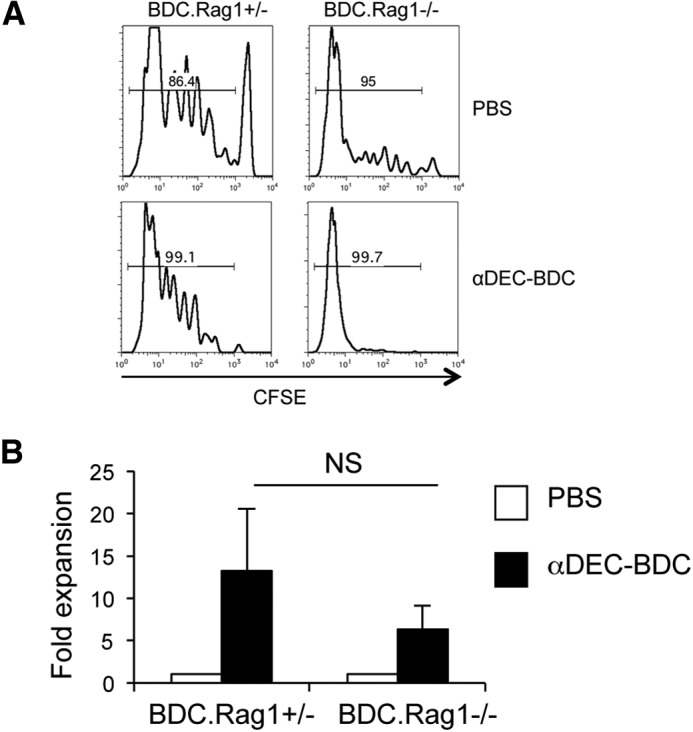
(A) CD4+ T cells were obtained from 2-week-old BDC2.5.Rag1+/− and BDC2.5.Rag1−/− mice, transferred to NOD.Thy1.1 mice, and then treated as indicated. After 10 days, LN cells were recovered, and BDC2.5 cell division was assessed by CFSE dilution. Percentage above the histogram is the percent of CFSElo BDC2.5 T cells. (B) Fold expansion of BDC2.5 T cells from Rag1+/− or Rag1−/− mice compared with cell numbers from PBS-treated mice. Data are the average of two experiments + sem.
T cell tolerance induction is not rescued in recipients that do not develop diabetes
DCs may be altered in NOD mice as a result of underlying genetics or inflammation associated with diabetes pathogenesis, which can result in the release of endogenous TLRL and inflammatory cytokines [2, 11, 12, 45]. Therefore, we transferred BDC2.5 T cells to NOD mice lacking the signaling molecule MyD88 (NOD.MyD88−/−), which is necessary for most TLR and IL-1R signaling. BDC2.5 T cells proliferated similarly in NOD and NOD.MyD88−/− mice after antigen targeting to CD8+ DCs, indicating that MyD88-dependent signals driven by TLR and IL-1R do not affect tolerance induction in this system (Fig. 4A).
Figure 4. Antigen-targeted CD8+ DCs from NOD.MyD88−/− or NOD.Idd3/5 mice induce similar T cell responses to CD8+ DCs in NOD mice.
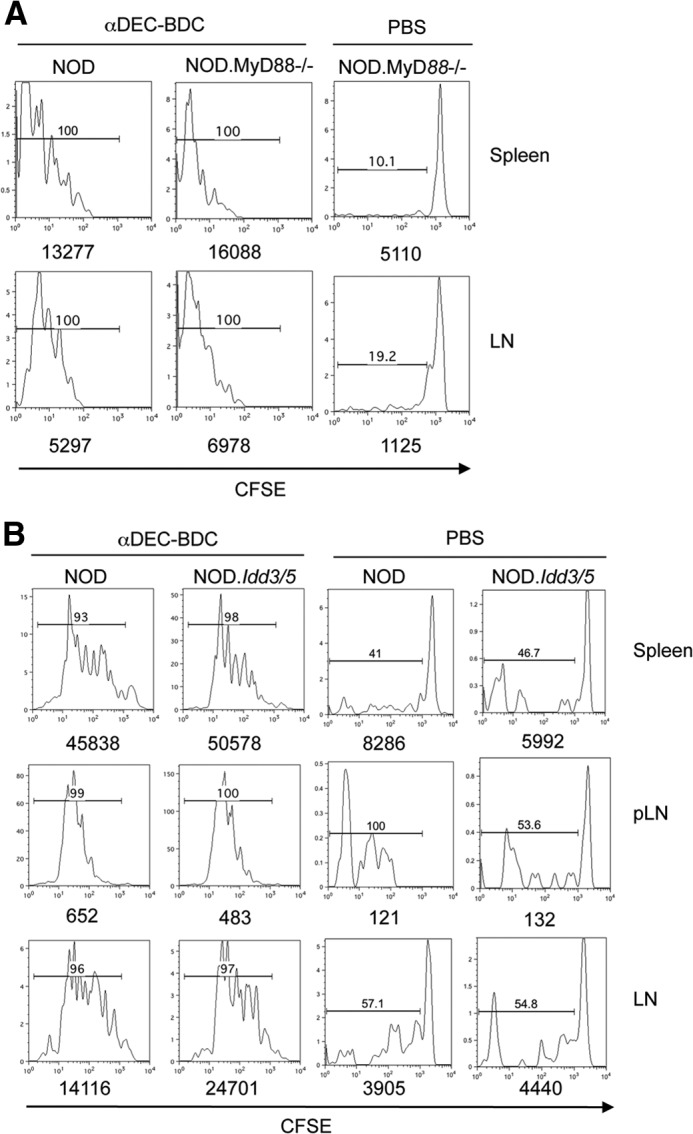
(A) BDC2.5 T cells were transferred to NOD or NOD.MyD88−/− mice, followed by PBS or αDEC-BDC treatment. After 10 days, BDC2.5 T cells in spleen and LNs were examined for proliferation based on CFSE dilution. The number above the bar is the percentage of BDC2.5 T cells that are CFSElo. Numbers below the graph are the total number of BDC2.5 T cells. (B) BDC2.5 T cells were transferred into NOD or diabetes-resistant NOD.Idd3/5 mice and then treated as indicated. Spleen, pLN, and LN cells were obtained at Day 10 and assessed for percentages of divided BDC2.5 T cells. Data are each representative of two independent experiments.
To determine if ongoing diabetogenic responses account for the loss of tolerance, we assessed the responses of BDC2.5 T cells in NOD.Idd3/5 mice. These mice are congenic strains that contain the Idd3 and Idd5 chromosomal regions from C57BL/6 and C57BL/10 mice, respectively, and rarely progress to diabetes (incidence <3%) [46]. Thus, we can use mice that have well-defined candidate genes that alter diabetes progression, including IL-2 (Idd3) and CTLA4 (Idd5). Proliferation of BDC2.5 T cells after stimulation by αDEC-BDC was similar to NOD mice in NOD.Idd3/5 mice (Fig. 4B). Similar experiments could not be performed in C57BL/6 mice congenic for the H-2g7 MHC haplotype because of deletion of NOD T cells in these mice, likely as a result of minor histocompatibility differences (data not shown). These data show that blocking endogenous TLRL or limiting autoimmune pathology does not alter the response of BDC2.5 T cells to antigen-specific stimulation by CD8+ DCs.
TLRLs increase T cell proliferation and IFN-γ secretion
Delivery of antigen to DCs, along with DC maturation factors, usually elicits a strong immune response and can bypass normally tolerogenic responses. DC defects have been observed previously in NOD mice in DC responses to stimulation and in their stimulatory capacity for T cells [15, 47]. We next examined if the lack of T cell tolerance after stimulation by CD8+ DCs in NOD mice was a result of maximally stimulating BDC2.5 T cells, even without exogenous maturation signals. CD8+ DCs preferentially express TLR3 [48], so the TLR3 ligand poly(I:C) RNA was administered during BDC2.5 T cell activation with agonistic αCD40 to further mature the DCs. poly(I:C) and αCD40 treatment induced increased expansion of BDC2.5 T cells in response to αDEC-BDC (Fig. 5A), as well as an increase in the percentage of BDC2.5 T cells that secrete IFN-γ (Fig. 5B). Costimulation is also important for the maintenance of Treg populations [49]. The percentage of FoxP3+ BDC2.5 T cells did not change after poly(I:C) and αCD40 administration, indicating that the increase in effector responses did not correspond with a change in regulatory populations (Fig. 5C). We can observe a greater T cell response when maturing CD8+ DCs, so CD8+ DCs in NOD mice have not maximized their T cell stimulatory capacity under untreated conditions.
Figure 5. DC-targeted antigen given with immune stimulation drives further BDC2.5 T cell expansion and effector function.
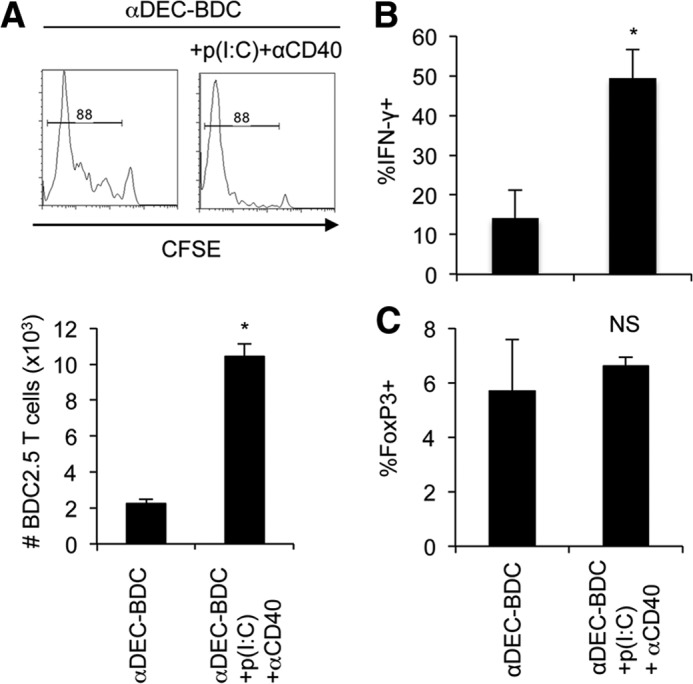
(A) BDC2.5 T cells were stimulated with αDEC-BDC, with or without poly(I:C) [p(I:C)] and αCD40, in vivo for 10 days. LN cells were isolated and BDC2.5 T cells were assessed for proliferation by dilution of CFSE. Bar graph (lower) indicates total number of BDC2.5 T cells after treatment with αDEC-BDC or αDEC-BDC + poly(I:C) + αCD40. (B) BDC2.5 T cells from A were stimulated with PMA and ionomycin and examined for IFN-γ by intracellular flow cytometry. (C) FoxP3 expression by intracellular flow cytometry was measured. Data are the average of two experiments + sem. *P < 0.05 compared with PBS control. NS, not significant.
αDEC-BDC treatment does not induce deletion or Treg induction in endogenous BDC-reactive T cells
We expanded our characterization of how stimulation by CD8+ DCs alters T cell responses by assessing the responses of endogenous BDC-reactive T cells. These T cells have a wider array of binding affinities for the BDC peptide on I-Ag7 rather than the single affinity of T cells isolated from BDC2.5 TCR Tg mice [50, 51]. NOD mice were treated with PBS or αDEC-BDC and then challenged 1 week later with αDEC-BDC plus TLRL and αCD40 to increase the number of rare tetramer+ cells for analysis. The challenge also gives a more stringent test of tolerance induction—cells in which tolerance is induced should not respond to even a strongly immunogenic systemic antigen delivery. T cells from lymphoid tissues and pancreas were interrogated with tetramers of BDC on I-Ag7. Pretreatment with αDEC-BDC did not lead to a decrease in BDC-reactive T cells; in fact, tetramer-positive cells were increased significantly in the pLN and spleen (Fig. 6A). The percentage of tetramer+ cells expressing FoxP3 was also measured, and initial treatment with αDEC-BDC led to a decrease in Tregs in the pancreas. In other lymphoid organs, Treg numbers were not affected significantly by treatment (Fig. 6B). These data show that the inability of αDEC-BDC to induce tolerance also occurs in the endogenous repertoire of BDC-reactive T cells and is not limited to TCR Tg cells.
Figure 6. αDEC-BDC fails to induce tolerance in endogenous CD4 T cells.

NOD mice were injected with PBS or αDEC-BDC, followed by a challenge with αDEC-BDC plus αCD40 and poly(I:C) 7 days later. Three to 4 days following the challenge the pancreas, pLN, LN, and spleen were analyzed for the percentage of BDC/I-Ag7 tetramer-positive cells among CD4+ T cells (A) and the percentage of Tregs (FoxP3+) among tetramer+ CD4+ T cells (B). Each point represents data from a single mouse; data compiled from two or three independent experiments. *P ≤ 0.05; **P ≤ 0.005.
Binding of αDEC-BDC does not alter DC phenotype
Previous studies have shown that antibody binding to DEC-205 does not alter DC phenotype [25], but the genetic differences and proinflammatory environment of NOD mice could change this. We tested whether αDEC-BDC alters DCs in NOD mice by treating the mice for 12 h with PBS, αDEC-BDC, or TLRL and αCD40 as a positive control for DC maturation. We examined the expression of the T cell-costimulatory proteins CD80 and CD86, as well as MHCII after αDEC-BDC treatment. αDEC-BDC did not induce maturation of CD8+ DCs, whereas TLRL treatment increased the amount on these DCs, as expected (Fig. 7A). We also assessed the percentage of CD8+ DCs that secreted the inflammatory cytokines TNF-α and IL-12 after in vivo injection of αDEC-BDC. Levels of cytokine produced were not affected by αDEC-BDC treatment, directly ex vivo or after stimulation with LPS in vitro (Fig. 7B). Therefore, αDEC-BDC does not induce TNF-α or IL-12 production by these DCs nor does it alter their capability to produce these cytokines. The phenotype of CD8+ DCs is not altered by binding of the chimeric antibodies in NOD mice.
Figure 7. CD8+ DCs from NOD mice are not matured by binding αDEC-BDC but do have higher expression of CD40 than DCs from C57BL/6 mice.

(A) NOD mice were treated with PBS, αDEC-BDC, or LPS + poly(I:C) + αCD40 for 12 h. Spleen DCs were recovered, and expression of maturation markers was assessed by flow cytometry for geometric MFI values, as expressed on CD11c+CD8+ DCs. Data are representative of three independent experiments. (B) After in vivo treatment with PBS or αDEC-BDC, splenocytes were cultured for 4 h in the presence or absence of LPS + poly(I:C) and then stained for intracellular cytokines in CD11c+CD8+ DCs. Average of three independent experiments + sem. (C) Expression of the costimulatory molecule CD40 on CD11c+CD8+ spleen DCs from NOD (solid line) and C57BL/6 (dashed line) mice or with isotype control (gray shaded). Each point in graph to the right represents data from a single mouse and shows the geometric MFI of CD40 or FMO control on CD11c+CD8+ DCs. Data are representative of four independent experiments. *P < 0.05; ***P < 0.0005.
CD8+ DCs in NOD mice have increased CD40 expression
As the examined alterations in DCs and T cells did not yield a tolerogenic signal for BDC2.5 T cells in NOD mice, we began to examine other costimulatory proteins involved in DC–T cell interactions. The CD40–CD40L pathway is a well-established DC:T cell costimulatory pathway that can alter diabetes progression in NOD mice [52]. CD40 and CD40L are up-regulated by BDC2.5 T cells after stimulation, and CD40 is a marker of activation of DCs [53]. We therefore compared the level of CD40 expression on CD8+ DCs in NOD and C57BL/6 mice, as previous studies have used C57BL/6 mice for CD4+ T cell tolerance induction. CD8+ DCs in NOD mice had a small but significant increase in CD40 levels (Fig. 7C). This increase was also observed in NOD.Idd3/5 mice and in NOD.MyD88−/− mice, indicating that the higher CD40 expression is not associated directly with diabetes pathogenesis and the related inflammatory state and correlates with our data showing that T cell responses after αDEC-BDC treatment are similar in NOD, NOD.Idd3/5 mice, and in NOD.MyD88−/− mice (Supplemental Fig. 3A and B). The increase in CD40 was also not indicative of a general activation of the DCs, as CD86 levels on DCs from NOD and C57BL/6 strains were similar (Supplemental Fig. 3C).
Blockade of CD40–CD40L interactions restores tolerance induction
We next tested the role of CD40–CD40L interactions in CD4+ T cell tolerance after antigen targeting to CD8+ DCs. We used a function-neutralizing antibody against CD40L to block this costimulatory pathway during BDC2.5 T cell stimulation by αDEC-BDC in vivo. With αCD40L treatment, BDC2.5 T cells still proliferate in response to αDEC-BDC, but total numbers of BDC2.5 T cells remaining at Day 10 are similar to that observed without stimulation, suggesting that some deletion is occurring (Fig. 8A and B). Blockade of CD40L had no effect on the percentage of Tregs observed among the BDC2.5 T cells (Fig. 8B, lower). We also examined cytokine secretion in BDC2.5 T cells from mice treated with αDEC-BDC and αCD40L. Secretion of IFN-γ by activated BDC2.5 T cells was reduced to background levels by blocking CD40L (Fig. 8C). BDC2.5 T cells in the spleen still produced IL-2 after CD40L blockade, but the percentage of IL-2+ cells was not significantly different from that observed in control mice (Fig. 8C). Blocking CD40–CD40L interactions delayed diabetes induction even without αDEC-BDC treatment; therefore, it was difficult to determine whether overactivation of this pathway was responsible for the inability of autoantigen targeted to CD8+ DCs to block diabetes development (Supplemental Fig. 2B; P=0.38 for PBS+anti(alpha)-CD40L vs. αDEC-BDC+αCD40L). We have shown that BDC2.5 T cells proliferate early after stimulation by CD8+ DCs without CD40–CD40L interactions, but fewer T cells survive, and these T cells do not produce IFN-γ. At a later time-point, we observed similar results as those found at Day 10: few BDC2.5 T cells remained in mice after 21 days of stimulation with αDEC-BDC, but the number remained significantly higher than PBS controls, whereas blocking CD40–CD40L interactions led to reduced numbers similar to PBS controls (Supplemental Fig. 4). Thus, this costimulatory pathway is necessary for full induction of effector responses.
Figure 8. Inhibiting CD40–CD40L interactions blocks T cell accumulation and cytokine secretion after stimulation by CD8+ DCs.

(A) BDC2.5 T cells were stimulated with PBS or αDEC-BDC and αCD40L or control IgG. Cells from lymphoid tissues were recovered at Day 10 and examined for cell number and proliferation. Numbers above the bar represent the percentage of divided BDC2.5 T cells, whereas numbers below the graph indicate the total number of BDC2.5 T cells in the lymphoid tissues. (B) Fold expansion of BDC2.5 T cells in spleen or LNs (upper) or percentage FoxP3+ of BDC2.5 T cells in spleen (lower) at Day 10 after the indicated stimulation compared with unstimulated cells with control IgG. Data are the average of three independent experiments + sem. *P < 0.05. (C) Cells from treated mice were assessed for IFN-γ and IL-2 production by intracellular flow cytometry. Data are the percent of BDC2.5 T cells positive for the indicated cytokines. Data are the average of three independent experiments + sem. *P < 0.05. NS, not significant.
DISCUSSION
Here, we have demonstrated that CD8+ DCs in NOD mice exhibit altered stimulation of CD4+ T cells. We have determined that autoreactive BDC2.5 T cells stimulated by antigen targeted to CD8+ DCs escape three major pathways of immune tolerance that are observed after stimulation of CD4+ T cells in nonautoimmune prone mice: T cell deletion, anergy, and Treg induction [21, 25, 26]. First, autoreactive T cell numbers increase and are maintained after stimulation by CD8+ DCs, so any cell deletion that may be occurring is offset by increased T cell proliferation to the autoantigen. Second, remaining BDC2.5 T cells are not anergic, as they produce robust IL-2 and IFN-γ responses. Importantly, no skewing of BDC2.5 phenotypes from a pathogenic Th1 response to Th2 or Th17 occurs after stimulation by CD8+ DCs. Third, BDC2.5 T cells did not convert into Tregs nor expand existing Treg populations after stimulation by CD8+ DCs, in contrast with studies performed in nonautoimmune mice [21]. In addition, αDEC-BDC was not able to induce deletion or Treg induction in endogenous autoreactive T cells, indicating that the lack of tolerance induction is not a result of the use of T cells of one particular affinity. The inability of αDEC-BDC to block diabetes induction gives further evidence for the lack of tolerance. Although one might expect such an activating signal to worsen disease, it may not be possible to see an acceleration of diabetes in the transfer model where homeostatic expansion expands the T cell populations rapidly.
BDC2.5 T cells have similar initial proliferative responses as nonautoreactive T cells in C57BL/6 mice but lack the later tolerogenic signals driven by DCs from nonautoimmune mice. Tolerance of CD4+ T cells following stimulation by CD8+ DCs in steady-state mice has been observed in several studies [23, 25]. Importantly, we have included several controls to show that our chimeric antibody is functioning to deliver the BDC peptide antigen as expected (Figs. 1 and 6) and that DCs are not phenotypically altered after targeting with our chimeric antibodies (Fig. 7). As NOD BDC2.5 T cells become tolerant in other studies when antigen is presented by cells other than DEC-205-expressing DCs [54–56], this suggests a specific defect in tolerance mediated by DEC-205+ DCs in autoimmune-prone NOD mice. Examination of stimulation of BDC2.5 T cells with other cell subsets in NOD mice or stimulation of T cells with alternate specificities will help clarify those characteristics that prevent tolerance.
We observed increased CD40 levels on CD8+ DCs in NOD mice and that inhibition of this pathway blocked continued expansion and cytokine production by BDC2.5 T cells after stimulation by CD8+ DCs (Figs. 7 and 8). Our data indicate that interaction between CD40 on CD8+ DCs and CD40L on T cells may lead to sustained T cell activation. Blocking CD40–CD40L interactions inhibits diabetes induction in NOD mice when given at early ages [52] and prevented diabetes development in NOD.scid mice after T cell transfer with or without treatment with αDEC-BDC (Supplemental Fig. 2B). Thus, we were unable to determine if antigenic stimulation alters diabetes progression following CD40L blockade. CD40 and CD40L are expressed by multiple immune cell types, including T and B lymphocytes, as well as DCs and macrophages; hence, systemic alterations in this costimulatory pathway could affect multiple aspects of diabetes pathogenesis. Other TNFR superfamily members expressed on DCs may also play a role in autoreactive T cell stimulation and likely represent one link between inflammatory factors in the immune cell microenvironment and autoreactive T cell activation. The cause of increased expression of CD40 on NOD CD8+ DCs needs further investigation but could be a result of genetic factors outside of the Idd3 and Idd5 loci and/or MyD88-independent inflammatory mediators.
Our data have several implications for tolerance induction of autoreactive T cells and diabetes progression and treatment. As autoreactive CD8+ T cells but not CD4+ T cells were deleted from NOD mice after stimulation by CD8+ DCs [28], our data suggest that CD4+ and CD8+ T cell have different requirements for tolerance in an autoimmune environment. In addition, CD4+ T cells expand in diabetes-resistant NOD.Idd3/5 mice after targeting of antigen to CD8+ DCs, suggesting that other diabetes susceptibility loci may play a role in this loss of tolerance. Importantly, these experiments were performed in the context of spontaneous ongoing autoimmunity with a naturally autoreactive T cell isolated from diabetic NOD mice. Such T cells display altered reactivity compared with T cells subject to less selection pressure against self in the thymus [57]. There are currently no I-Ag7-restricted TCR Tg lines in NOD mice that are not specific for a β-cell antigen, so it is necessary to use T cells, such as BDC2.5, which will respond to DCs presenting diabetogenic antigens at the site of disease and are under increased selective pressure in the thymus. We were able to demonstrate similar responses when endogenous islet-reactive T cells are stimulated by DEC-205+ DCs, indicating that the aberrant responses that we observe are not confined to T cells from BDC2.5 TCR Tg mice. Responses of CD4+ T cells specific for other β-cell antigens, such as insulin or glutamic acid decarboxylase, can also be tested using TCR Tg mice and/or tetramers.
In conclusion, we show that NOD mice have a defect in the ability of CD8+ DCs to induce CD4+ T cell tolerance that is corrected by blocking CD40–CD40L interactions. This suggests that requirements for DC-mediated T cell tolerance induction are altered in an autoimmune context. Therefore, antigen-specific therapy for autoimmune diseases, such as type 1 diabetes, may require a combination of antigen delivery and inhibition of inflammatory pathways.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Intramural Research Programs of the NIDDK and the Juvenile Diabetes Research Foundation.
We acknowledge Mrs. Alice Franks (Diabetes, Endocrinology, and Obesity Branch, NIDDK) for help with mouse husbandry, Ms. Jennifer King (Jennifer King Creative) for technical graphics support, and Drs. Michael Lenardo (National Institute of Allergy and Infectious Disease) and Giorgio Trinchieri (National Cancer Institute) for helpful discussions.
The online version of this paper, found at www.jleukbio.org, includes supplemental information.
- CD40L
- CD40 ligand
- DCIR2
- DC inhibitory receptor 2
- Flt3L
- Fms-related tyrosine kinase 3 ligand
- FMO
- fluorescence minus one
- FoxP3
- forkhead box P3
- Idd
- insulin-dependent diabetes
- MFI
- mean fluorescence intensity
- NIDDK
- National Institute of Diabetes and Digestive and Kidney Diseases
- pLN
- pancreatic LN
- poly(I:C)
- polyinosinic:polycytidylic acid
- Tg
- transgenic
- TLRL
- TLR ligand
- Treg
- regulatory T cell
AUTHORSHIP
J.D.P. and K.V.T. designed the project, collected and analyzed data, and wrote the manuscript. N.M.B., G.R., Y.Z., C.C.R., and A.W.L-K. collected and analyzed data and edited the manuscript.
DISCLOSURES
The authors declare no conflict of interest.
REFERENCES
- 1. Steinman R. M., Hawiger D., Nussenzweig M. C. (2003) Tolerogenic dendritic cells. Ann. Rev. Immunol. 21, 685–711 [DOI] [PubMed] [Google Scholar]
- 2. Turley S. J. (2002) Dendritic cells: inciting and inhibiting autoimmunity. Curr. Opin. Immunol. 14, 765–770 [DOI] [PubMed] [Google Scholar]
- 3. Turley S., Poirot L., Hattori M., Benoist C., Mathis D. (2003) Physiological β cell death triggers priming of self-reactive T cells by dendritic cells in a type-1 diabetes model. J. Exp. Med. 198, 1527–1537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hoglund P., Mintern J., Waltzinger C., Heath W., Benoist C., Mathis D. (1999) Initiation of autoimmune diabetes by developmentally regulated presentation of islet cell antigens in the pancreatic lymph nodes. J. Exp. Med. 189, 331–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bluestone J. A., Herold K., Eisenbarth G. (2010) Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature 464, 1293–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang L., Eisenbarth G. S. (2011) Prediction and prevention of type 1 diabetes mellitus. J. Diabetes 3, 48–57 [DOI] [PubMed] [Google Scholar]
- 7. Voorbij H. A., Jeucken P. H., Kabel P. J., De Haan M., Drexhage H. A. (1989) Dendritic cells and scavenger macrophages in pancreatic islets of prediabetic BB rats. Diabetes 38, 1623–1629 [DOI] [PubMed] [Google Scholar]
- 8. Wicker L. S., Todd J. A., Peterson L. B. (1995) Genetic control of autoimmune diabetes in the NOD mouse. Ann. Rev. Immunol. 13, 179–200 [DOI] [PubMed] [Google Scholar]
- 9. Concannon P., Chen W. M., Julier C., Morahan G., Akolkar B., Erlich H. A., Hilner J. E., Nerup J., Nierras C., Pociot F., Todd J. A., Rich S. S. (2009) Genome-wide scan for linkage to type 1 diabetes in 2,496 multiplex families from the Type 1 Diabetes Genetics Consortium. Diabetes 58, 1018–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lau-Kilby A. W., Kretz C. C., Pechhold S., Price J. D., Dorta S., Ramos H., Trinchieri G., Tarbell K. V. (2011) Interleukin-2 inhibits FMS-like tyrosine kinase 3 receptor ligand (flt3L)-dependent development and function of conventional and plasmacytoid dendritic cells. Proc. Natl. Acad. Sci. USA 108, 2408–2413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang X., Jia S., Geoffrey R., Alemzadeh R., Ghosh S., Hessner M. J. (2008) Identification of a molecular signature in human type 1 diabetes mellitus using serum and functional genomics. J. Immunol. 180, 1929–1937 [DOI] [PubMed] [Google Scholar]
- 12. Lorini R., Montagna D., Lanfranchi A., Cortona L., Livieri C., Larizza D., d'Annunzio G., Severi F. (1989) Alterations of in vitro interleukin 1 and 2 in diabetic children. Eur. J. Pediatr. 148, 732–734 [DOI] [PubMed] [Google Scholar]
- 13. Yaacob N. S., Kaderi M. A., Norazmi M. N. (2004) The expression of cytokine genes in the peritoneal macrophages and splenic CD4- and CD8-positive lymphocytes of the nonobese diabetic mice. J. Clin. Immunol. 24, 177–184 [DOI] [PubMed] [Google Scholar]
- 14. Lee L. F., Xu B., Michie S. A., Beilhack G. F., Warganich T., Turley S., McDevitt H. O. (2005) The role of TNF-α in the pathogenesis of type 1 diabetes in the nonobese diabetic mouse: analysis of dendritic cell maturation. Proc. Natl. Acad. Sci. USA 102, 15995–16000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Diana J., Gahzarian L., Simoni Y., Lehuen A. (2011) Innate immunity in type 1 diabetes. Discov. Med. 11, 513–520 [PubMed] [Google Scholar]
- 16. Poligone B., Weaver D. J., Jr., Sen P., Baldwin A. S., Jr., Tisch R. (2002) Elevated NF-κB activation in nonobese diabetic mouse dendritic cells results in enhanced APC function. J. Immunol. 168, 188–196 [DOI] [PubMed] [Google Scholar]
- 17. Vasquez A. C., Feili-Hariri M., Tan R. J., Morel P. A. (2004) Qualitative and quantitative abnormalities in splenic dendritic cell populations in NOD mice. Clin. Exp. Immunol. 135, 209–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Van Belle T. L., Juntti T., Liao J., von Herrath M. G. (2010) Pre-existing autoimmunity determines type 1 diabetes outcome after Flt3-ligand treatment. J. Autoimmun. 34, 445–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Iyoda T., Shimoyama S., Liu K., Omatsu Y., Akiyama Y., Maeda Y., Takahara K., Steinman R. M., Inaba K. (2002) The CD8+ dendritic cell subset selectively endocytoses dying cells in culture and in vivo. J. Exp. Med. 195, 1289–1302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu K., Iyoda T., Saternus M., Kimura Y., Inaba K., Steinman R. M. (2002) Immune tolerance after delivery of dying cells to dendritic cells in situ. J. Exp. Med. 196, 1091–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yamazaki S., Dudziak D., Heidkamp G. F., Fiorese C., Bonito A. J., Inaba K., Nussenzweig M. C., Steinman R. M. (2008) CD8+ CD205+ splenic dendritic cells are specialized to induce Foxp3+ regulatory T cells. J. Immunol. 181, 6923–6933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kamphorst A. O., Guermonprez P., Dudziak D., Nussenzweig M. C. (2010) Route of antigen uptake differentially impacts presentation by dendritic cells and activated monocytes. J. Immunol. 185, 3426–3435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dudziak D., Kamphorst A. O., Heidkamp G. F., Buchholz V. R., Trumpfheller C., Yamazaki S., Cheong C., Liu K., Lee H. W., Park C. G., Steinman R. M., Nussenzweig M. C. (2007) Differential antigen processing by dendritic cell subsets in vivo. Science 315, 107–111 [DOI] [PubMed] [Google Scholar]
- 24. Jiang W., Swiggard W. J., Heufler C., Peng M., Mirza A., Steinman R. M., Nussenzweig M. C. (1995) The receptor DEC-205 expressed by dendritic cells and thymic epithelial cells is involved in antigen processing. Nature 375, 151–155 [DOI] [PubMed] [Google Scholar]
- 25. Hawiger D., Inaba K., Dorsett Y., Guo M., Mahnke K., Rivera M., Ravetch J. V., Steinman R. M., Nussenzweig M. C. (2001) Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J. Exp. Med. 194, 769–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bonifaz L., Bonnyay D., Mahnke K., Rivera M., Nussenzweig M. C., Steinman R. M. (2002) Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in the steady state leads to antigen presentation on major histocompatibility complex class I products and peripheral CD8+ T cell tolerance. J. Exp. Med. 196, 1627–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Soares H., Waechter H., Glaichenhaus N., Mougneau E., Yagita H., Mizenina O., Dudziak D., Nussenzweig M. C., Steinman R. M. (2007) A subset of dendritic cells induces CD4+ T cells to produce IFN-γ by an IL-12-independent but CD70-dependent mechanism in vivo. J. Exp. Med. 204, 1095–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mukhopadhaya A., Hanafusa T., Jarchum I., Chen Y. G., Iwai Y., Serreze D. V., Steinman R. M., Tarbell K. V., DiLorenzo T. P. (2008) Selective delivery of β cell antigen to dendritic cells in vivo leads to deletion and tolerance of autoreactive CD8+ T cells in NOD mice. Proc. Natl. Acad. Sci. USA 105, 6374–6379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bruder D., Westendorf A. M., Hansen W., Prettin S., Gruber A. D., Qian Y., von Boehmer H., Mahnke K., Buer J. (2005) On the edge of autoimmunity: T-cell stimulation by steady-state dendritic cells prevents autoimmune diabetes. Diabetes 54, 3395–3401 [DOI] [PubMed] [Google Scholar]
- 30. Hawiger D., Masilamani R. F., Bettelli E., Kuchroo V. K., Nussenzweig M. C. (2004) Immunological unresponsiveness characterized by increased expression of CD5 on peripheral T cells induced by dendritic cells in vivo. Immunity 20, 695–705 [DOI] [PubMed] [Google Scholar]
- 31. Stadinski B. D., Delong T., Reisdorph N., Reisdorph R., Powell R. L., Armstrong M., Piganelli J. D., Barbour G., Bradley B., Crawford F., Marrack P., Mahata S. K., Kappler J. W., Haskins K. (2010) Chromogranin A is an autoantigen in type 1 diabetes. Nat. Immunol. 11, 225–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Katz J. D., Wang B., Haskins K., Benoist C., Mathis D. (1993) Following a diabetogenic T cell from genesis through pathogenesis. Cell 74, 1089–1100 [DOI] [PubMed] [Google Scholar]
- 33. Haskins K., Portas M., Bradley B., Wegmann D., Lafferty K. (1988) T-lymphocyte clone specific for pancreatic islet antigen. Diabetes 37, 1444–1448 [DOI] [PubMed] [Google Scholar]
- 34. Roederer M. (2001) Spectral compensation for flow cytometry: visualization artifacts, limitations, and caveats. Cytometry 45, 194–205 [DOI] [PubMed] [Google Scholar]
- 35. Kraal G., Breel M., Janse M., Bruin G. (1986) Langerhans' cells, veiled cells, and interdigitating cells in the mouse recognized by a monoclonal antibody. J. Exp. Med. 163, 981–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hathcock K. S., Laszlo G., Pucillo C., Linsley P., Hodes R. J. (1994) Comparative analysis of B7-1 and B7-2 costimulatory ligands: expression and function. J. Exp. Med. 180, 631–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Clynes R. A., Towers T. L., Presta L. G., Ravetch J. V. (2000) Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat. Med. 6, 443–446 [DOI] [PubMed] [Google Scholar]
- 38. Judkowski V., Pinilla C., Schroder K., Tucker L., Sarvetnick N., Wilson D. B. (2001) Identification of MHC class II-restricted peptide ligands, including a glutamic acid decarboxylase 65 sequence, that stimulate diabetogenic T cells from transgenic BDC2.5 nonobese diabetic mice. J. Immunol. 166, 908–917 [DOI] [PubMed] [Google Scholar]
- 39. Fu W., Wojtkiewicz G., Weissleder R., Benoist C., Mathis D. (2012) Early window of diabetes determinism in NOD mice, dependent on the complement receptor CRIg, identified by noninvasive imaging. Nat. Immunol. 13, 361–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Falcone M., Sarvetnick N. (1999) The effect of local production of cytokines in the pathogenesis of insulin-dependent diabetes mellitus. Clin. Immunol. 90, 2–9 [DOI] [PubMed] [Google Scholar]
- 41. Long M., Slaiby A. M., Wu S., Hagymasi A. T., Mihalyo M. A., Bandyopadhyay S., Vella A. T., Adler A. J. (2007) Histone acetylation at the Ifng promoter in tolerized CD4 cells is associated with increased IFN-γ expression during subsequent immunization to the same antigen. J. Immunol. 179, 5669–5677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lesage S., Hartley S. B., Akkaraju S., Wilson J., Townsend M., Goodnow C. C. (2002) Failure to censor forbidden clones of CD4 T cells in autoimmune diabetes. J. Exp. Med. 196, 1175–1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gonzalez A., Andre-Schmutz I., Carnaud C., Mathis D., Benoist C. (2001) Damage control, rather than unresponsiveness, effected by protective DX5+ T cells in autoimmune diabetes. Nat. Immunol. 2, 1117–1125 [DOI] [PubMed] [Google Scholar]
- 44. You S., Slehoffer G., Barriot S., Bach J. F., Chatenoud L. (2004) Unique role of CD4+CD62L+ regulatory T cells in the control of autoimmune diabetes in T cell receptor transgenic mice. Proc. Natl. Acad. Sci. USA 101 (Suppl. 2), 14580–14585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bertin-Maghit S., Pang D., O'Sullivan B., Best S., Duggan E., Paul S., Thomas H., Kay T. W., Harrison L. C., Steptoe R., Thomas R. (2011) Interleukin-1β produced in response to islet autoantigen presentation differentiates T-helper 17 cells at the expense of regulatory T-cells: implications for the timing of tolerizing immunotherapy. Diabetes 60, 248–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hill N. J., Lyons P. A., Armitage N., Todd J. A., Wicker L. S., Peterson L. B. (2000) NOD Idd5 locus controls insulitis and diabetes and overlaps the orthologous CTLA4/IDDM12 and NRAMP1 loci in humans. Diabetes 49, 1744–1747 [DOI] [PubMed] [Google Scholar]
- 47. Feili-Hariri M., Morel P. A. (2001) Phenotypic and functional characteristics of BM-derived DC from NOD and non-diabetes-prone strains. Clin. Immunol. 98, 133–142 [DOI] [PubMed] [Google Scholar]
- 48. Edwards A. D., Diebold S. S., Slack E. M., Tomizawa H., Hemmi H., Kaisho T., Akira S., Reis e Sousa C. (2003) Toll-like receptor expression in murine DC subsets: lack of TLR7 expression by CD8 α+ DC correlates with unresponsiveness to imidazoquinolines. Eur. J. Immunol. 33, 827–833 [DOI] [PubMed] [Google Scholar]
- 49. Salomon B., Lenschow D. J., Rhee L., Ashourian N., Singh B., Sharpe A., Bluestone J. A. (2000) B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity 12, 431–440 [DOI] [PubMed] [Google Scholar]
- 50. Stratmann T., Martin-Orozco N., Mallet-Designe V., Poirot L., McGavern D., Losyev G., Dobbs C. M., Oldstone M. B., Yoshida K., Kikutani H., Mathis D., Benoist C., Haskins K., Teyton L. (2003) Susceptible MHC alleles, not background genes, select an autoimmune T cell reactivity. J. Clin. Invest. 112, 902–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jang M. H., Seth N. P., Wucherpfennig K. W. (2003) Ex vivo analysis of thymic CD4 T cells in nonobese diabetic mice with tetramers generated from I-A(g7)/class II-associated invariant chain peptide precursors. J. Immunol. 171, 4175–4186 [DOI] [PubMed] [Google Scholar]
- 52. Balasa B., Krahl T., Patstone G., Lee J., Tisch R., McDevitt H. O., Sarvetnick N. (1997) CD40 ligand-CD40 interactions are necessary for the initiation of insulitis and diabetes in nonobese diabetic mice. J. Immunol. 159, 4620–4627 [PubMed] [Google Scholar]
- 53. Baker R. L., Wagner D. H., Jr., Haskins K. (2008) CD40 on NOD CD4 T cells contributes to their activation and pathogenicity. J. Autoimmun. 31, 385–392 [DOI] [PubMed] [Google Scholar]
- 54. Masteller E. L., Warner M. R., Ferlin W., Judkowski V., Wilson D., Glaichenhaus N., Bluestone J. A. (2003) Peptide-MHC class II dimers as therapeutics to modulate antigen-specific T cell responses in autoimmune diabetes. J. Immunol. 171, 5587–5595 [DOI] [PubMed] [Google Scholar]
- 55. Tarbell K. V., Yamazaki S., Olson K., Toy P., Steinman R. M. (2004) CD25+ CD4+ T cells, expanded with dendritic cells presenting a single autoantigenic peptide, suppress autoimmune diabetes. J. Exp. Med. 199, 1467–1477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gardner J. M., Metzger T. C., McMahon E. J., Au-Yeung B. B., Krawisz A. K., Lu W., Price J. D., Johannes K. P., Satpathy A. T., Murphy K. M., Tarbell K. V., Weiss A., Anderson M. S. (2013) Extrathymic aire-expressing cells are a distinct bone marrow-derived population that induce functional inactivation of CD4 T cells. Immunity 39, 560–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Schubert D. A., Gordo S., Sabatino J. J., Jr., Vardhana S., Gagnon E., Sethi D. K., Seth N. P., Choudhuri K., Reijonen H., Nepom G. T., Evavold B. D., Dustin M. L., Wucherpfennig K. W. (2012) Self-reactive human CD4 T cell clones form unusual immunological synapses. J. Exp. Med. 209, 335–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.