

Targeted mutation of STAT3 in myeloid cells dramatically altered the relative abundance of specific immune cell types within tumors, with no impact on tumor incidence.
Keywords: myeloid-derived suppressor cell, STAT3, immunosuppression
Abstract
Although the immune system may provide early protection against cancer, tumors may exploit the healing arm of the immune system to enhance their growth and metastasis. For example, myeloid derived suppressor cells (MDSCs) are thought to promote tumor growth by several mechanisms, including the suppression of T cell activity. It has been suggested that STAT3 activation in myeloid cells modulates multiple aspects of MDSC physiology, including their expansion and activity. Whereas most animal studies investigating tumor immunology have used tumor implants, we used transgenic mice (Smo*) that spontaneously develop medulloblastoma brain tumors to investigate the temporal accumulation of MDSCs within tumors and how myeloid STAT3 disruption affects MDSC and other immune cell types. We found distinct populations of MDSC in medulloblastoma tumors, with a high prevalence of CD11b+Ly6G+Ly6Clow/− cells, described previously by others as G-MDSCs. These were found early in tumor development, in premalignant lesions located on the surface of the cerebellum of 28-day-old mice. In fully developed tumors, pSTAT3 was found in the majority of these cells. Conditional STAT3 gene disruption in myeloid cells resulted in an enhanced proinflammatory phenotype of macrophages in Smo* mice. Moreover, a significant reduction in the abundance of G-MDSCs and Tregs was observed within tumors along with an increased presence of CD4+ and CD8+ cells. Despite these alterations in immune cells induced by myeloid STAT3 disruption, we found no effect on tumor incidence in Smo* mice with this deletion.
Introduction
Innate and adaptive immune surveillance is thought to limit the development of cancer at early stages. However, tumor attack by immune cells is compromised by the existence of immunosuppressive cellular and molecular mechanisms. Cells from lymphoid and myeloid origin serve this purpose. MDSCs are considered to be a heterogeneous subset of myeloid cells within tumors that exhibit an immature phenotype and immunosuppressive properties [1–3]. Two types of morphologically different MDSCs have been described: G-MDSCs and M-MDSCs [4, 5]. Although both cell types express Gr1, they exhibit different cellular surface expression of the two Gr-1 subcomponents, Ly6C and Ly6G, which allows their discrimination by flow cytometry [4]. G-MDSCs have thus been characterized as Ly6G+Ly6Clow/− cells and M-MDSC as Ly6G−Ly6C+ cells [4]. Although the mechanisms maintaining these cellular populations within tumors are not well understood, considerable evidence indicate that the STAT3 pathway in myeloid cells may be crucial for MDSC differentiation, survival, and function (ref. [6] and reviewed in refs. [7, 8]). STAT3 is an intracellular signaling protein that participates in normal cellular responses to certain cytokines and growth factors. Constitutive activation of STAT3 (nuclear tyrosine pSTAT3) has been demonstrated in a number of human tumor cells and cancer cell lines, and its inhibition can suppress growth of cancer cells by promoting apoptosis and inhibiting cell proliferation [9, 10]. In fact, activated STAT3 modulates the expression of target genes involved in various functions, including apoptosis (e.g., survivin and Bcl-xL), cell-cycle regulation (e.g., cyclin D1 and c-Myc), tumor invasiveness (matrix metalloproteinases), and angiogenesis (e.g., VEGF) [11–13].
Activation of STAT3 has been also reported in cells of myeloid origin within tumors [11]. In general, STAT3 signaling in myeloid cells has been associated with suppression of proinflammatory activity and promotion of tolerance [14, 15]. Targeted deletion of the STAT3 gene in these cells resulted in enhanced production of proinflammatory cytokines and Th1 activity and loss of tolerogenic features [16, 17]. A number of factors frequently present within the tumor environment, including VEGF, IL-6, and IL-10, can activate STAT3 signaling in invading DCs and macrophages cells, subverting them to a tolerogenic/suppressor phenotype [18]. For example, blockade of tumor-induced STAT3 activity in APCs increased their expression levels of MHC-II, B7 costimulatory molecules, IL-12 and IL-23, the latter being recently implicated in tumor growth [6, 19, 20].
Although different aspects of anti- and protumor immune responses have been investigated in multiple human and murine models of cancer, there is relatively scarce information regarding the dynamics of MDSCs in brain tumors. The understanding of the ongoing immunological processes in brain tumors is extremely important, as these account for high rates of morbidity and mortality, as they are the most common solid tumors in children [21–23]. Current therapy is complicated by the development of resistance and the fact that the blood brain barrier (BBB) limits the penetration of many chemotherapy agents. Although immunotherapy may offer an attractive, alternative treatment, the success of this modality depends on our ability to understand the ongoing mechanisms of immune regulation. Orthotopic syngeneic graft models have been used to study this but are hampered by the fact that implanted cells lack normal tumor stroma, derive from tumors that have already learned to avoid immunoeradication, are altered by long-term culture, and have surpassed the stepwise genetic changes that occur in human tumors. Moreover, the immune BBB is breached at the outset of implantation by the injection procedure, artificially creating an injury inflammatory response and an entry site for immune cells. Here, we first investigated the temporal accumulation of MDSCs in Smo* mutant mice, which develop spontaneous cerebellar tumors that resemble pediatric medulloblastoma [24]. Smo* mice harbor a transgene that targets the expression of a constitutively active form of Smo, specifically to the cerebellar granule precursor cells thought to give rise to MB, leaving immune and other stromal cells unperturbed. Nearly all Smo* mice exhibit preneoplastic lesions on the cerebellar surface at 1 month of age and in ∼50% of the cases, develop symptomatic MB [24, 25]. We found that MDSCs (particularly G-MDSCs) are already present in premalignant lesions in these mice and are highly abundant in fully developed tumors. We conditionally deleted STAT3 in myeloid cells in Smo* mice by means of a Cre/LoxP system, where the expression of Cre recombinase was driven by a LysM promoter. We found that deletion of STAT3 in myeloid cells increased the proinflammatory phenotype of peripheral macrophages and resulted in a strong reduction in the abundance of G-MDSCs within tumors. In parallel, the ratio of Teff/Tregs was increased in these mice. Despite these changes, we did not find differences in tumor incidence between WT and STAT3-cKO mice.
MATERIALS AND METHODS
Mice
Transgenic male and female mice on a C57BL/6 background (backcrossed for at least eight generations) were used in these studies. Smo* mice and LysM-Cre mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA) and STAT3fl/fl mice obtained from Dr. Michael Sofroniew (UCLA, CA, USA) [26], generated previously by Takeda et al. [27]. Smo*, LysM-Cre, and STAT3fl/fl mice were interbred to obtain Smo*.STAT3fl/fl (referred to as WT) and Smo*.LysM-Cre STAT3fl/fl (referred to as STAT3-cKO) mice. Mice were housed with standard light-dark cycles with food and water ad libidum, according to UCLA Institutional Guidelines and were monitored daily for signs of illness or tumor effects, such as lethargy, ataxia, balance defects, and dehydration. Euthanasia was performed when such symptoms appeared as recommended by UCLA Division of Laboratory Animal Medicine veterinarians. Mice were handled in accordance with the UCLA animal care policy and approved animal protocols.
H&E staining
Mice were anesthetized and perfused intracardially, first with 0.1 M phosphate buffer, pH 7.4, and then by 4% PFA, and brains were removed, postfixed in 4% PFA overnight, and embedded in paraffin. Sections (4 μm) were prepared with a microtome, deparaffined and rehydrated, and stained with standard H&E procedures.
Immunofluorescence
Mice were perfused as above, and brains were removed, postfixed in 4% PFA overnight, and cryoprotected with PBS containing 20% sucrose. Tissues were embedded in optimal cutting temperature compound, and 15-μm sections were incubated with rat anti-CD11b (1:200; BD Biosciences, San Diego, CA, USA), rabbit anti-pSTAT3 (1:200; Cell Signaling Technology, Beverly, MA, USA), and/or rat anti-FITC-conjugated Ly6G (1:200; BioLegend, San Diego, CA, USA) in PBS solution containing 1% BSA and 0.3% Triton X-100 at 4°C overnight. For pSTAT3 staining, an antigen-retrieval method with pH 9.0 EDTA at 60°C was applied. Then, sections were washed and incubated with appropriate fluorochrome-conjugated secondary antibodies from Invitrogen (Grand Island, NY, USA) for 40 min at room temperature. Slides were mounted using Vectashield with DAPI (Vector Laboratories, Burlingame, CA, USA), and sections were examined with an Olympus upright fluorescence microscope equipped with a Hamamatsu ORCA-3CCD camera.
To determine angiogenesis, coronal brain sections (30 μm thickness) were incubated with rabbit anti-laminin (1:200; Sigma-Aldrich, St. Louis, MO, USA), followed by secondary antibodies. After taking photographs, the areas of vessels were measured by using Scion Image 4.0 software (Scion, Frederick, MD, USA).
Isolation of immune cells from tumors and flow cytometry
Mononuclear cells were isolated from cerebellar tumors, as described previously [28]. Briefly, mice were anesthetized and perfused with PBS, and cerebella were dissected. Tissues were minced in small pieces and digested in HBSS containing 0.1 mg/ml collagenase and 0.1 mg/ml DNase I (Roche, Mannheim, Germany) for 40 min at 37°C with continuous shaking. The suspension was passed through a 40-μm mesh, and cells were separated on a discontinuous 40%/80% (v/v) Percoll gradient after centrifugation at 500 g for 30 min with no brake. Mononuclear cells were obtained from the 40%/80% interphase and washed in PBS. All antibodies for flow cytometry analysis were purchased from eBioscience (San Diego, CA, USA). Antibodies were diluted in PBS/1% BSA, as recommended by the manufacturer, and cells were incubated with them for 20 min at 4°C. For MDSC staining, cells were incubated with allophycocyanin-anti-CD11b, FITC-anti-Ly6G, and PerCP-Cy5.5-anti-Ly6C antibodies. For analysis of apoptosis in MDSCs, cells were stained additionally with 5 μl 7-AAD and 5 μl PE-Annexin V for 15 min at room temperature and analyzed within 15 min of the staining, according to the manufacturer's instructions (BD Biosciences). For all experiments, cells were analyzed in a FACSCalibur flow cytometer.
Treg staining was performed with the murine Foxp3+ staining kit from eBioscience following the manufacturer's instructions. Briefly, cells were first incubated with PerCP-Cy5.5-anti-CD4, PE-anti-CD8, and FITC-anti-CD25 antibodies for 15 min at 4°C, fixed/permeabilized overnight, and then stained with allophycocyanin-anti-Foxp3 antibody in permeabilization buffer.
Preparation of spleen and peritoneal cell suspensions
For preparation of spleen suspensions, spleens were dissected and tapped through a 40-μm nylon mesh. Cell suspensions were treated with red blood lysis buffer and washed with PBS before staining. For isolation of peritoneal cells, mice were injected with 4% thioglicollate. Three days later, mice were injected in the peritoneum with 2 ml PBS, and cells were collected from the abdomen after massage.
Cell culture and ELISA
Peritoneal cells were distributed at 1 × 106 cells/ml in 96-well plates in triplicate and cultured in RPMI 1640 with 2% FBS (HyClone, Logan, UT, USA) and 1% penicillin/streptomycin for 24 h with 100 ng/ml LPS (Escherichia coli; Sigma-Aldrich) at 37°C with 5% CO2 and humidified atmosphere. To measure the levels of TNF-α and IL-6 in the culture supernatants, we used sandwich ELISA kits from PeproTech (Rocky Hill, NJ, USA), according to the manufacturer's instructions. Briefly, plates were coated with primary antibodies overnight and then washed and blocked with PBS/1% BSA for 2 h. Next, plates were incubated with samples and standards overnight and washed, and Avidin-HRP was added for 40 min at room temperature. Then, substrate was added, and the absorbance at 405 nm was measured using a standard ELISA microplate reader.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 4.01 software (GraphPad Software, San Diego, CA, USA). Student's t-tests were used to determine significant differences between groups, except for the mortality curve, for which the log-rank test was used.
RESULTS
MDSCs are present in medulloblastoma tumors from Smo* mice and exhibit activation of the STAT3 pathway
It has been shown that MDSCs can be found within various types of tumors in different experimental animal models. However, there is little information concerning the presence of MDSCs in brain tumors. Transgenic Smo* mice exhibit a mutation in the hedgehog pathway transducer Smo in granule neural precursors, leaving immune cells unperturbed [24]. This mutation leads to hyperactivation of the hedgehog pathway, which causes the spontaneous transformation of the granule cells and subsequently, medulloblastoma-like tumors. To determine whether MDSCs are present in medulloblastoma tumors from Smo* mice, we prepared a mononuclear suspension highly enriched in immune cells from tumors, and we used specific markers of MDSC to identify these cells by flow cytometry analysis. A distinct population of CD11b+ cells was found in the tumors (Fig. 1A). The majority of these cells was Gr1+ (90±2.5% of CD11b+ cells). To identify putative G-MDSCs and M-MDSCs, we used Ly6G and Ly6C markers as described [4]. It has been shown that whereas G-MDSCs are Ly6G+Ly6Clow/−, M-MDSCs are Ly6G−Ly6C+. We found that the great majority of CD11b+ cells in Smo* mice tumors was Ly6G+Ly6Clow/− (86.7±2.3%), and a much lower proportion of the CD11b+ cells was Ly6G−Ly6C+ (4.3±1%). In addition, we found by immunofluorescence staining that whereas these cells were completely absent in the normal cerebellum, they were present in early asymptomatic, premalignant lesions located in the surface of the cerebellum in 28-day-old mice (Fig. 1B). Thereafter, the numbers of cells increased gradually. In fully developed tumors, these cells were highly abundant, and practically all CD11b+ cells were Ly6G+, in accordance with the flow cytometry data.
Figure 1. MDSCs are present in medulloblastoma tumors from Smo* mice.
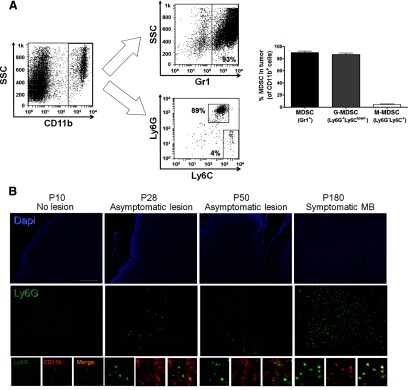
(A) Flow cytometry analysis of immune cell suspensions prepared from adult Smo* mice medulloblastoma tumors (n=9) using CD11b, Gr1, Ly6G, and Ly6C markers. Cells were first gated on the CD11b+ population and then analyzed for Gr1 or Ly6C/Ly6G expression. Representative FACS plots (left) and average quantification (right) are shown. Graph shows percent of cells within the CD11b+ population. SSC, Side-scatter. (B) Immunofluorescence analysis of CD11b+Ly6G+ cell presence in cerebellar lesions of Smo* mice collected at different stages: Postnatal Day (P) 10 (no lesions observed, preserving normal cerebellar structures), P28 (cell-dense, ectopic lesions are observed), P50, and P180 (symptomatic, fully developed tumor is present). CD11b, red; Ly6G, green; nuclei (DAPI), blue. One experiment from three is shown. Original scale bar, 100 μm.
Constitutive activation of the STAT3 signaling pathway in tumor MDSCs has been described in different tumor models. To test whether this is the case in our model, we stained medulloblastoma tumors with antibodies for Ly6G as a marker for G-MDSCs, the most prevalent population of MDSCs that we found in our model, and pSTAT3. As shown in Fig. 2, pSTAT3 was found in almost all of the Ly6G+ cells, providing evidence that this signaling pathway is active in almost all G-MDSCs within these tumors.
Figure 2. STAT3 pathway activation in Ly6G+ cells from medulloblastoma in Smo* mice.

Symptomatic tumors in Smo* mice show coexpression of activated pSTAT3 (red) and Ly6G (green) within the tumors. Representative images of three experiments are shown. Original scale bar, 100 μm.
Ablation of STAT3 in myeloid cells leads to increased proinflammatory responses
It has been shown previously that STAT3 regulates cytokine expression, promoting an immunosuppressive profile [29]. Here, we used a Cre/LoxP system, used previously by others, to delete STAT3 specifically in myeloid cells expressing LysM [16]. The LysM gene encodes a glycanhydrolase used in the breakdown of the tissue matrix in the process of tissue remodeling and regeneration and is expressed exclusively in myeloid cells, including monocytes and their precursors [30]. Conditional disruption of STAT3 in myeloid cells using this promoter has been shown by others to promote a proinflammatory phenotype [16, 31]. In concurrence, we found that phenotypic characteristics of CD11b+ splenocytes from LysM-Cre-STAT3fl/fl mice (STAT3-cKO mice) assessed by flow cytometry exhibited signs of a higher basal activation status. In this regard, we found that cKO CD11b+ splenocytes expressed higher CD80 than WT cells (Fig. 3A). However, a mild reduction in CD86 and no difference in MHC class II were found in these cells. Conditional deletion of STAT3 did not significantly affect the proportions of CD11b splenocytes that expressed Ly6G or Ly6C (see Supplemental Fig. 2). We also tested the functional response of myeloid cells from these mice, assessing cytokine production after LPS stimulation in vitro. For that purpose, we isolated peritoneal cells (highly enriched in macrophages by previous thioglycollate injection) and measured the levels of IL-6 and TNF-α produced by these cells by ELISA after in vitro stimulation with LPS. We found that cells from mice with the conditional STAT3 disruption produced significantly higher levels of these proinflammatory cytokines than cells from WT mice (*P<0.05; Fig. 3B). This suggests that deletion of STAT3 in myeloid cells leads to a stronger proinflammatory phenotype in these cells or increased numbers of cells with a proinflammatory phenotype.
Figure 3. Specific reduction of STAT3 in myeloid cells leads to an enhanced proinflammatory phenotype.

A flow cytometry analysis of spleen CD11b+ cells from WT versus STAT3-cKO mice with CD80, CD86, and MHC class II markers (n=3). STAT3-cKO myeloid cells presented higher CD80, slightly reduced CD86, and no different MHC class II expressions. Representative FACS plots and histograms (white, WT; gray, STAT3-cKO) are shown. (B) In vitro response to LPS of peritoneal cells from WT versus STAT3-cKO mice. Cells were cultured in complete RPMI-1640 medium with/without LPS (100 ng/ml; E. coli), and cytokine content in supernatants was determined by ELISA. STAT3-cKO myeloid cells produced higher levels of TNF-α and IL-6 than WT cells. Student's t-test, *P < 0.05; ***P < 0.001. A representative experiment of three is shown.
STAT3-cKO mice present reduced G-MDSC in tumors
We then evaluated the impact of STAT3 deletion in myeloid cells in the population of MDSCs within medulloblastoma tumors in Smo* mice. By immunofluorescence staining, we found that the deletion of STAT3 in LysM-expressing cells led to a pronounced reduction of pSTAT3 in Ly6G+ cells in medulloblastoma tumors, although pSTAT3 staining was not completely absent (Fig. 4). Moreover, the relative abundance of Ly6G+ cells was diminished in tumors from cKO mice (Fig. 4). In addition to immunofluorescence, we used CD11b, Ly6G, and Ly6C markers to determine the relative proportions of G-MDSC (Ly6G+Ly6Clow/−) and M-MDSC (Ly6G−Ly6C+) within the population of CD11b+ cells in tumors by flow cytometry (Fig. 5). We found a significant decrease in the relative abundance of CD11b+ cells among mononuclear cells isolated from tumors of cKO mice (*P<0.05). In addition, the proportion of Ly6G+ cells (G-MDSC) was reduced profoundly in tumors from these mice. CD11b+ cells that were Ly6G+Ly6Clow/− represented 86.7 ± 2.3% in WT mice and 59.9 ± 4.4% (***P<0.001) in STAT3-cKO mice (Fig. 5). Conversely, a significant increase was found in the proportion of CD11b+ cells that was Ly6G−Ly6C+ (M-MDSC). Likewise, the proportions of non-MDSCs within the CD11b+ population (Ly6G−Ly6C− and Ly6G−Ly6Cint) were also increased in STAT3-KO mice (Supplemental Fig. 1A). A high percentage of these cells expressed F4/80, a macrophage marker, and thus may correspond to differentiated macrophages (Supplemental Fig. 1B). Moreover, the proportion of these cells within the total CD11b+ population was higher in STAT3-KO mice than in WT mice (Supplemental Fig. 1C).
Figure 4. Conditional deletion of STAT3 in myeloid cells leads to diminished pSTAT3 in Ly6G+ cells within the tumors of Smo* mice.
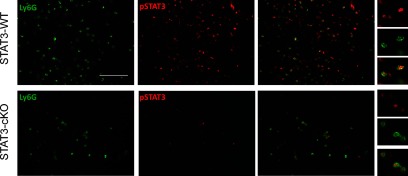
Sections from symptomatic Smo* WT (upper) and STAT3-cKO (lower) cerebellar tumors were stained with anti-Ly6G (green) and pSTAT3 (red) antibodies. Ly6G immunoreactive cells were abundant in tumors from WT mice but scarce in STAT3-cKO mice. pSTAT3 staining was present in most of Ly6G+ cells in WT mice tumors but lost in the same cells from STAT3-cKO mice. Original scale bar, 100 μm.
Figure 5. The prevalence of G-MDSC is reduced, but M-MDSC increased within the tumor myeloid population of STAT3-cKO versus WT mice.

A cellular suspension highly enriched in immune cells was isolated from Smo* WT or STAT3-cKO tumors, as described in Materials and Methods. Cells were stained with CD11b, Ly6G, and Ly6C antibodies and analyzed in a FACSCalibur. Within the CD11b+ population, G-MDSCs were gated as Ly6G+Ly6Clow/−, and M-MDSCs were gated as Ly6G−Ly6C+ cells. (A) Representative FACS plots of Smo* WT and STAT3-cKO tumors. (B) Average proportions of CD11b+ and MDSC subsets (n=8). Student's t-test, *P < 0.05; **P < 0.01; and ***P < 0.001.
As MDSCs in other tumor models also accumulate in peripheral immune organs, we also determined the proportions of MDSC populations in spleens of WT and STAT3-cKO mice with and without tumors (Supplemental Fig. 2). The proportion of G-MDSCs was higher in both WT and STAT3-cKO mice with tumors than without tumors (***P<0.001 for both genotypes). However, there was also a trend to lower G-MDSCs (and higher M-MDSCs) in STAT3-cKO mice with tumors, although the differences here were not statistically significant.
A major purported function of STAT3 in myeloid cells is to promote their survival. Thus, the reduction in G-MDSCs could be a result of increased apoptosis in these cells. We measured the apoptotic index of Smo* tumor CD11b+Ly6G+ cells by flow cytometry using 7-AAD and Annexin V markers (Fig. 6). We found that G-MDSCs from STAT3-cKO mice had a tendency to exhibit higher early- and late-apoptotic rates (36.3±6.4% vs. 26.9±3.6 in STAT3-cKO vs. WT for early apoptosis, with P=0.13, and 16.5±1.9% vs. 13.1±0.6% in STAT3-cKO vs. WT for late apoptosis, with P=0.07). As the differences here were not statistically significant, other mechanisms may be involved in the reduction of G-MDSCs in tumors from STAT3-cKO mice, for example, impaired production or recruitment.
Figure 6. G-MDSCs from STAT3-cKO mice tend to have increased apoptotic index compared with those from WT mice.
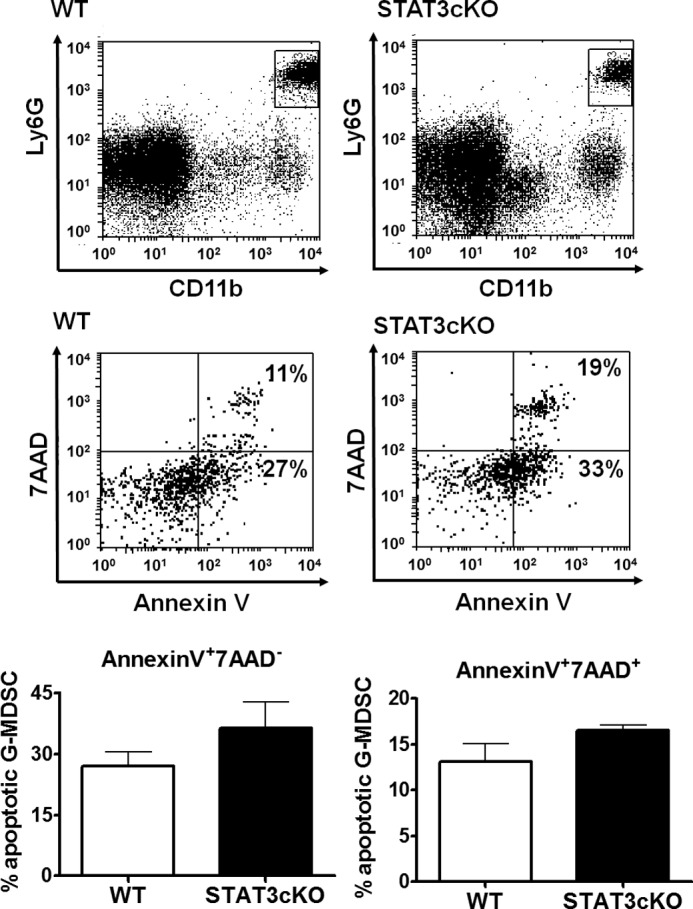
Apoptosis in G-MDSCs from tumors in Smo* WT or STAT3-cKO mice was measured by flow cytometry using 7-AAD and Annexin V as markers. Cells were stained with CD11b and Ly6G, and apoptotic populations were depicted within the CD11b+Ly6G+ gate. 7-AAD−Annexin V+ cells were defined as early apoptotic cells and 7-AAD+Annexin V+ cells as late apoptotic/necrotic cells. Representative FACS plots (upper) and average proportions (lower) are shown.
Proportions of Tregs are reduced in STAT3-cKO mice
In addition to myeloid cells, T cells are key components of the anti-tumoral immune response and putatively suppressed in the tumor microenvironment by MDSCs. This may involve direct effects on anti-tumor T cells, but it has been suggested recently that MDSCs may induce or support the population of Tregs within tumors, which in turn, suppresses anti-tumor lymphocyte responses. Thus, we hypothesized that alterations in MDSCs caused by STAT3 deletion in these cells may ultimately affect the types and/or numbers of T cells present in tumors. Here, we found that in tumors of STAT3-cKO mice, the proportions of CD4 and CD8 cells among mononuclear cells were increased by more than threefold compared to those in mice with intact STAT3 in myeloid cells (Fig. 7). In addition, the proportion of Foxp3+ cells within the CD4+ population was profoundly diminished in tumors from STAT3-cKO mice (Fig. 7). In fact, the Teff/Treg ratio was, on average, higher in STAT3-cKO mice tumors (3.7 in WT vs. 13.7 in STAT3-cKO tumors), which could potentially lead to a reduced suppression of Teff activity by Tregs. In the spleen, we did not find any noticeable differences in the proportions of CD4+, CD8+, and Tregs between WT and STAT3-cKO mice with tumors (Supplemental Fig. 3).
Figure 7. CD4+ and CD8+ cells are increased, but Tregs are reduced in STAT3-cKO mice compared with WT mice tumors.

The proportions of CD4+, CD8+, and CD25+Foxp3+ cells in Smo* WT versus STAT3-cKO tumors were determined by flow cytometry. For Tregs, cells were first gated on the CD4+ population. (A) Representative FACS plots and (B) the average proportions of these cells (n=8). Student's t-test, *P < 0.05; **P < 0.01.
Conditional deletion of STAT3 in myeloid cells does not alter tumor incidence
The above studies demonstrate a strong reduction in the numbers of G-MDSCs and Tregs in tumors of STAT3-cKO Smo* compared with Smo* mice without the condition STAT3 deletion. The lack of these immunosuppressive cell types could lead, in principle, to reinforced immune responses against tumor development and therefore, reduced tumor incidence in STAT3-cKO mice. However, we found that the rate of tumor development and overall incidence were very similar between the two murine stains. Two types of representations of tumor incidence are shown in Fig. 8. In Fig. 8A, the cumulative incidence of tumors over mouse age is shown, whereas in Fig. 8B, a Kaplan-Meier analysis of the data reflects the number of death events/age group. In the analysis of cumulative tumor formation, we found that the proportion of mice with tumors increased similarly in WT or STAT3-cKO mice, with almost 75–80% incidence in mice older than 6 months in either genotype. In addition, survival decreased at a similar rate in both mice groups.
Figure 8. Tumor incidence is not altered in selective myeloid STAT3-cKO.
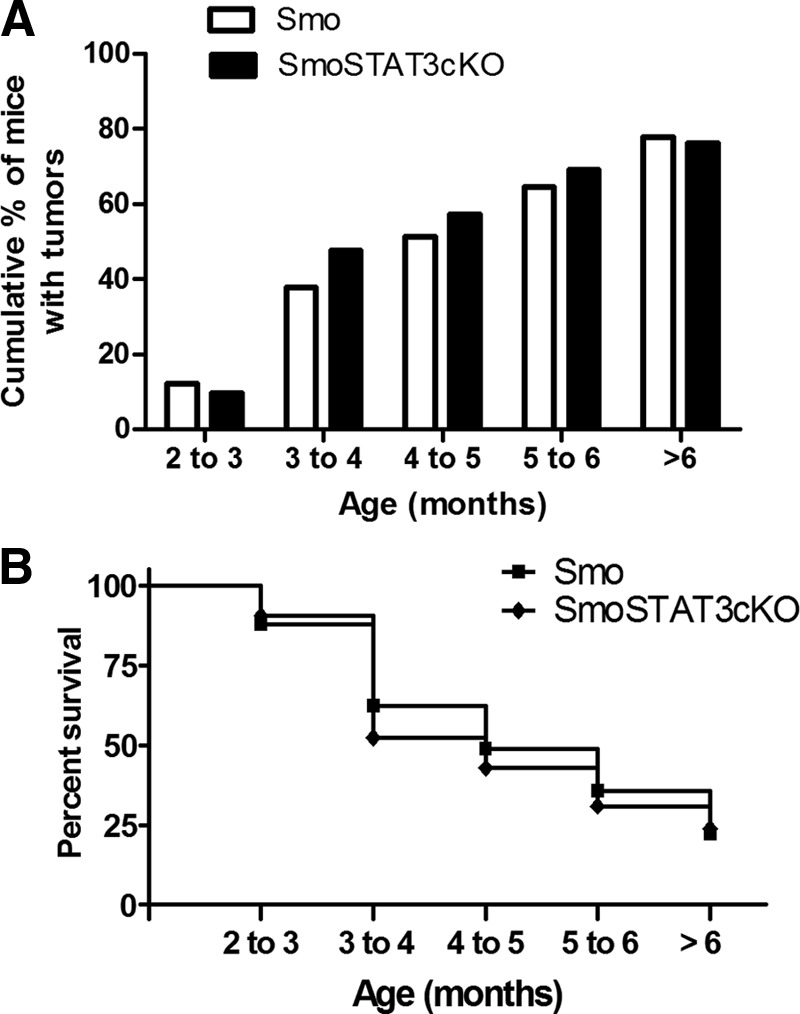
The development of medulloblastoma was monitored in Smo* WT (n=90) and Smo* STAT3-cKO (n=41) mice. (A) The cumulative percent of mice with tumors and (B) a Kaplan-Meyer representation of mouse survival. Comparison of survival curves performed using the log rank test showed no statistical differences between the survivals of the two groups of mice.
To investigate differences in the tumor characteristics of WT and STAT3-KO mice at the microscopic level, we stained tumors sections with H&E. We did not observe any clear differences in cellular morphology, the presence of mitotic figures, apoptotic bodies, or tumor invasion, the latter being very infrequent in this model (data not shown).
Angiogenesis did not differ significantly in tumors from Smo* versus Smo* STAT3-cKO mice
Angiogenesis is essential for cancer growth and metastasis. It has been shown that one of the mechanisms by which MDSCs promote tumor growth is by producing endothelial growth factors and thus, promoting angiogenesis. Endothelial factors, such as VEGF, are downstream factors of activated STAT3 [32]. A study showed that MDSCs and macrophages isolated from mouse B16 melanoma tumors displayed activated STAT3 and induced angiogenesis in an in vitro tube formation assay via STAT3 induction of angiogenic factors, including VEGF and bFGF [11]. In addition, STAT3-deleted myeloid cells were deficient in their ability to induce blood-vessel formation in vivo in melanoma-implanted tumors compared with WT MDSC [11]. Thus, we would expect that the lack of STAT3 activation in MDSCs may lead to lower production of growth factors and reduced angiogenesis in our model, potentially impairing tumor growth.
We thus compared the degree of angiogenesis in tumor sections from Smo* WT versus STAT3-cKO mice by immunofluorescence (not shown). Surprisingly, there was a trend for a higher (rather than lower) level of angiogenesis in tumors from Smo* STAT3-cKO than control Smo* mice, although this was not statistically significant (16.1%±2.7 vs. 11.5%±3.7; P>0.05).
DISCUSSION
The brain is considered to be an immune privilege organ because of the existence of a BBB that is relatively impermeable to peripheral immune cells in the healthy state, a limited lymphatic system that can drain antigens to LNs, and the still debatable absence of professional APCs. However, it is now well-known that the CNS is constantly under the surveillance of patrolling immune cells, including T cells. In addition, pathological conditions, including cancer, may cause cellular and molecular alterations to promote immune cell infiltration further. In this regard, innate and adaptive immune cells have been demonstrated within human brain tumors [33–36]. Enhancement of T cell anti-tumor activity can halt cancer progression in some cases and is a major focus of immune-based anti-cancer research. However, the strong immunosuppressive mechanisms existing in tumors may reduce or even impede the action of T cells. Myeloid cells with reported immunosuppressive properties, MDSCs, accumulate in large numbers in different models of cancer, inflammation, and other conditions (reviewed in refs. [37–41]). These have been characterized as a heterogeneous population of myeloid cells with an immature phenotype, which, in tumor conditions, expands and migrates from the bone marrow into the blood, spleen, and the tumors themselves. Although the knowledge on MDSCs is gradually increasing, there are relatively few reports demonstrating the presence of MDSCs in the context of brain tumors. For example, they have been found in the blood of glioblastoma patients [42]. In addition, tumor-infiltrating MDSCs were found to be present in murine models where glioblastoma cell lines were s.c. or intracraneally implanted [43, 44]. Studies of the immune response in brain tumors by using models based on the injection of tumor cell lines have provided essential data regarding the pathogenesis of tumor formation and growth and about the ongoing immune response. However, the fact that the mice used here develop spontaneous tumors may be advantageous in the sense that tumors develop slowly in situ with normal stroma, and there is no breakdown of the BBB caused by an injection, a technique that may alter the physiological immune response.
Although medulloblastoma is the most common form of malignant brain tumor in children, the immune response in human tumors is poorly understood, and it has not been studied previously in an animal model. Myeloid and lymphoid immune cells have been shown to be present in human primitive neuroectodermal tumors and medulloblastomas [45, 46]. With the use of KP1 and Mac387 antibodies, monocyte/macrophages were detected in human medulloblastoma tumors [47]. However, whether the myeloid cells in these tumors exhibit a MDSC phenotype or express pSTAT3 has not been reported. Although G-MDSCs constituted the majority of MDSCs in peripheral organs in a study with 10 different cancer types implanted in mice [4], the proportions of G-MDSCs and M-MDSCs in the tumors themselves vary considerably in different models. Here, we found a distinct population of MDSCs in medulloblastoma tumors from Smo* mice. In the medulloblastoma model used in our study, most of the MDSCs exhibited phenotypic markers of G-MDSCs. These cells appeared to arrive to the tumors at early stages of tumor development, as they were found in ectopic cerebellar lesions in asymptomatic mice. These ectopic cerebellar lesions, characterized as hyperplastic patches of the external granule layer, are frequent in several murine models of medulloblastoma at early age [24]. Similar to our result, tumor infiltration by T cells, including a significant proportion of Tregs, was also found at early asymptomatic stages in a spontaneous model of astrocytoma [48]. In this model, however, MDSCs were not present at this time. The finding of immunosuppressive cell types in the initial stages of tumor formation may have two important implications. First, it hints that the immune system is alerted of ongoing pathogenic changes at early stages of tumorigenesis through as-yet unknown mechanisms. Second, it suggests that the prompt setting of an immunosuppressive environment may impair an effective anti-tumor immune response from the beginning, promoting its escape from immune-system attack.
Several signaling pathways have been implicated in MDSC expansion and functionality in tumors. Strong evidence suggests that STAT3-pathway activation is key. We found that cerebellar tumors of STAT3-cKO mice, which exhibit a strong reduction of STAT3 in myeloid cells, presented considerably reduced amounts of MDSC of the granulocytic subtype. The LysM-Cre mice used here have been shown to direct LoxP-mediated recombination specifically in macrophages and neutrophils. However, it has also been reported that a residual STAT3fl/fl could be found in peritoneal macrophages, suggesting that the STAT3 deletion in these cells is not complete [16]. Nevertheless, it was found that these mice presented enhanced Th1 responses and chronic enterocolitis, and their peritoneal macrophages produced higher levels of proinflammatory cytokines than WT mice [16, 31]. We found a similar colitis phenotype in our mice (data not shown) and reduced LPS induction of proinflammatory cytokines and a large reduction in pSTAT3 in CD11b+ cells within medulloblastoma.
There are several mechanisms by which STAT3 in myeloid cells may be required to maintain this population within tumors. In myeloid cells, STAT3 signaling drives the expression of Bcl-xL, c-myc, cyclin D1, or survivin, which prevents cell apoptosis and promotes cell proliferation [49]. In our study, G-MDSCs had a tendency to exhibit higher apoptosis indexes in STAT3-cKO tumors, although the differences were not significant. In addition, STAT3 has been associated with other aspects of MDSC expansion. Although different mechanisms have been suggested, this may occur, at least in part, by STAT3-mediated blockade of immature myeloid cell differentiation. For example, it has been shown that blockade of STAT3 signaling in DCs abrogated the tumor-induced inhibition of their functional maturation, decreasing the number of immature CD11c+ MHC class IIlowCD86low DCs [18]. Likewise, in another study, chemical inhibition of the Jak2/STAT3 pathway induced the maturation of CD11b+Gr1+ cells isolated from CT-26 colon adenocarcinomas to DCs in vitro and caused a reduction in the numbers of MDSCs in vivo in several models of cancer, leading to an accumulation of mature DCs [50]. The precise molecular mechanisms by which STAT3 blocks myeloid cell maturation are still being investigated. One of these may include the STAT3-mediated up-regulation of the proinflammatory proteins S100A9 and S100A8, which may inhibit myeloid differentiation by promoting ROS formation [51].
In addition to their own activity, MDSCs modulate other cell types within the tumor microenvironment to reinforce immunosuppression and promote tumor growth. For example, there is evidence suggesting that STAT3 activation in MDSCs from tumors can potentially induce or maintain the population of Tregs. In this sense, deletion of STAT3 specifically in hematopoietic cells with an Mx1-Cre-STAT3fl/fl system was associated with a reduction of Tregs in the melanoma tumor model based on s.c. implantation of the B16 cell line [6]. Here, we found that the proportion of Tregs was reduced, and the ratio Teff/Treg was increased in the cKO, supporting a role for STAT3 in myeloid cells to maintain the Treg population within meduloblastoma tumors. Although not completely understood, the mechanisms by which STAT3 may promote the Treg population have been started to be elucidated by others. For example, the promoter of the gene encoding TGF-β, a factor promoting the expansion of existing Tregs and inducible Tregs de novo, contains two potential STAT3-binding sites [52]. It has been shown that deletion of suppressor of cytokine signaling-3, an inhibitor of the STAT3 signaling pathway, in DCs increased the production of TGF-β in DCs, and through this cytokine, they selectively expanded the population of Foxp3+ Tregs in vitro [53]. However, in a model of B cell lymphoma, MDSCs induced Tregs in a process dependent on arginase and independent of TGF-β [54].
We predicted that the deletion of STAT3 in myeloid cells might ultimately lead to a decrease in medulloblastoma incidence as a result of reinforced anti-tumor immunity. It has been demonstrated in several models of cancer that a reduction or elimination of MDSCs led to an improvement of anti-tumor immune responses [55–57]. However, we did not find differences in the incidence of tumors between WT and STAT3-cKO mice despite the significant reduction that we found in tumor-infiltrating MDSCs and Tregs. It might be necessary to block STAT3 signaling in multiple cell types to reduce tumor growth. STAT3 activity may be important in multiple tumor-infiltrating immune cell types other than MDSCs, including DCs, T cells (including Tregs), NK cells, and neutrophils in a melanoma model [6, 19, 58]. Pan-hematopoietic deletion of STAT3 in mice led to enhanced anti-tumor responses and reduced melanoma growth and metastases [6]. Moreover, ablation of STAT3 in CD8+ cells improved their proliferation and anti-tumor activity upon transfer to tumor-carrying mice [59]. Another possibility that could contribute to the lack of clinical differences between WT and myeloid STAT3-KO mice in our study may be an up-regulation of other tumor escape mechanisms of an immunological or nonimmunological nature. For example, we observed a trend for higher angiogenesis in STAT3-KO mice, although the significance of this finding remains to be elucidated. Alternatively, despite the reduction of immunosuppression, the anti-tumor T cell response may not be strong enough to eliminate the tumor completely. In either case, it would be of interest to determine if inhibition of STAT3 in myeloid cells would improve the efficiency of other tumor therapies as chemotherapy or existing cancer T cell-based immunotherapies. For example, it could potentially reinforce immune responses in therapies based on anti-tumor T cell transfer, providing an additional tool for the treatment of brain tumors.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Brain Tumor Society (grant 20092804) and U.S. National Institutes of Health HD068686 and HD046112. Flow cytometry was performed at the UCLA JCCC and Center for AIDS Research Flow Cytometry Core Facility, which is supported by U.S. National Institutes of Health awards CA-16042 and AI-28697 and by the JCCC, the UCLA AIDS Institute, and the David Geffen School of Medicine at UCLA.
The online version of this paper, found at www.jleukbio.org, includes supplemental information.
- 7-AAD
- 7-amino-actinomycin D
- BBB
- blood brain barrier
- cKO
- conditional knockout
- Foxp3
- forkhead box p3
- G-MDSC
- granulocytic myeloid-derived suppressor cell
- JCCC
- Jonsson Comprehensive Cancer Center
- KO
- knockout
- LysM
- lysozyme M
- M-MDSC
- monocytic myeloid-derived suppressor cell
- MB
- Medulloblastoma
- MDSC
- myeloid-derived suppressor cell
- pSTAT
- phosphorylated STAT
- Smo
- smoothened
- Teff
- effector T cell
- Treg
- regulatory T cell
- UCLA
- University of California, Los Angeles
REFERENCES
- 1. Condamine T., Gabrilovich D. I. (2011) Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 32, 19–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Filipazzi P., Huber V., Rivoltini L. (2012) Phenotype, function and clinical implications of myeloid-derived suppressor cells in cancer patients. Cancer Immunol. Immunother. 61, 255–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ueha S., Shand F. H., Matsushima K. (2011) Myeloid cell population dynamics in healthy and tumor-bearing mice. Int. Immunopharmacol. 11, 783–788 [DOI] [PubMed] [Google Scholar]
- 4. Youn J. I., Nagaraj S., Collazo M., Gabrilovich D. I. (2008) Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J. Immunol. 181, 5791–5802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Movahedi K., Guilliams M., Van den Bossche J., Van den Bergh R., Gysemans C., Beschin A., De Baetselier P., Van Ginderachter J. A. (2008) Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood 111, 4233–4244 [DOI] [PubMed] [Google Scholar]
- 6. Kortylewski M., Kujawski M., Wang T., Wei S., Zhang S., Pilon-Thomas S., Niu G., Kay H., Mule J., Kerr W. G., Jove R., Pardoll D., Yu H. (2005) Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat. Med. 11, 1314–1321 [DOI] [PubMed] [Google Scholar]
- 7. Drake C. G., Jaffee E., Pardoll D. M. (2006) Mechanisms of immune evasion by tumors. Adv. Immunol. 90, 51–81 [DOI] [PubMed] [Google Scholar]
- 8. Kusmartsev S., Gabrilovich D. I. (2006) Role of immature myeloid cells in mechanisms of immune evasion in cancer. Cancer Immunol. Immunother. 55, 237–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lee H., Pal S. K., Reckamp K., Figlin R. A., Yu H. (2011) STAT3: a target to enhance antitumor immune response. Curr. Top. Microbiol. Immunol. 344, 41–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. He G., Karin M. (2011) NF-κB and STAT3—key players in liver inflammation and cancer. Cell Res. 21, 159–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kujawski M., Kortylewski M., Lee H., Herrmann A., Kay H., Yu H. (2008) Stat3 mediates myeloid cell-dependent tumor angiogenesis in mice. J. Clin. Invest. 118, 3367–3377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Amin H. M., McDonnell T. J., Ma Y., Lin Q., Fujio Y., Kunisada K., Leventaki V., Das P., Rassidakis G.Z., Cutler C., Medeiros L.J., Lai R. (2004) Selective inhibition of STAT3 induces apoptosis and G(1) cell cycle arrest in ALK-positive anaplastic large cell lymphoma. Oncogene 23, 5426–5434 [DOI] [PubMed] [Google Scholar]
- 13. Tsareva S. A., Moriggl R., Corvinus F. M., Wiederanders B., Schutz A., Kovacic B., Friedrich K. (2007) Signal transducer and activator of transcription 3 activation promotes invasive growth of colon carcinomas through matrix metalloproteinase induction. Neoplasia 9, 279–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. O'Farrell A. M., Liu Y., Moore K. W., Mui A. L. (1998) IL-10 inhibits macrophage activation and proliferation by distinct signaling mechanisms: evidence for Stat3-dependent and -independent pathways. EMBO J. 17, 1006–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lang R., Patel D., Morris J. J., Rutschman R. L., Murray P. J. (2002) Shaping gene expression in activated and resting primary macrophages by IL-10. J. Immunol. 169, 2253–2263 [DOI] [PubMed] [Google Scholar]
- 16. Takeda K., Clausen B. E., Kaisho T., Tsujimura T., Terada N., Forster I., Akira S. (1999) Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity 10, 39–49 [DOI] [PubMed] [Google Scholar]
- 17. Cheng F., Wang H. W., Cuenca A., Huang M., Ghansah T., Brayer J., Kerr W. G., Takeda K., Akira S., Schoenberger S. P., Yu H., Jove R., Sotomayor E. M. (2003) A critical role for Stat3 signaling in immune tolerance. Immunity 19, 425–436 [DOI] [PubMed] [Google Scholar]
- 18. Wang T., Niu G., Kortylewski M., Burdelya L., Shain K., Zhang S., Bhattacharya R., Gabrilovich D., Heller R., Coppola D., Dalton W., Jove R., Pardoll D., Yu H. (2004) Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat. Med. 10, 48–54 [DOI] [PubMed] [Google Scholar]
- 19. Kortylewski M., Xin H., Kujawski M., Lee H., Liu Y., Harris T., Drake C., Pardoll D., Yu H. (2009) Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor microenvironment. Cancer Cell 15, 114–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Langowski J. L., Zhang X., Wu L., Mattson J. D., Chen T., Smith K., Basham B., McClanahan T., Kastelein R. A., Oft M. (2006) IL-23 promotes tumour incidence and growth. Nature 442, 461–465 [DOI] [PubMed] [Google Scholar]
- 21. Buckner J. C., Brown P. D., O'Neill B. P., Meyer F. B., Wetmore C. J., Uhm J. H. (2007) Central nervous system tumors. Mayo Clin. Proc. 82, 1271–1286 [DOI] [PubMed] [Google Scholar]
- 22. Huse J. T., Holland E. C. (2010) Targeting brain cancer: advances in the molecular pathology of malignant glioma and medulloblastoma. Nat. Rev. Cancer. 10, 319–331 [DOI] [PubMed] [Google Scholar]
- 23. Pollack I. F., Jakacki R. I. (2011) Childhood brain tumors: epidemiology, current management and future directions. Nat. Rev. Neurol. 7, 495–506 [DOI] [PubMed] [Google Scholar]
- 24. Hatton B. A., Villavicencio E. H., Tsuchiya K. D., Pritchard J. I., Ditzler S., Pullar B., Hansen S., Knoblaugh S. E., Lee D., Eberhart C. G., Hallahan A. R., Olson J. M. (2008) The Smo/Smo model: hedgehog-induced medulloblastoma with 90% incidence and leptomeningeal spread. Cancer Res. 68, 1768–1776 [DOI] [PubMed] [Google Scholar]
- 25. Hallahan A. R., Pritchard J. I., Hansen S., Benson M., Stoeck J., Hatton B. A., Russell T. L., Ellenbogen R. G., Bernstein I. D., Beachy P. A., Olson J. M. (2004) The SmoA1 mouse model reveals that Notch signaling is critical for the growth and survival of sonic hedgehog-induced medulloblastomas. Cancer Res. 64, 7794–7800 [DOI] [PubMed] [Google Scholar]
- 26. Herrmann J. E., Imura T., Song B., Qi J., Ao Y., Nguyen T. K., Korsak R. A., Takeda K., Akira S., Sofroniew M. V. (2008) STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J. Neurosci. 28, 7231–7243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Takeda K., Kaisho T., Yoshida N., Takeda J., Kishimoto T., Akira S. (1998) Stat3 activation is responsible for IL-6-dependent T cell proliferation through preventing apoptosis: generation and characterization of T cell-specific Stat3-deficient mice. J. Immunol. 161, 4652–4660 [PubMed] [Google Scholar]
- 28. Abad C., Tan Y. V., Lopez R., Nobuta H., Dong H., Phan P., Feng J. M., Campagnoni A. T., Waschek J. A. (2010) Vasoactive intestinal peptide loss leads to impaired CNS parenchymal T-cell infiltration and resistance to experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 107, 19555–19560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yu H., Kortylewski M., Pardoll D. (2007) Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 7, 41–51 [DOI] [PubMed] [Google Scholar]
- 30. Cross M., Mangelsdorf I., Wedel A., Renkawitz R. (1988) Mouse lysozyme M gene: isolation, characterization, and expression studies. Proc. Natl. Acad. Sci. USA 85, 6232–6236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Reindl W., Weiss S., Lehr H. A., Forster I. (2007) Essential crosstalk between myeloid and lymphoid cells for development of chronic colitis in myeloid-specific signal transducer and activator of transcription 3-deficient mice. Immunology 120, 19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Niu G., Wright K. L., Huang M., Song L., Haura E., Turkson J., Zhang S., Wang T., Sinibaldi D., Coppola D., Heller R., Ellis L. M., Karras J., Bromberg J., Pardoll D., Jove R., Yu H. (2002) Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene 21, 2000–2008 [DOI] [PubMed] [Google Scholar]
- 33. Kim Y. H., Jung T. Y., Jung S., Jang W. Y., Moon K. S., Kim I. Y., Lee M. C., Lee J. J. (2012) Tumour-infiltrating T-cell subpopulations in glioblastomas. Br. J. Neurosurg. 26, 21–27 [DOI] [PubMed] [Google Scholar]
- 34. Metelitsa L. S., Wu H. W., Wang H., Yang Y., Warsi Z., Asgharzadeh S., Groshen S., Wilson S. B., Seeger R. C. (2004) Natural killer T cells infiltrate neuroblastomas expressing the chemokine CCL2. J. Exp. Med. 199, 1213–1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Weber F., Volgmann T., Menzel J. (1993) Tumor infiltrating lymphocytes in malignant brain tumors. Arch. Immunol. Ther. Exp. (Warsz). 41, 41–44 [PubMed] [Google Scholar]
- 36. Yang I., Han S. J., Sughrue M. E., Tihan T., Parsa A. T. (2011) Immune cell infiltrate differences in pilocytic astrocytoma and glioblastoma: evidence of distinct immunological microenvironments that reflect tumor biology. J. Neurosurg. 115, 505–511 [DOI] [PubMed] [Google Scholar]
- 37. Cuenca A. G., Delano M. J., Kelly-Scumpia K. M., Moreno C., Scumpia P. O., Laface D. M., Heyworth P. G., Efron P. A., Moldawer L. L. (2011) A paradoxical role for myeloid-derived suppressor cells in sepsis and trauma. Mol. Med. 17, 281–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Youn J. I., Gabrilovich D. I. (2010) The biology of myeloid-derived suppressor cells: the blessing and the curse of morphological and functional heterogeneity. Eur. J. Immunol. 40, 2969–2975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Peranzoni E., Zilio S., Marigo I., Dolcetti L., Zanovello P., Mandruzzato S., Bronte V. (2010) Myeloid-derived suppressor cell heterogeneity and subset definition. Curr. Opin. Immunol. 22, 238–244 [DOI] [PubMed] [Google Scholar]
- 40. Dilek N., van Rompaey N., Le Moine A., Vanhove B. (2010) Myeloid-derived suppressor cells in transplantation. Curr. Opin. Organ Transplant. 15, 765–768 [DOI] [PubMed] [Google Scholar]
- 41. Ostrand-Rosenberg S., Sinha P. (2009) Myeloid-derived suppressor cells: linking inflammation and cancer J. Immunol. 182, 4499–4506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Raychaudhuri B., Rayman P., Ireland J., Ko J., Rini B., Borden E. C., Garcia J., Vogelbaum M. A., Finke J. (2011) Myeloid-derived suppressor cell accumulation and function in patients with newly diagnosed glioblastoma. Neuro. Oncol. 13, 591–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Umemura N., Saio M., Suwa T., Kitoh Y., Bai J., Nonaka K., Ouyang G. F., Okada M., Balazs M., Adany R., Shibata T., Takami T. (2008) Tumor-infiltrating myeloid-derived suppressor cells are pleiotropic-inflamed monocytes/macrophages that bear M1- and M2-type characteristics. J. Leukoc. Biol. 83, 1136–1144 [DOI] [PubMed] [Google Scholar]
- 44. Jia W., Jackson-Cook C., Graf M. R. (2010) Tumor-infiltrating, myeloid-derived suppressor cells inhibit T cell activity by nitric oxide production in an intracranial rat glioma + vaccination model. J. Neuroimmunol. 223, 20–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Strik H. M., Stoll M., Meyermann R. (2004) Immune cell infiltration of intrinsic and metastatic intracranial tumours. Anticancer Res. 24, 37–42 [PubMed] [Google Scholar]
- 46. Salsman V. S., Chow K. K., Shaffer D. R., Kadikoy H., Li X. N., Gerken C., Perlaky L., Metelitsa L. S., Gao X., Bhattacharjee M., Hirschi K., Heslop H. E., Gottschalk S., Ahmed N. (2011) Crosstalk between medulloblastoma cells and endothelium triggers a strong chemotactic signal recruiting T lymphocytes to the tumor microenvironment. PLoS One 6, e20267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rossi M. L., Buller J. R., Heath S. A., Carey M. P., Carboni P., Jr., Koutsoubelis G., Coakham H. B. (1991) The monocyte/macrophage infiltrate in 35 medulloblastomas: a paraffin-wax study. Tumori 77, 36–40 [DOI] [PubMed] [Google Scholar]
- 48. Tran Thang N. N., Derouazi M., Philippin G., Arcidiaco S., Di Berardino-Besson W., Masson F., Hoepner S., Riccadonna C., Burkhardt K., Guha A., Dietrich P. Y., Walker P. R. (2010) Immune infiltration of spontaneous mouse astrocytomas is dominated by immunosuppressive cells from early stages of tumor development. Cancer Res. 70, 4829–4839 [DOI] [PubMed] [Google Scholar]
- 49. Yu H., Pardoll D., Jove R. (2009) STATs in cancer inflammation and immunity: a leading role for STAT3. Nat. Rev. Cancer 9, 798–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Nefedova Y., Nagaraj S., Rosenbauer A., Muro-Cacho C., Sebti S. M., Gabrilovich D. I. (2005) Regulation of dendritic cell differentiation and antitumor immune response in cancer by pharmacologic-selective inhibition of the Janus-activated kinase 2/signal transducers and activators of transcription 3 pathway. Cancer Res. 65, 9525–9535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cheng P., Corzo C. A., Luetteke N., Yu B., Nagaraj S., Bui M. M., Ortiz M., Nacken W., Sorg C., Vogl T., Roth J., Gabrilovich D. I. (2008) Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J. Exp. Med. 205, 2235–2249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kinjyo I., Inoue H., Hamano S., Fukuyama S., Yoshimura T., Koga K., Takaki H., Himeno K., Takaesu G., Kobayashi T., Yoshimura A. (2006) Loss of SOCS3 in T helper cells resulted in reduced immune responses and hyperproduction of interleukin 10 and transforming growth factor-β 1. J. Exp. Med. 203, 1021–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Matsumura Y., Kobayashi T., Ichiyama K., Yoshida R., Hashimoto M., Takimoto T., Tanaka K., Chinen T., Shichita T., Wyss-Coray T., Sato K., Yoshimura A. (2007) Selective expansion of Foxp3-positive regulatory T cells and immunosuppression by suppressors of cytokine signaling 3-deficient dendritic cells. J. Immunol. 179, 2170–2179 [DOI] [PubMed] [Google Scholar]
- 54. Serafini P., Mgebroff S., Noonan K., Borrello I. (2008) Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Res. 68, 5439–5449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Roth F., De La Fuente A. C., Vella J. L., Zoso A., Inverardi L., Serafini P. (2012) Aptamer-mediated blockade of IL4Rα triggers apoptosis of MDSCs and limits tumor progression. Cancer Res. 72, 1373–1383 [DOI] [PubMed] [Google Scholar]
- 56. Bunt S. K., Yang L., Sinha P., Clements V. K., Leips J., Ostrand-Rosenberg S. (2007) Reduced inflammation in the tumor microenvironment delays the accumulation of myeloid-derived suppressor cells and limits tumor progression. Cancer Res. 67, 10019–10026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Srivastava M. K., Zhu L., Harris-White M., Kar U., Huang M., Johnson M. F., Lee J. M., Elashoff D., Strieter R., Dubinett S., Sharma S. (2012) Myeloid suppressor cell depletion augments antitumor activity in lung cancer. PLoS One 7, e40677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yu H., Jove R. (2004) The STATs of cancer—new molecular targets come of age. Nat. Rev. Cancer 4, 97–105 [DOI] [PubMed] [Google Scholar]
- 59. Kujawski M., Zhang C., Herrmann A., Reckamp K., Scuto A., Jensen M., Deng J., Forman S., Figlin R., Yu H. (2010) Targeting STAT3 in adoptively transferred T cells promotes their in vivo expansion and antitumor effects. Cancer Res. 70, 9599–9610 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.