

Abstract
Inhibition of the extracellular process of collagen fibril formation represents a new approach to limiting posttraumatic or postsurgical localized fibrosis. It has been demonstrated that employing a monoclonal antibody that targets the C-terminal telopeptide of the α2 chain of collagen I blocks critical collagen I-collagen I interaction, thereby reducing the amount of collagen deposits in vitro and in animal models. Here, we developed a chimeric variant of a prototypic inhibitory antibody of mouse origin. The structure of this novel antibody was analyzed by biochemical and biophysical methods. Moreover, detailed biochemical and biological studies were employed to test its antigen-binding characteristics. The ability of the chimeric variant to block formation of collagen fibrils was tested in vitro and in high-density cultures representing fibrotic processes occurring in the skin, tendon, joint capsule, and gingiva. The potential toxicity of the novel chimeric antibody was analyzed through its impact on the viability and proliferation of various cells and by testing its tissue cross-reactivity in sets of arrays of human and mouse tissues. Results of the presented studies indicate that engineered antibody-based blocker of localized fibrosis is characterized by the following: (i) a correct IgG-like structure, (ii) a high-affinity and high-specificity for a defined epitope, (iii) a great potential to limit accumulation of collagen-rich deposits, and (iv) a lack of cytotoxicity and non-specific tissue reactivity. Together, the presented study shows the great potential of the novel chimeric antibody to limit localized fibrosis, thereby setting ground for critical preclinical tests in a relevant animal model.
Keywords: localized fibrosis, chimeric antibody, collagen fibrils, extracellular matrix, therapeutic antibody, protein engineering
INTRODUCTION
The potential formation of human anti-mouse antibodies (HAMA) limits the clinical utility of the mouse-derived monoclonal antibodies developed against a specific therapeutic target. Because of this limitation, various techniques have to be employed to minimize the HAMA response. For example, protein engineering approaches are utilized to reduce the content of mouse-derived antibody fragments. These approaches include (i) engineering chimeric antibodies consisting of mouse-derived variable regions fused with the human-derived constant regions, (ii) production of humanized antibodies in which the majority of variable regions and the entire constant regions are of human origin, and (iii) creation of single chain variable fragments (scFv) in which the constant regions of the native antibody are removed and the variable regions are connected via a short peptide-linker [1–6].
In this study, we focused on developing a clinically-relevant chimeric antibody variant designed to inhibit excessive accumulation of collagen-rich fibrotic deposits via blocking collagen-collagen interactions critical for the formation of collagen fibrils, a main component of fibrotic tissues [7]. Previously, we demonstrated that the mouse monoclonal antibody which targets the C-terminal telopeptide of the α2 chain (α2Ct) of the human procollagen I (anti-α2Ct) is effective in blocking collagen fibril formation in vitro and in keloid-like constructs [7]. Because of its mouse origin, this archetypical mouse antibody of the IgA type, however, cannot be considered an acceptable candidate for human-relevant tests. Consequently, we engineered a chimeric antibody variant (chIgG) consisting of the variable domains derived from the prototypic IgA-type anti-α2Ct antibody and the constant domains of the human IgG (hIgG). Detailed analyses of the recombinant chIgG variant demonstrated its correct structure. Moreover, this novel antibody was also tested for its ability to inhibit collagen fibril formation in vivo and in cultures of various cells associated with the fibrotic process. Results demonstrate a great potential of the chIgG variant for preclinical tests with the use of relevant biological models.
MATERIALS AND METHODS
Amplification of DNA sequence encoding the VH region of the mouse α chain (mVH)
Total RNA was isolated from mouse-derived hybridoma cells producing the original anti-α2Ct antibody of the IgA (mIgA) class [7]. Subsequently, hybridoma-derived RNA was employed as a template to generate a PCR product spanning the sequence encoding the mVH region of the α chain of the mIgA antibody (Fig. 1). The Ig-Primer Sets (EMD Millipore/Novagen) were employed to target the 5′ end of the sequence of interest while the 3′ end was targeted with custom-designed primers (Table I) [8].
Figure 1.
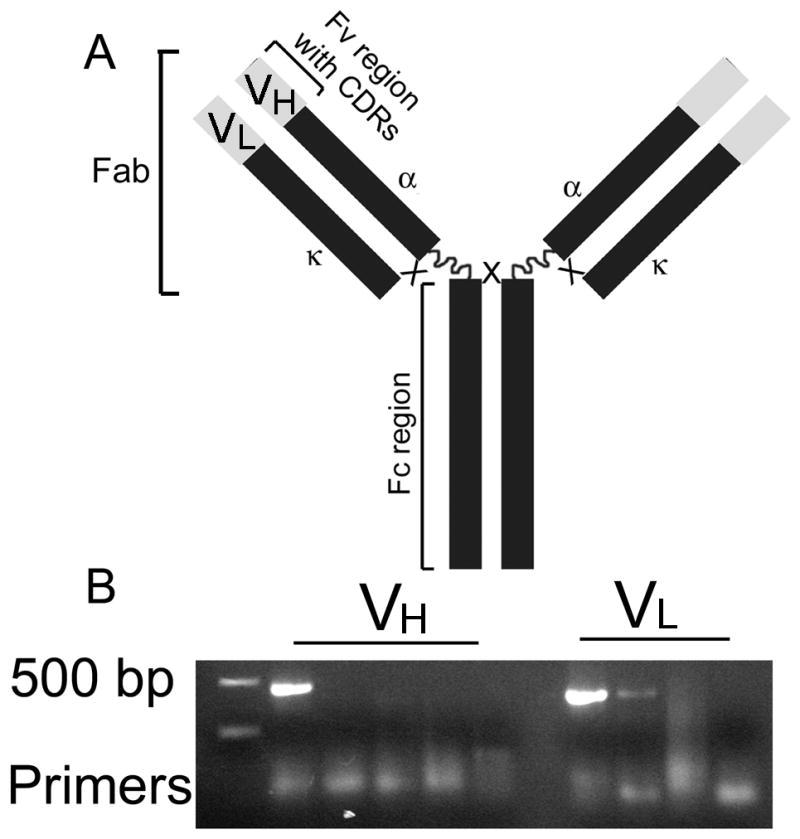
PCR products corresponding to the variable regions of the mIgA-type anti-α2Ct antibody. A; A schematic illustrating relevant domains of the mIgA variant. B; PCR products encompassing DNA sequences that encode the variable regions of the heavy α (VH) and the light κ (VL) chains of the mIgA-type anti-α2Ct antibody. Note: Some sets of the PCR primers did not create any products due to the lack of homology with the variable regions of the template. Symbols: Fv; Fragment variable regions, CDR; Complementarity determining regions, α and κ; constant regions of the heavy α and light κ chains, respectively, Fab; Fragment antigen-binding, Fc; Fragment crystallizable region, Vα and Vκ; PCR products corresponding to the Fv regions of the α and κ chains, respectively.
Table I.
Sets of primers employed to generate PCR products spanning the sequence encoding the VH of the heavy α chain of the mouse IgA.
| a Sets of forward PCR primers targeting the 5′ end of the sequence encoding the VH of the mouse heavy μ chain (Ig-Primer Set EMD Millipore/Novagen) | b Reverse PCR primers targeting the 5′ end of the sequence encoding the constant region of the mouse heavy α chain a |
|---|---|
| MuIgH5′-A | α1_1 GGGAGTGTCAGTGGGTAGATGGTG* |
| MuIgH5′-B | α1_2 TTCCCACTCTTTCCCCAGGTCACA |
| MuIgH5′-C* | α1_3 AGAGGCGAGGGCAGGTGGAAAGTT |
| MuIgH5′-D | α1_4 CAGAGGCGAGGGCAGGTGGAAAGT |
| MuIgH5′-E | α1_5 TCACGGATCTCCTTCTGGGCACTG |
| MuIgH5′-F |
Detailed information on these primers can be found in the manual for the Ig-Primer Set (EMD Millipore/Novagen).
Primers were designed based on the DNA sequence of the mouse IgA heavy α chain published by Tucker et al. [8].
A primer set that resulted in a PCR product encompassing the VH region of the α chain of mouse IgA (see Fig. 1).
Amplification of DNA sequence encoding the VL region of the mouse κ chain (mVL)
A commercial Ig-Primer Set (EMD Millipore/Novagen) was employed to generate a PCR product corresponding to the mVL of the κ chain.
Cloning the PCR fragments
The PCR products for the mVH and the mVL regions were cloned into the pETBlue-1 Perfectly Blunt vector (EMD Millipore/Novagen).
DNA constructs for the chimeric mVH-human γ chain (mVH-hγ)
To facilitate cloning into the pFUSE-CHIg-hG1 vector that includes the sequence encoding the constant region of the heavy γ chain of the human IgG1 and the Zeocin-resistance gene (InvivoGen), the EcoRI and NheI restriction sites were introduced via PCR to the original DNA sequence encoding the mVH region. Subsequently, an insert encoding the mVH region of the mouse α chain was cloned into the corresponding restriction sites of the pFUSE-CHIg-hG1 vector. The fidelity of the mVH-hγ construct was confirmed by DNA sequencing.
DNA constructs for chimeric mVL-human light κ chain (mVL-hκ)
To enable cloning into the pFUSE2-CLIg-hk vector that includes the sequence encoding the constant region of the human light κ chain and the Blasticidin-resistance gene (InvivoGen), the AgeI and BsiWI restriction sites were introduced via PCR to the original DNA sequence encoding the mVL region of the light κ chain. Subsequently, the insert encoding the mVL region was cloned into the corresponding restriction sites of the pFUSE2-CLIg-hk vector. The fidelity of the mVL-hκ construct was confirmed by DNA sequencing.
Selection of clones co-expressing chimeric mVH-hγ and mVL-hκ variants in mammalian cells
Chinese Hamster Ovary cells (CHO; ATTC# CCL-61) were transfected with DNA constructs encoding the mVH-hγ and mVL-hκ variants. Subsequently, Zeocin and Blasticidin-resistant clones were selected and screened for the presence of the mVH-hγ and mVL-hκ chains by Western blot. For these assays the goat anti-human γ chain antibodies and goat anti-human κ chain antibodies conjugated with HRP were employed (Sigma-Aldrich).
Purification of the chIgG consisting of the mVH-hγ and mVL-hκ chains
Selected CHO clones were cultured in serum-free conditions in the FiberCell System (FiberCell Systems Inc.). The chIgG was purified by affinity chromatography using Protein-L agarose, a resin that specifically binds to the light κ chain (Thermo Scientific). The molecular mass of purified chIgG, was analyzed by size-exclusion chromatography (SEC) using the BioBasic SEC-300 column (Thermo Scientific). For these assays, chIgG concentrated to 20 mg/ml was injected to the column equilibrated with PBS. Subsequently, the mass of protein eluted in a single peak was calculated based on the elution profiles of protein mass markers (Sigma-Aldrich).
Structural assays of the chIgG
Circular dichroism (CD) spectra in the far-UV region were recorded for the inhibitory chIgG variant and non-inhibitory control hIgG (Bethyl Laboratories) using the Jasco J-810 spectropolarimeter equipped with a thermostated cell holder and interfaced with a Peltier temperature controller (Jasco Products Co.). Scans were performed at 4°C with 2 μM final protein concentration. The molar mean residue ellipticity [θ] in deg cm2 dmol−1 was obtained from the measured raw data after subtracting the buffer spectrum, taking into account the measured ellipticity in millidegrees (θ), the molar protein concentration in mol/l (c), the optical pathlength in cm (l), the number of amino acid residues (N) (1364 residues), and molecular mass (Mw) (150 kDa) according to the relation:[θ]/θ/(10.c.l). A mean residue molecular mass of 109.97 Da was used for both samples. Typically, CD spectra were measured in the wavelength range of 200–260 nm, and rectangular quartz cuvette with a path length of 0.1 cm was used. Low-noise circular dichroism spectra were measured by averaging five scans. Data were collected at 0.5-nm intervals using a bandwidth of 2 nm, response time of 1 s and averaging time of 30 s for each measurement. Secondary structure contents were calculated by the K2d method available on the DichroWeb Server [9].
Generation of the F(ab′)2 fragment of the chIgG
The F(ab′)2 fragment (Fig. 1) was generated by cleaving the purified chIgG variant with IdeS, a cysteine proteinase secreted by the Streptococcus pyogenes which specifically cleaves the heavy chain of IgG (FabRICATOR, Genovis AB) [10, 11].
Western blot assays of antigen specificity of intact chIgG and its F(ab′)2 fragment
To detect the binding of the intact chIgG to the pro-α2 chain target electroblotted onto a nitrocellulose membrane, the anti-human γ-chain antibodies conjugated with HRP were employed. In similar assays, the binding of the F(ab′)2 fragment was detected with the HRP-conjugated anti-human κ chain antibodies (Sigma-Aldrich). Following Western blots, procollagen and procollagen-derived bands were visualized with the Ponceau S by the direct staining of nitrocellulose membranes. Such staining was helpful in precisely determining the patterns of electrophoretic migration of analyzed protein bands.
Biosensor-based binding assays
The ability of the prototypic mIgA anti-α2Ct antibody to specifically bind the-α2Ct in both procollagen I and collagen I has been previously demonstrated [7]. Here, binding interactions between intact procollagen I and the mIgA, the chIgG or the F(ab′)2 variants were studied with the use of the SensiQ Pioneer biosensor (SensiQ Technologies Inc.). In addition, binding interactions of control human IgG and control mouse IgA were also studied. In brief, procollagen I was immobilized on a sensor, as described [12, 13]. Following procollagen immobilization, free antibody variants solubilized in HEPES-buffered saline (HBS, 10 mM HEPES, 150 mM NaCl, and 0.005% Tween 20, pH 7.4) were injected in separate cycles at concentrations ranging from 12.5 to 200 nM. After collecting data representing the association phase, antibody-free HBS was injected to the sensor, and the dissociation phase was recorded. After each assay, the surface of the sensor was regenerated by washing with 10 mM HCl, followed by equilibration with HBS. Data from the biosensor were analyzed by the global fitting method with the use of the QDat software (SensiQ Technologies Inc.) [14]. For each assay, the association rate constants (kon) and the dissociation rate constants (koff) were obtained, and the equilibrium dissociation constants (KD) values were calculated from the koff/kon ratios.
Inhibition of collagen fibril formation in vitro
The potential of the chIgG variant of the anti-α2Ct antibody to block formation of collagen fibrils in vitro was examined, as described [7]. In brief, the mIgA or chIgG variant of the anti-α2Ct antibody was added to the collagen I samples at 1:1 collagen: antibody molar ratio. Subsequently, collagen fibril formation was triggered by incubating the samples at 37°C. Next, at predefined time points, the samples were centrifuged to separate collagen fibrils (pellet fractions) from collagen I monomers (supernate fractions). Later, the pellet and the supernate fractions were analysed by gel electrophoresis. Samples without antibodies and those containing hIgG isolated from normal human serum were employed as controls. Note that collagen fibrils formed using this method have chracteristics of native fibrils preserved and retain epitopes recognized by studied anti-α2Ct antibodies [7]. Still, proper assembly of fibrils formed in control groups was monitored by transmission electron microscopy, as described [7]. In brief, 10-μl samples containing fibrils formed for 24 h were transferred onto 200-mesh Formvar-supported and carbon-coated copper grids (Ted Pella, Inc.). Collagen fibrils deposited on the grids were stained negatively with 1% phosphotungstate (pH 7.0) and visualized by a transmission electron microscope operating at 75 kV (model H-7000, Hitachi Ltd.).
Inhibition of formation of collagen deposits in layers of cells cultured in the presence of the anti-α2Ct chIgG
Note: patient-derived primary cells were collected from discarded tissues with the permission of the Institutional Review Boards of the participating institutions.
Assays of inhibition of collagen fibril deposition were done in cultures of dermal fibroblasts, keloid-derived fibroblasts, tenocytes, cells from the shoulder capsule derived from a patient with adhesive capsulitis, cells isolated from the tissue associated with the palmar fibromatosis, and in cultures of gingival fibroblasts isolated from patients diagnosed with gingival fibromatosis. In brief, for the Western blot assays, 5 × 104 cells were seeded into a 24-well plate, while for the immunostaining assays, 1.5 × 104 cells were seeded into wells of an 8-well slide. The chIgG was added to the medium at a concentration of 70 μg/ml and the medium was changed every 2 days. Control groups consisting of cells treated with hIgG and non-treated cells were also prepared.
For the Western blot assays, cell layers were lysed and then proteins were analyzed; collagen I was detected with the use of polyclonal anti-collagen I antibodies (AFCol-1) prepared for us by Abgent Inc. while glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was detected with the anti-GAPDH antibody (Santa Cruz Biotechnology Inc.).
For immunostaining assays, the cell layers were fixed with methanol and then collagen fibrils were stained with the primary AFCol-1 rabbit anti-collagen I antibodies and the secondary anti-rabbit-IgG antibodies conjugated to the Alexa Fluor 594 (Invitrogen). Nuclei were visualized with 4′,6-diamidino-2-phenylindole (DAPI) staining. Immunostained cell layers were observed with a fluorescence microscope (Eclipse E600, Nikon Inc.).
Cytotoxicity assays
Cytotoxicity assays for the chIgG were carried out with human dermal fibroblasts and with cells isolated from the human tendon sheath. These assays were done with the use of the WST-8 compound whose cell-mediated catabolism is observed colorimetrically (Sigma-Aldrich). First, the ability of the selected assay to detect changes in the viability of cells in response to a cytotoxic agent was experimentally tested. In brief, fibroblasts seeded in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) at a density of 10,000 cells/microplate-well were treated with increasing concentrations of tissue necrosis factor α (TNFα) for 24 h in the presence of 1.0 μg/ml of actinomycin-D (Sigma-Aldrich). After that time, the changes in response to a varying amount of TNFα were recorded. Next, the potential of the anti-α2Ct antibody to induce the acute cytotoxicity was analyzed by measuring the viability of cells treated for 24 h with increasing concentrations, up to 200 μg/ml, of the tested antibody variants. Control groups treated with hIgG and PBS only were also employed. In these assays, data were presented by employing a linear fit (GraphPad Prism software, GraphPad Software Inc.)
The possible toxicity of the chIgG was also tested by analyzing its impact on cell proliferation. In brief, analyzed cells were cultured for 6 days in DMEM supplemented with 10% FBS in the presence of the chIgG added to the media at a concentration of 100 μg/ml. At the specific time intervals, the relative amounts of the metabolic product of WST-8 catabolism were assayed colorimetrically. Employing control groups consisting of cells cultured in the presence of 2 ng/ml of pro-proliferative platelet-derived growth factor (PDGF; Sigma-Aldrich), and those cultured in the presence of 1.0 μg/ml of anti-proliferative actinomycin-D, validated the described test. Additional control groups included cells treated with hIgG and PBS only. In these assays, data were presented by employing an exponential fit (GraphPad Prism software, GraphPad Software Inc.)
Cross-reactivity assays
To analyze the possible cross-reactivity of the anti-α2Ct antibody, we employed a Food and Drug Administration-recommended method in which the tissue cross-reactivity of a tested agent is analyzed by immunohistology done on arrays of various tissues. The cross-reactivity assays in human tissues (USBiomax, Inc.) were carried out with the use of the original mIgA variant of the anti-α2Ct antibody, whose antigen specificity is identical to that of its chIgG counterpart [7]. For these assays, biotinylated rat anti-mouse-IgA antibodies were used as the secondary antibodies (eBioscience). The cross-reactivity of the chIgG variant was analyzed with the use of the mouse-derived tissue arrays (US Biomax, Inc.). For these assays, biotinylated goat anti-human-IgG antibodies were employed as the secondary antibodies (Vector Laboratories, Inc.). Following immunostaining, all samples were counterstained with Methyl Green (Vector Laboratories, Inc.). Moreover, the general morphology of the analyzed tissues was observed after staining corresponding samples with hematoxylin and eosin.
RESULTS
PCR products encompassing DNA sequences for the mVH and the mVL of the prototypic mIgA-type anti-α2Ct antibody
Based on the PCR results, a set of primers including MuIgVH5′-C and α1_1 primers (Table I) was selected to generate DNA fragment encoding the mVH (Fig. 1). The forward MuIgκVL5′-G and the reverse MuIgκVL3′-1 primers were selected to generate a DNA fragment encompassing the sequence for the mVL of the mouse light κ chain (Fig. 1 and Table I).
Composition and binding-specificity of the intact chIgG
The specific chain composition of purified chIgG was confirmed by Western blot (Fig. 2A and 2B). A distinct protein band seen after electrophoresis of the purified chIgG in non-reducing conditions indicates that this variant produced by CHO cells forms a disulfide bond-stabilized mVH-hγ/mVH-hκ complex (Fig. 2B). As depicted in Fig. 2C, the anti-α2Ct chIgG antibody specifically recognized only the pro-α2 chain and a product of its partial processing, both including the α2Ct epitope. In contrast, no pro-α1 chain was detected, thereby indicating the high specificity of this anti-α2Ct variant.
Figure 2.

Assays of the chIgG variant of the anti-α2Ct antibody and its F(ab′)2 fragment. A; Purified chIgG variant produced by CHO cells was analyzed in reducing conditions for the presence of the γ (upper panel) and κ chains (lower panel). Specific antibodies against particular chains confirm the production of the γ and κ chains by CHO cells, B; Purified chIgG variant was analyzed in non-reducing conditions for the ability of the γ and κ chains to assemble into disulfide bonded molecules. The presence of the high molecular band, detected with both the anti-γ and the anti-κ antibodies, indicates correct folding of the chIgG molecules. The hIgG variant which contains the κ chain was used as a positive marker. C; Western blot assays of the specificity of the chIgG variant for binding the α2Ct antigen. The two bands visible in the blot represent an intact and partially processed pro-α2 chain both specifically recognized by the chIgG. D; Non-reducing electrophoretic separation of the intact chIgG and its F(ab′)2 and Fc fragments generated by the enzymatic cleavage with IdeS. E; Western blot assays (left panel) of the binding of the F(ab′)2 fragment to the pro-α2 chain or its partially degraded form (a few different concentrations of procollagen I were applied onto separate lines). Corresponding staining of a nitrocellulose membrane with Ponceau S (right panel) identifies bands positively stained with the use of the F(ab′)2 fragment of the chIgG variant. Symbols: hIgG(k) and chIgG; control human IgG with the κ light chain and the chIgG variant, respectively, ch-γ and ch-κ; chimeric γ and κ chains of the chIgG, respectively, Anti-γ and Anti-κ; specific antibodies employed to detect the ch-γ and ch-κ chains of the chIgG, anti-α2Ct chIgG; the chIgG variant tested for its ability to detect the α2Ct, Fab; Fragment antigen-binding, Fc; Fragment crystallizable region, anti-a2Ct F(ab)2; the F(ab′)2 fragment of the chIgG variant tested for its ability to detect the α2Ct Coomassie blue, Ponceau S; dyes employed to stain indicated samples, −/+ indicates the samples that were non-treated or treated with IdeS.
F(ab′)2 fragments
Treatment of the recombinant chIgG with IdeS that specifically cleaves IgG at a site present within its hinge region has generated fragments of the predicted molecular masses (Fig. 2D). Specifically, in the non-reducing electrophoretic conditions the IdeS-generated products were apparent as a 100 kDa band corresponding to the F(ab′)2 fragment and a band representing the Fc fragment migrating in the range of 27 kDa (Fig. 2D). As demonstrated in Fig. 2E, the F(ab′)2 fragment specifically recognizes the pro-α2 chain and a product of its partial processing, both including the α2Ct epitope. Similar to the intact chIgG, no pro-α1 chain was detected, thereby indicating an epitope specificity of the F(ab′)2 fragment. Individual lanes seen in Fig. 4E represent samples loaded at increasing concentrations.
Figure 4.

Kinetics of the binding of the anti-α2Ct antibody variants to procollagen I. In each panel, the curves represent association and dissociation events during analyzed binding interactions between procollagen I and antibody variants present at concentrations ranging from 12.5 nM to 200 nM. For each assay, the association rate constants (kon) and the dissociation rate constants (koff) were obtained, and the equilibrium dissociation constants (KD) values were calculated from a ratio of koff/kon.
Structural characteristics of the chIgG
Essential structural characteristics of the chIgG were analyzed with the use of CD spectroscopy. Analysis of data from the CD assays (Fig. 3A) demonstrated that α-helices, β-sheets, and random-coil conformations contribute 3%, 50%, and 47%, respectively, to the structure of control hIgG. The corresponding values for the inhibitory chIgG variant were 7%, 45%, and 48%, thereby further indicating the native-like structure of the therapeutic chIgG antibody. In addition, the SEC assays (Fig. 3B) have determined that 100% of the chIgG variant was produced by CHO cells in a monomeric form of the expected molecular mass of 150 kDa.
Figure 3.
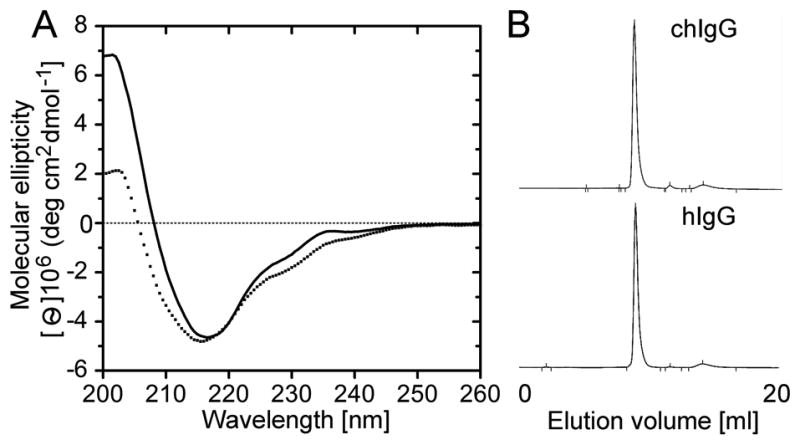
Structural assays of the recombinant chIgG variant. A; CD assays done on the chIgG (dotted line) and hIgG control (solid line). B; Chromatography profiles illustrating elution characteristics of the analyzed IgG variants.
Biosensor binding assays
As indicated in Fig. 4, the mIgA-procollagen I binding is characterized by the equilibrium dissociation constants (KD) of 0.2 nM, while the corresponding value for the chIgG-procollagen I and F(ab′)2-procollagen I binding is 3.2 nM and 2.8 nM, respectively. The lack of binding of the control mouse IgA (not shown) and control hIgG to procollagen I further corroborate binding specificities of the analyzed anti-α2Ct variants (Fig. 4).
Inhibition of fibril formation in vitro
As evident by the lack of collagen in the fibril/pellet fractions, the chIgG variant applied at a 1:1 molar ratio with procollagen I has completely blocked the fibril formation process (Fig. 5A). In applied experimental conditions, the inhibitory activity of the chIgG was similar to that seen for the prototypic anti-α2Ct mIgA variant (Fig. 5B). In contrast, the fibril formation process was not affected in control groups prepared in the absence of the inhibitory antibodies (Fig. 5C) or in the presence of non-inhibitory hIgG (Fig. 5D). In these groups, the presence of fibrils in the pellet fractions was readily detectable, and as indicated in Fig. 5E, their overall morphology and apparent periodicity resembled that of a typical collagen fibril.
Figure 5.

Effects of the chIgG on collagen fibril formation in vitro. Collagen fibrils were formed in the presence of tested antibodies added to collagen I in a 1:1 molar ratio. At the indicated points, collagen fibrils were separated from free collagen monomers by centrifugation. Subsequently pellet (fibrils) and supernatant (monomers) fractions were analyzed by gel electrophoresis. A and B: Experimental groups in which collagen fibrils were formed in the presence of the chIgG or the mIgA variants of the anti-α2-Ct antibody,, respectively. C and D; Control groups in which collagen fibrils were formed in the absence of antibodies or in the presence of non-inhibitory hIgG, respectively. E; EM images of representative fibrils formed in a control group. Symbols: P and S; pellet and supernatant fractions, respectively, α, h-γ, and ch-γ; heavy α chain of the mIgA variant, heavy γ chain of the hIgG variant, and heavy γ chain of the chIgG, Coll-I; α1 and α2 chains of collagen I.
Inhibition of formation of collagen deposits in cell layers in the presence of the chIgG variant
As indicated in Figure 6, in the presence of control hIgG, all analyzed cells deposited collagen-rich matrices around them. The gross morphologies of deposited collagen fibrils and their densities varied among analyzed cells. For instance, the apparent density of readily visible fibrils was relatively high in matrices formed by the dermal fibroblasts and tenocytes. In contrast, the apparent overall density of thin fibrils formed by cells isolated from fibrotic gingiva was relatively low. In all analyzed cell types, however, a reduction in the relative amounts of accumulated collagen-rich deposits in the presence of the chIgG was readily visible (Fig. 6). More quantitative Western blot assays done to detect collagen I present in cell layers further demonstrate the reduction of accumulated collagen I-rich deposits in cells cultured in the presence of the chIgG variant (Fig. 7). Specifically, densitometric assays of the fully processed α1 chain extracted from the cell layers demonstrated that in comparison to the control treated with hIgG, the reduction of deposited collagen ranged from about 60% in keloid-derived fibroblasts, tenocytes, and cells from the joint capsule to about 90% in dermal fibroblasts, cells isolated from palmar fibromatosis tissue, and gingival fibroblasts. Note, that multiple bands seen above the α1 chain of collagen I most likely represent the intermediate products of the processing of pro-α1 chain, and some of them may also represent the crosslinked forms of the α1 chain.
Figure 6.
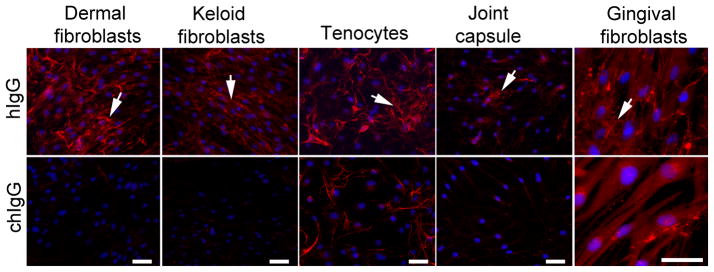
Microscopic assays of collagen fibrils deposited in layers of cells cultured in the presence of control hIgG (upper row) or the inhibitory chIgG variant of the anti-α2Ct antibody (bottom row). Because collagen fibrils produced by gingival fibroblasts isolated from a patient with the idiopathic form of gingival fibromatosis appear as thin filaments, larger images are presented to visualize them. Arrows indicate extracellular collagen deposits. Bars = 50 μm.
Figure 7.
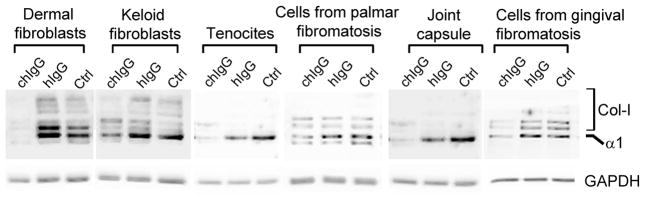
Western blot assays of collagen I extracted from cell layers similar to those seen in Fig. 6. Note that in the presence of the inhibitory chIgG, the amount of deposited collagen I is reduced in all analyzed cell lines. Symbols: chIgG, inhibitory chIgG variant; hIgG, control non-inhibitory hIgG; Ctrl, samples not treated with any of the IgG variants, Col-I partially processed pro-α1 chains and cross-linked forms of α1 chains, GAPDH; glyceraldehyde 3-phosphate dehydrogenase; indicates uniform gel loading and provides a reference for the α1 collagen chain.
Effects of the chIgG on the viability of cells
As seen in Fig. 8A, increasing the concentration of the chIgG variant to 200 μg/ml, a 2.5-fold concentration of that employed for the cell-based inhibitory assays (Figs. 6), did not have any significant acute toxic effects on the viability of tested cells. The ability of the employed test to detect changes in the viability of cells treated with increasing concentrations of TNFα, validates its utility (Fig. 8B). The effects of the chIgG on the viability of selected cells were also analyzed by monitoring their proliferation. As shown in Fig. 8C and 8D, the presence of tested antibodies added to the cell culture media at a concentration of 100 μg/ml did not have any effect on proliferation. The proliferation characteristics, illustrated by the fitted curves, of cells cultured in the presence of chIgG or hIgG were similar to these observed for cells cultured in the absence of IgG variants. An accelerated proliferation of cells in the presence of PDGF and the lack of proliferation in the presence of actinomycin D validate the utility of the applied assay (Fig. 8C and 8D). Although the above proliferative behavior seen in response to various factors was similar in tested dermal fibroblasts (Fig. 8C) and cells isolated from the tendon sheaths (Fig. 8D), the specific dynamics of proliferation differed in these cell types. These differences are most likely attributed to broad biological differences expected in primary cells isolated from distant tissues.
Figure 8.
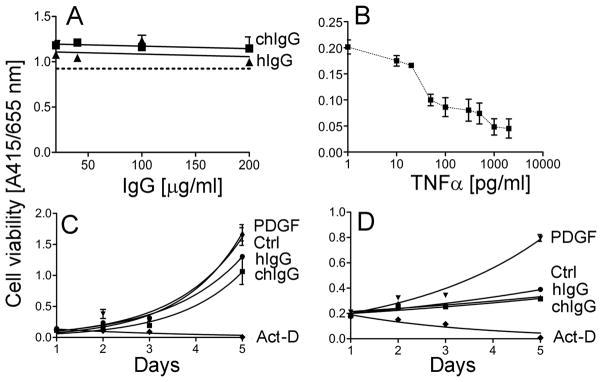
Analysis of the effects of the chIgG variant on the viability of cells. A; Assays of the viability of dermal fibroblasts as the function of a 24-h treatment with increasing concentrations of the chIgG variant or control hIgG. The dotted line represents data for cells cultured in the absence of antibodies. B; The ability of WST-8-based assays to detect changes in the viability of fibroblastic cells was monitored in dermal fibroblasts cultured in the presence of various concentrations of TNFα. C and D; Proliferation assays of dermal fibroblasts (C) and cells isolated from the tendon sheaths (D) cultured in the presence of 100 μg/ml of tested antibodies. In addition, proliferation of cells cultured in the presence of PDGF or actinomycin D (Act-D) was also monitored. Moreover, a control (Ctrl) group of cells cultured in the absence of tested agents was also analyzed.
Cross-reactivity assays
As indicated in Fig. 9A, the mIgA variant specifically recognized the collagen I-rich dermis of the human skin; in the same conditions, no staining of epidermis, a skin layer that lacks collagen I, was observed. In a similar set of experiments, the chIgG variant was tested with the mouse-derived tissues. A positive staining of the collagen I-rich dermis and a negative result of staining of epidermis was observed (Fig. 9B). Note that in contrast to the human skin, the mouse epidermis consists only of a thin layer of keratinocytes, thereby making them less clearly visible (Fig. 9B). Moreover, in both skin samples, some nonspecific weak staining of keratinized epidermis is observed. The lack of staining of tissues in which collagen I is poorly represented was also determined. For instance, the expected positive staining of the interlobular regions of a mouse thymus was observed under a high magnification, while collagen I was not detected in the lobular regions of this tissue (Fig. 9B). Together, the cross-reactivity assays demonstrate the high specificity of the mIgA and the chIgG variants for the binding of the collagen I target.
Figure 9.

Cross-reactivity assays of the mIgA and chIgG variants of the anti-α2Ct antibody. The human skin sample (A) was immunostained with the mIgA variant, while the mouse skin and thymus were immunostained with the chIgG form of the anti-α2Ct antibody (B). Corresponding panels were stained with H&E. As depicted in panel A, staining of the human skin (h-Skin) has revealed that collagen I -specific staining was limited to the collagen-rich dermis while the epidermis was collagen I-negative. Panel B demonstrates collagen I-positive staining of the dermal layer of the mouse skin (m-Skin), whereas the epidermis is collagen I-negative. In the mouse thymus (m-Thymus), collagen I-positive staining is apparent only in the interlobular regions (arrows). Bars = 50 μm.
DISCUSSION
Excessive scar formation and fibrosis is a common clinical problem. A hallmark of such a tissue is the presence of collagen-rich deposits formed in the process that involves inflammation and activation of cells. To date, therapeutic approaches to limit fibrosis have included anti-inflammatory and anti-proliferative agents such as steroids, mitomycin C, 5-fluorouracil, among many others [15–17]. All of these approaches have been met with little clinical success [18]. Recently, our group has proposed that the extracellular process of collagen fibril formation may serve as a valid target to reduce the mass of collagen-rich fibrotic tissues [7, 19]. We postulated that targeting the specific process of fibril formation may offer not only an effective but also a safe way to reduce the growth of fibrotic tissue at the site of accidental or postoperative trauma. Since fibril formation is driven by the telopeptide-mediated collagen-collagen interaction, we generated the anti-α2Ct antibody that specifically binds to the telopeptide of the α2 chain of human collagen I, thereby blocking its critical binding interactions [7, 19–21]. Consequently, the anti-α2Ct antibody was tested as the inhibitor of localized fibrosis in a keloid-like model [7]. As described by Chung et al., these initial tests indicated the great potential of the anti-α2Ct antibody to reduce the mass of the formed fibrotic tissue [7].
Because the prototypic anti-α2Ct antibody is of mouse origin and it belongs to the IgA class, its utility to serve as a therapeutic agent in human patients is limited [22, 23]. To circumvent this limitation, we rationally engineered the chimeric anti-α2Ct antibody variant consisting of the murine variable domains of the α and κ chains and the constant regions of the human γ and κ chains. Our proposed strategy is supported by the fact that to date a number of chimeric therapeutic antibodies exist in the market. Examples of such antibodies include Basiliximab, Cetuximab, and Infliximab [24]. Biophysical and biochemical assays of the chIgG variant indicate its native-like structure characterized by the specific assembly of the light and heavy chains and the content of the α-chains and β-sheets similar to that of the wild type hIgG. Moreover, lack of non-specific aggregation of the chIgG variant, demonstrated by a single chromatographic peak corresponding to the predicted molecular mass of a monomer, further indicates the proper structure of the chIgG variant. As formation of soluble and insoluble antibody aggregates is indicative of abnormal folding of engineered antibodies, a feature that may reduce their bioactivity and increase the risk for immunogenicity, our data indicate the promising potential of the chIgG variant for preclinical tests in relevant biological models [25–27]. Furthermore, preservation of the therapeutic utility of the engineered chIgG form of the anti-α2Ct antibody is indicated by the results of Western blot assays which indicate that the chIgG variant retains its antigen specificity. Of interest is the observation that the antigen specificity has been preserved in the F(ab′)2 fragment of the chIgG. This observation is particularly critical in the context of the intended use of the chIgG. Specifically, the proposed action of the anti-α2Ct antibody is based on its antigen-binding function rather than on the effector function. In fact, it is conceivable that the potential Fc-mediated effector function of the anti-α2Ct antibody could indeed be a limiting factor for its envisioned anti-fibrotic applications. Thus, the ability of the F(ab′)2 fragment to bind the α2Ct target furthers the prospect of the anti-α2Ct antibody for being used as a safe inhibitor of the formation of fibrotic deposits.
One of the challenges in engineering of therapeutic antibodies is to maintain their high affinity for binding to the designated target. Thus, preserving the specific, high-affinity binding of an antibody is critical not only for achieving the therapeutic objective but also for reaching this objective in a safe way with the smallest possible concentration of administered antibody [5]. Analyses of the binding kinetics of the chIgG and its F(ab′)2 fragment demonstrated a nM-range of the KD values, thereby indicating a strong binding affinity. Of note is the fact that the specific KD values reported in the presented study were obtained with the use of intact procollagen I molecules serving as binding partners for the tested antibody variants. In our previously reported work, we suggested that a bulky C propeptide of procollagen I may partially restrict the access of the anti-telopeptide antibodies to their specific epitopes, thereby influencing the binding kinetics [7]. Consequently, we expect that the affinities of the tested anti-α2Ct variants for the binding of the α2Ct present in the fully processed collagen I may be greater than those reported for the binding interactions with intact procollagen I. Still, KD values for the chIgG and its F(ab′)2 fragment were higher than that calculated for the original mIgA variant. Such changes in binding affinities are frequently seen in engineered antibody variants and they are explained by the differences in the primary structures of antibody variants and by changes in the posttranslational modifications such as glycosylation [5].
Similar to its original mIgA counterpart, the chIgG version of the anti-α2Ct was also active in inhibiting collagen self-assembly in vitro. In addition, the efficacy of the chIgG variant to limit the formation of collagen-rich deposits was demonstrated in cell culture conditions. Although the employed system does not fully represent the native tissues, it provides an opportunity to test the blocking potential of the analyzed antibody in a complex environment created by cultured cells. The inhibitory characteristics of the chIgG variant seen in cultures of cells isolated from tissues such as skin, tendon, tendon sheath, and gingiva point toward the therapeutic utility of this antibody in a broad range of tissues prone to developing localized fibrotic deposits in response to trauma. The above in vitro and cell-based assays do not clearly answer, however, to what extent the proposed antibodies are able to reach their intended target in vivo. In comparison to the in vitro and cell-based experimental systems, the access of a 150,000-Da antibody to the sites of collagen fibril formation may be hampered by a compact structure of a targeted connective tissue. In addition, in some tissues, the access to specific target sites may be limited due to the presence of cell membrane indentations reported to serve as sites for the initial stages of procollagen processing and collagen fibril formation in a developing tendon of a chick embryo [28]. Indirect evidence for the ability of antibodies to penetrate the structure of connective tissues is provided by some pathological situations associated with the presence of auto-antibodies. For instance, in epidermolysis bullosa acquisita, a blistering disease, the formation of the anchoring fibrils is altered by the auto-antibodies that interfere with collagen VII-collagen VII interaction. Similarly, the alteration of the formation of the collagen IV network via auto-antibodies that bind to a specific region of this collagen type is a fundamental cause of Goodpasture’s syndrome.
Although the above considerations about the accessibility of target sites have to be addressed in relevant animal models, we postulate that the potential problems with the access of inhibitory antibodies to the target sites could be circumvented by, for example, optimizing the antibody-delivery methods. Moreover, engineering relatively small antibody variants, e.g. a 25,000-Da scFv construct, could improve their diffusion characteristics, thereby enhancing their potential therapeutic utility.
One of the critical considerations for the use of a therapeutic antibody is its potential cytotoxicity and cross-reactivity. As demonstrated by viability assays of cells treated with varying concentrations of the chIgG, the cytotoxic effects of the tested antibody have not been observed in the applied range of antibody concentrations. Although monoclonal antibodies are developed to recognize a specific epitope, they may cross-react with different epitopes and cause toxicity [23]. To test for such a possibility, it is recommended to check the immunoreactivity of a candidate antibody in a panel of various tissues [23]. In the presented study, these tests were done with the original mIgA and the chIgG variants with the use of human and mouse-derived tissues, respectively. Results of immunostaining assays indicate the great specificity of the tested antibody variants. Although we demonstrated the binding of inhibitory antibodies to the collagen-rich tissues, their ability to bind the established collagen fibrils in vivo is not currently determined. We expect, however, that binding of these antibodies to established fibrils in vivo is limited. Specifically, we predict that, unlike in immunohistological assays done on slices of tissues in which antibody-binding sites are exposed due to the histological processing of samples, in the native fibrils the access to the α2-Ct telopeptide may be restricted [29, 30]. Still, considering the recent report by Antipova and Orgel it is evident that anti-biglycan antibodies are able to penetrate collagen fibers and partially dissociate them by disrupting biglycan-mediated inter-fibrillar interactions [31]. Considering these recent observations, further studies are needed to clearly establish whether the anti-α2Ct antibody is able to gain the access to its telopeptide target present in the gap region of mature fibrils. Such information will be helpful to determine whether the proposed antibody is only able to block the formation of newly created fibrils or whether it is also capable of decomposing already-formed fibrotic deposits.
The described study presents a critical step in a quest to implement a novel approach to inhibit localized fibrotic responses via blocking the formation of the collagen-rich deposits, namely by developing a therapeutically-relevant form of the anti-α2Ct antibody [7]. Overall, the presented study demonstrates the great potential of the bioengineered chIgG variant of the anti-α2Ct antibody to be considered for preclinical tests in a relevant animal model. The most prominent characteristics that support this notion include a proper antibody structure, a high antigen specificity, a high antigen-binding affinity, and a low risk for toxicity. Planned future studies will provide critical preclinical data that will help to determine the prospects and limits of the proposed approach to limit localized fibrosis in injured connective tissues.
Acknowledgments
This work was supported by grants from NIH to A.F. (AR061118 and AR048544). The authors are grateful to Drs. Katarzyna Gawron, Katarzyna Lazarz-Bartyzel, Mieczyslaw Lazarz, and Maria Chomyszyn-Gajewska (Jagiellonian University, Krakow, Poland) for generous gift of gingival fibroblasts. Moreover, the authors thank Anthony Kasinskas and Annie Ashok for help with biochemical and cell-based assays.
Footnotes
DECLARATION OF INTEREST
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- 1.Beck A, Wurch T, Bailly C, Corvaia N. Strategies and challenges for the next generation of therapeutic antibodies. Nature reviews Immunology. 2010;10:345–352. doi: 10.1038/nri2747. [DOI] [PubMed] [Google Scholar]
- 2.Chadd HE, Chamow SM. Therapeutic antibody expression technology. Curr Opin Biotechnol. 2001;12:188–194. doi: 10.1016/s0958-1669(00)00198-1. [DOI] [PubMed] [Google Scholar]
- 3.Cheng JD, Rieger PT, von Mehren M, Adams GP, Weiner LM. Recent advances in immunotherapy and monoclonal antibody treatment of cancer. Semin Oncol Nurs. 2000;16:2–12. doi: 10.1053/sonu.2000.19775. [DOI] [PubMed] [Google Scholar]
- 4.Holliger P, Hudson PJ. Engineered antibody fragments and the rise of single domains. Nat Biotechnol. 2005;23:1126–1136. doi: 10.1038/nbt1142. [DOI] [PubMed] [Google Scholar]
- 5.Kim SJ, Park Y, Hong HJ. Antibody engineering for the development of therapeutic antibodies. Molecules and cells. 2005;20:17–29. [PubMed] [Google Scholar]
- 6.Nelson AL, Reichert JM. Development trends for therapeutic antibody fragments. Nat Biotechnol. 2009;27:331–337. doi: 10.1038/nbt0409-331. [DOI] [PubMed] [Google Scholar]
- 7.Chung HJ, Steplewski A, Chung KY, Uitto J, Fertala A. Collagen fibril formation. A new target to limit fibrosis. J Biol Chem. 2008;283:25879–25886. doi: 10.1074/jbc.M804272200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tucker PW, Slightom JL, Blattner FR. Mouse IgA heavy chain gene sequence: implications for evolution of immunoglobulin hinge axons. Proc Natl Acad Sci U S A. 1981;78:7684–7688. doi: 10.1073/pnas.78.12.7684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Whitmore L, Wallace BA. DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 2004;32:W668–673. doi: 10.1093/nar/gkh371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Akesson P, Moritz L, Truedsson M, Christensson B, von Pawel-Rammingen U. IdeS, a highly specific immunoglobulin G (IgG)-cleaving enzyme from Streptococcus pyogenes, is inhibited by specific IgG antibodies generated during infection. Infect Immun. 2006;74:497–503. doi: 10.1128/IAI.74.1.497-503.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vincents B, von Pawel-Rammingen U, Bjorck L, Abrahamson M. Enzymatic characterization of the streptococcal endopeptidase, IdeS, reveals that it is a cysteine protease with strict specificity for IgG cleavage due to exosite binding. Biochemistry. 2004;43:15540–15549. doi: 10.1021/bi048284d. [DOI] [PubMed] [Google Scholar]
- 12.Brittingham R, Uitto J, Fertala A. High-affinity binding of the NC1 domain of collagen VII to laminin 5 and collagen IV. Biochem Biophys Res Commun. 2006;343:692–699. doi: 10.1016/j.bbrc.2006.03.034. [DOI] [PubMed] [Google Scholar]
- 13.Sieron AL, Louneva N, Fertala A. Site-specific interaction of bone morphogenetic protein 2 with procollagen ii. Cytokine. 2002;18:214–221. doi: 10.1006/cyto.2002.1035. [DOI] [PubMed] [Google Scholar]
- 14.Myszka DG, Morton TA. CLAMP: a biosensor kinetic data analysis program. Trends Biochem Sci. 1998;23:149–150. doi: 10.1016/s0968-0004(98)01183-9. [DOI] [PubMed] [Google Scholar]
- 15.Wang XQ, Liu YK, Qing C, Lu SL. A review of the effectiveness of antimitotic drug injections for hypertrophic scars and keloids. Ann Plast Surg. 2009;63:688–692. doi: 10.1097/SAP.0b013e3181978753. [DOI] [PubMed] [Google Scholar]
- 16.Shin SJ, Lee SY. Efficacies of corticosteroid injection at different sites of the shoulder for the treatment of adhesive capsulitis. Journal of shoulder and elbow surgery/American Shoulder and Elbow Surgeons [et al] 2012 doi: 10.1016/j.jse.2012.06.015. [DOI] [PubMed] [Google Scholar]
- 17.Hsu JE, Anakwenze OA, Warrender WJ, Abboud JA. Current review of adhesive capsulitis. Journal of shoulder and elbow surgery/American Shoulder and Elbow Surgeons [et al] 2011;20:502–514. doi: 10.1016/j.jse.2010.08.023. [DOI] [PubMed] [Google Scholar]
- 18.Gaujoux-Viala C, Dougados M, Gossec L. Efficacy and safety of steroid injections for shoulder and elbow tendonitis: a meta-analysis of randomised controlled trials. Ann Rheum Dis. 2009;68:1843–1849. doi: 10.1136/ard.2008.099572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Steplewski A, Fertala A. Inhibition of collagen fibril formation. Fibrogenesis Tissue Repair. 2012;5(Suppl 1):S29. doi: 10.1186/1755-1536-5-S1-S29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prockop DJ, Fertala A. Inhibition of the self-assembly of collagen I into fibrils with synthetic peptides. Demonstration that assembly is driven by specific binding sites on the monomers. J Biol Chem. 1998;273:15598–15604. doi: 10.1074/jbc.273.25.15598. [DOI] [PubMed] [Google Scholar]
- 21.Prockop DJ, Fertala A. The collagen fibril: the almost crystalline structure. Journal of structural biology. 1998;122:111–118. doi: 10.1006/jsbi.1998.3976. [DOI] [PubMed] [Google Scholar]
- 22.Lobo ED, Hansen RJ, Balthasar JP. Antibody pharmacokinetics and pharmacodynamics. J Pharm Sci. 2004;93:2645–2668. doi: 10.1002/jps.20178. [DOI] [PubMed] [Google Scholar]
- 23.Dubel S. Handbook of Therapeutic Antibodies. 1. Weinheim: Wiley-VCH Verlag GmbH&Co. KGaA; 2007. [Google Scholar]
- 24.Wang W, Wang EQ, Balthasar JP. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clinical pharmacology and therapeutics. 2008;84:548–558. doi: 10.1038/clpt.2008.170. [DOI] [PubMed] [Google Scholar]
- 25.Jenkins N, Murphy L, Tyther R. Post-translational modifications of recombinant proteins: significance for biopharmaceuticals. Mol Biotechnol. 2008;39:113–118. doi: 10.1007/s12033-008-9049-4. [DOI] [PubMed] [Google Scholar]
- 26.Rosenberg AS. Effects of protein aggregates: an immunologic perspective. Aaps J. 2006;8:E501–507. doi: 10.1208/aapsj080359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brych SR, Gokarn YR, Hultgen H, Stevenson RJ, Rajan R, Matsumura M. Characterization of antibody aggregation: role of buried, unpaired cysteines in particle formation. J Pharm Sci. 2010;99:764–781. doi: 10.1002/jps.21868. [DOI] [PubMed] [Google Scholar]
- 28.Canty EG, Lu Y, Meadows RS, Shaw MK, Holmes DF, Kadler KE. Coalignment of plasma membrane channels and protrusions (fibripositors) specifies the parallelism of tendon. J Cell Biol. 2004;165:553–563. doi: 10.1083/jcb.200312071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holmes DF, Gilpin CJ, Baldock C, Ziese U, Koster AJ, Kadler KE. Corneal collagen fibril structure in three dimensions: Structural insights into fibril assembly, mechanical properties, and tissue organization. Proc Natl Acad Sci U S A. 2001;98:7307–7312. doi: 10.1073/pnas.111150598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Malone JP, Veis A. Heterotrimeric type I collagen C-telopeptide conformation as docked to its helix receptor. Biochemistry. 2004;43:15358–15366. doi: 10.1021/bi048304b. [DOI] [PubMed] [Google Scholar]
- 31.Antipova O, Orgel JP. Non-enzymatic decomposition of collagen fibers by a biglycan antibody and a plausible mechanism for rheumatoid arthritis. PLoS One. 2012;7:e32241. doi: 10.1371/journal.pone.0032241. [DOI] [PMC free article] [PubMed] [Google Scholar]