

Abstract
Cation exchange high performance liquid chromatography (CE HPLC) provides an excellent tool for accurate and reliable diagnosis of various hemoglobin (Hb) disorders. HbQ India is a rare alpha chain variant that usually presents in the heterozygous state. Its presence in double heterozygous state with HbD Punjab is extremely rare. The double heterozygosity for α and β chain variants leads to formation of abnormal heterodimer hybrids, which can lead to diagnostic dilemmas. We report two rare cases of double heterozygous HbQ India/HbD Punjab where the hybrid Hb was seen to elute at retention time similar to HbC on CE HPLC. The first case had unconjugated hyperbilirubinemia at presentation; while, the second case was asymptomatic.
Keywords: Double heterozygous, hemoglobin Q Punjab, hemoglobin Q India, high performance liquid chromatography
Introduction
Abnormalities of hemoglobin (Hb) synthesis include hemoglobinopathies, which arise from changes in the amino sequence of either globin chain and thalassemia, which are due to a decrease in globin chain production.[1] Common hemoglobinopathies in India include thalassemia along with HbS, HbE, and HbD and their combinations.[2] Hemoglobinopathies can occur in the heterozygous and homozygous states. A change in amino acid sequence in both α and β chains, termed double heterozygosity, in the same individual is very unusual. Here we describe two extremely rare cases of double heterozygous hemoglobinopathy, HbQ India/HbD Punjab (ααQ India ββD Punjab), the second report in the literature to date.
Case Reports
Case 1
A 26-year-old male of Indian Punjabi origin, resident of Delhi, presented with complaints of weakness, epigastric pain, and jaundice for 2 weeks. General physical examination revealed icterus. Systemic examination was unremarkable. Liver function tests revealed predominantly unconjugated hyperbilirubinemia (total bilirubin: 3.2 mg/dL, unconjugated: 2.5 mg/dL). Transaminases and serology for viral hepatitis were normal. Molecular studies for Gilbert's syndrome were negative. In view of the unconjugated hyperbilirubinemia, a complete hemolytic work up was performed. Hb high performance liquid chromatorphy (HPLC) of proband performed on Bio-Rad variant; β thalassemia short program, showed four major peaks (retention time in minutes) which were identified as HbA (α2 β2) (2.30), HbD Punjab (α2 ββD Punjab) (4.11), HbQ India (ααQ India β2) (4.72), HbD Punjab/HbQ India (ααQ India ββD Punjab) (4.96) [Figure 1]. The variant α and β chain hybrid was seen to elute in the HbC window. The patient had normal Hb values with raised mean corpuscular hemoglobin concentration (MCHC) (Hb: 16.1 g/dL, mean corpuscular volume: 85.5 fL, mean corpuscular hemoglobin: 32.8 pg, MCHC: 38.3 g/dL, red cell count: 4.91 × 1012/L). The peripheral blood smear showed normocytic normochromic picture and reticulocyte count was 4%. Work up for other hemolytic anemia including glucose-6-phosphate dehydrogenase deficiency and osmotic fragility was normal. Agarose gel electrophoresis at alkaline pH showed bands in the A, S, and C positions [Figure 2]. This was interpreted as HbA in the A position, HbD Punjab in the S position, and HbD Punjab/HbQ India in the C position.
Figure 1.
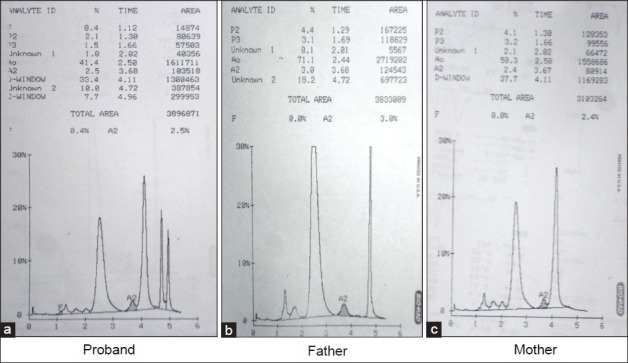
Cation exchange high performance liquid chromatography of case 1 (proband and parents). The patient's sample (a) demonstrates four prominent peaks: Hemoglobin (Hb) A, HbD Punjab, HbQ India, and the hybrid HbQ India/HbD Punjab eluting in HbC window (retention time 4.96 - 4.97 min). The parent's samples (b and c) demonstrate two prominent peaks of HbA and HbQ India and HbA and HbD Punjab, respectively
Figure 2.
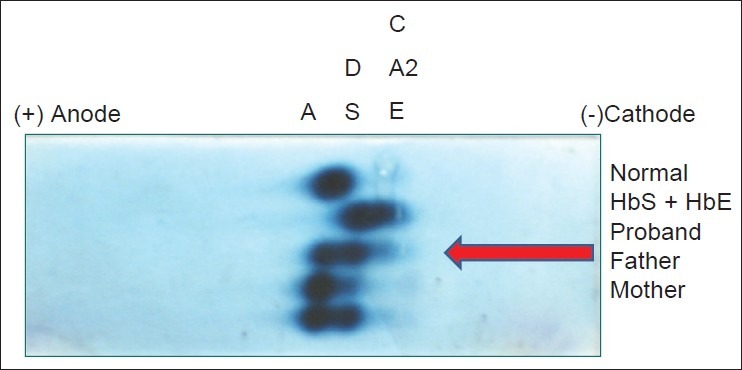
Agarose gel electrophoresis at alkaline pH showed bands in the A, S, and C positions. This was interpreted as HbA in the A position, HbD Punjab in the S position, and HbD Punjab/HbQ India in the C position
Case 2
A 29-year-old asymptomatic male of Indian Punjabi origin, resident of Meerut, was evaluated as part of family study, as his 1-year-old son had microcytic hypochromic anemia and associated HbQ India hemoglobinopathy. The Hb profiles and globin genotypes of the family members are summarized in Figure 3. The Hb values and red cell indices were similar to case 1 [Table 1]. Hb HPLC findings are depicted in Figure 4.
Figure 3.
Pedigree of family with HbQ India/HbD Punjab double heterozygosity. The arrow indicates the case. The father was heterozygote for HbD Punjab and mother was HbQ India heterozygote. His son has inherited only the heterozygous HbQ India and his wife is normal
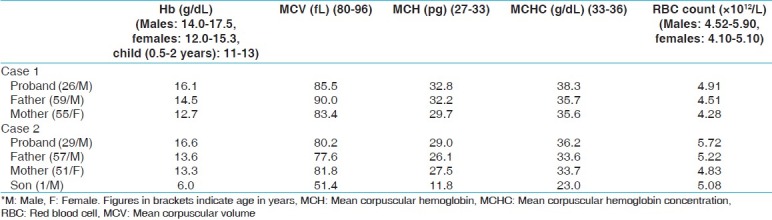
Table 1.
Hematological parameters of cases and families
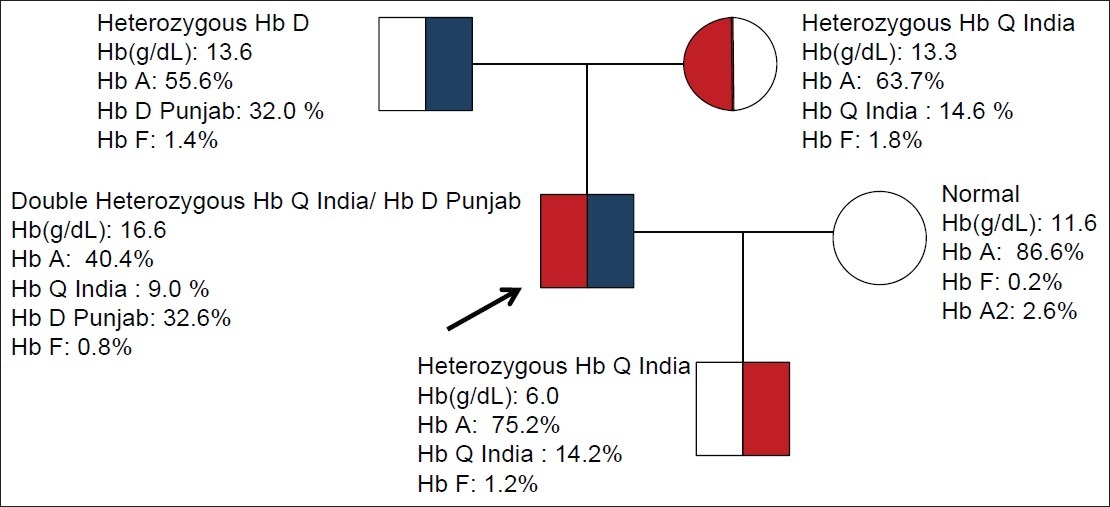
Figure 4.
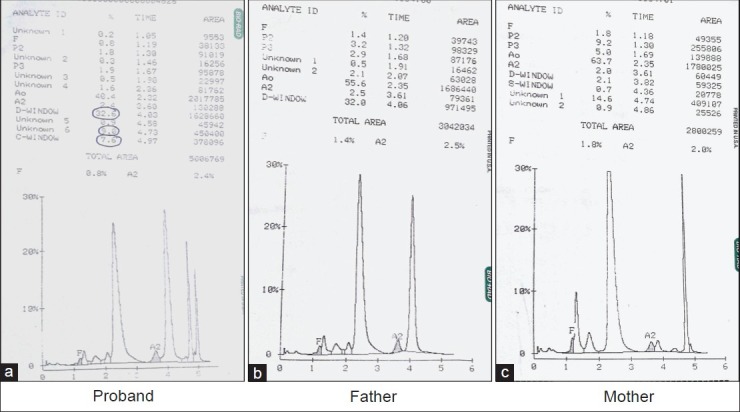
CE HPLC of case 2 (proband and parents). The patient's sample (a) demonstrates four prominent peaks: HbA, HbD Punjab, HbQ India, and the hybrid HbQ India/HbD Punjab eluting in Hb C window (retention time 4.96 - 4.97 min). The parents’ samples (b and c) demonstrate two prominent peaks of HbA and HbD Punjab and HbA and HbQ India, respectively
Discussion
HbQ India is a rare α chain structural variant caused by a mutation in the position of codon 64 of α-1 gene with a change of Asp→His. The prevalence of HbQ India in India is 0.4%, found predominantly in Sindhi families and in individuals from western and northern India. HbQ India levels in heterozygotes are normally below 20% and reduce further in interactions with β thalassemia.[3]
HbD Punjab (also known as HbD Los Angeles) is a β chain variant caused by a point mutation on codon 121 with change of Glu→Gln. The greatest prevalence of HbD Punjab is among Sikhs in Punjab, India where it is reported to be around 2%. Heterozygous HbD is a clinically silent condition, with levels of HbD of less than 40%.[4]
The Hb HPLC in both cases 1 and 2 in this report showed four peaks. HbD Punjab (retention time of 4.03-4.13 min) showed the characteristic rising baseline which suppresses the HbA2.[4] HbQ India showed a sharp, narrow peak (retention time of 4.75 min). The peak in HbC window (retention time 4.97 min) represented the hybrid of HbQ India/HbD Punjab (ααQ India ββD Punjab). The first case in this report presented with unconjugated hyperbilirubinemia; however, the second case was clinically asymptomatic. Both the cases had normal Hb values and mild raised MCHC. The clinical impact of this entity is not known. In the absence of molecular studies, whether hemolysis in the first case could be attributed to the variant Hb still remains a dilemma.
The hybrids of abnormal α and β chains have unpredictable elution times on HPLC and uncharacterized bands on Hb electrophoresis. It is important to identify and characterize these abnormal peaks on Hb HPLC. The hybrid HbQ India/HbD Punjab (ααQ India ββD Punjab) eluted in the HbC window and its mobility was similar to HbC on Hb electrophoresis on alkaline pH. The identification of this hybrid was based on parental study in both cases, as both sets of parents had heterozygous HbQ India and HbD Punjab, respectively. Only a single case report of double heterozygosity for HbQ India and HbD Punjab has been reported previously.[5] The said case, also of Indian origin did not have any hematological abnormality and the hybrid variant was also seen to elute in the HbC window on Hb HPLC.
The double heterozygosity for α and β chain variants leads to formation of heterodimer hybrids. These may cause unidentifiable peaks on Hb HPLC. This is the second report of coinheritance of HbQ India and HbD Punjab. The HbQ India/HbD Punjab hybrid elutes in the HbC window on HPLC and moves with C band on alkaline electrophoresis.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
References
- 1.Kutlar F. Diagnostic approach to hemoglobinopathies. Hemoglobin. 2007;31:243–50. doi: 10.1080/03630260701297071. [DOI] [PubMed] [Google Scholar]
- 2.Agarwal MB. The burden of haemoglobinopathies in India-0time to wake up? J Assoc Physicians India. 2005;53:1017–8. [PubMed] [Google Scholar]
- 3.Phanasgaonkar S, Colah R, Ghosh K, Mohanty D, Gupte S. Hb Q (India) and its interaction with beta-thalassaemia: A study of 64 cases from India. Br J Biomed Sci. 2007;64:160–3. doi: 10.1080/09674845.2007.11732780. [DOI] [PubMed] [Google Scholar]
- 4.Pandey S, Mishra RM, Pandey S, Shah V, Saxena R. Molecular characterization of hemoglobin D Punjab traits and clinical-hematological profile of the patients. Sao Paulo Med J. 2012;130:248–51. doi: 10.1590/S1516-31802012000400008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Higgins T, Schnabl K, Savoy M, Rowe P, Flamini M, Bananda S. A novel double heterozygous, HbD Punjab/HbQ India, hemoglobinopathy. Clin Biochem. 2012;45:264–6. doi: 10.1016/j.clinbiochem.2011.11.012. [DOI] [PubMed] [Google Scholar]