

Abstract
Objectives/Hypothesis
To clone and characterize the integration site of an insertional inner ear mutation, produced in one of fourteen transgenic mouse lines. The insertion of the transgene led to a mutation in a gene(s) necessary for normal development of the vestibular labyrinth.
Study Design
Molecular genetic analysis of a transgene integration site.
Methods
Molecular cloning, Southern and northern blotting, DNA sequencing and genetic database searching were the methods employed.
Results
The integration of the transgene resulted in a dominantly inherited waltzing phenotype and in degeneration of the pars superior. During development, inner ear fluid homeostasis was disrupted. The integration consisted of the insertion of a single copy of the transgene. Flanking DNA was cloned, and mapping indicated that the genomic DNA on either side of the transgene was not contiguous in the wild-type mouse. Localization of unique markers from the two flanks indicated that both were in the proximal region of mouse chromosome 1. However, in the wild-type mouse the markers were separated by 6.3 cM, indicating a sizable rearrangement. Analysis of the mutant DNA indicated that the entire region between the markers was neither deleted nor simply inverted.
Conclusions
These results are consistent with a complex rearrangement, including at least four breakpoints and spanning at least 6.3 cM, resulting from the integration of the transgene. This genomic rearrangement disrupted the function of one or more genes critical to the maintenance of fluid homeostasis during development and the normal morphogenesis of the pars superior.
Keywords: Transgenic, insertional mutant, inner ear
Introduction
Animal models have provided much of the foundation for our current understanding of inner ear genetics. Over 100 inner ear mutants are grouped in the shaker/waltzer class of mutations constituting approximately one-tenth of the murine mutants identified thus far.1–8 Complex inner ear mutations have been shown to follow simple Mendelian inheritance patterns, suggesting single gene defects.
The mouse has proven to be an excellent animal model for the study of human deafness.9 The adult mouse cochlea is structurally and functionally similar to the human, and each share similar developmental processes.10 Furthermore, hereditary disorders of the inner ear have been shown to be similar in the two species.11 The modeling of human genetic disorders in the mouse is further facilitated by the high degree of homology between the human and mouse genomes with regions of each demonstrating highly conserved gene content genetic linkage.12 There are approximately 100 naturally occurring mouse mutations with hearing impairment.13,14 Further support for the strength of the mouse model for human hereditary hearing loss is the recent identification of delayed hearing loss (non-syndromic) in several inbred mouse strains.12
The analysis of inner ear mutations in mice has proven invaluable in the identification of four genes involved in human non-syndromic hearing loss and at least three genes involved in human sensorineural hearing loss (SHL). Mice homozygous for the shaker-1 mutation (sh1) demonstrated a waltzing phenotype consisting of head-bobbing, hyperactivity, circling and deafness secondary to a neuroepithelial defect in the inner ear. Positional cloning identified a mutation in an unconventional myosin gene, Myo7A, as causative in this mouse.15 The homologous gene in humans, MYO7A, has subsequently been implicated in two forms of non-syndromic hearing loss (DFNA11 and DFNB2) and in one form of SHL, Usher syndrome 1B (USH1B).16,17
Mice homozygous for the shaker-2 mutation (sh2) demonstrated a similar phenotype to those with sh1. The causative gene mutation, a newly described unconventional myosin, Myo15, was recently described.18 Utilizing this information and the candidate gene approach, a mutation in the homologous gene in humans, MYO15, was subsequently identified in an Israeli kindred with autosomal dominant non-syndromic hearing loss (DFNA15).19
In addition to USH1B, mouse mutants have played pivotal roles in the elucidation of the molecular defects in two forms of SHL associated with pigmentary cell defects, Waardenburg syndromes type 1 and type 2. A mutation in the Pax 3 gene was identified in mice with the Splotch mutation and mutations in the human homologue were subsequently identified in several families with Waardenburg syndrome type 1.20,21 Mutations at the mouse microphthalmia locus in a transgenic mouse line led to the identification of a novel basic-helix-loop-helix-zipper transcription factor encoding gene (MITF) leading to loss of pigmentation, reduced eye size, disorders of connective tissues and early onset deafness.22 Mutations in the human homologue have been implicated in Waardenburg syndrome type 2.23
In this manuscript we describe one such inner ear mutant resulting from the insertion of a foreign piece of DNA, a transgene. The genetics and phenotypic abnormalities of this mutant, Wocko, have been described previously.24 In brief, transgenic mice were generated by microinjection of a chimeric gene consisting of the promoter (5′ flanking region) of the human vasopressin gene fused to the v-src gene of Rous Sarcoma Virus and the termination (splicing/polyadenylation) signals of the Simian Virus 40. Fourteen pedigrees of transgenic mice were generated. One of the fourteen demonstrated bi-directional circling, hyperactivity, and vertical head bobbing, typical of the waltzing phenotype, and was named Wocko. The waltzing phenotype cosegregated with the transgene in more than 220 animals (100%). These data strongly suggest that the waltzing phenotype is the result of transgene integration and is not the result of a spontaneous mutation. Because transgene expression in the inner ear of the Wocko strain appeared to be similar to that in other, non-waltzing, strains with the same transgene, it was concluded that Wocko represents an insertional mutation. Analysis of heterozygote intercross matings suggested that the mutation was homozygous lethal.
Gross morphological changes in the adult (3–5 mo) Wocko inner ear consisted of collapse of the utricle and all three semicircular canals. The cochlea and saccule appeared grossly normal. The vestibular ganglion appeared normal and nerve fibers entered the vestibule where they projected in a disorganized fashion. The saccular sensory epithelium was present, but displayed a reduced population of both sensory and supporting cells. In several mutants, the otoconia of the saccular macula were giant and spherical. The endolymphatic sac showed mild abnormalities, including vacuolization and distortion of epithelial cells and intraluminal precipitates, in some mutant animals. In the cochlea, the apical turn of all mutants displayed loss of spiral ganglion cells and their associated nerve fibers. Additionally, the spiral limbus was reduced in size and disorganized throughout the cochlea.
Morphological abnormalities of the inner ear were first noted in the Wocko mouse at embryonic day 16, evidenced by constriction of the lumens of the semicircular canals, and the absence of otoconia on the utricular macula. At birth the semicircular canal lumens were constricted and the diameter of their arcs were smaller than normal. Their sensory epithelia were present, as were the saccular and utricular maculae. No loss of spiral ganglion cells in the cochlear apex was apparent at birth. By 1 to 2 weeks of age, edema was present in the dark cell areas of the vestibular labyrinth, and in the stria vascularis. At 2 to 3 weeks of age hydrops was present in both the cochlear and vestibular labyrinths, but the edema had resolved. Shortly thereafter, the utricle and semicircular canals collapsed and degenerated along with resolution of the cochlear hydrops.
In this paper we describe the cloning of the integration site in this complex inner ear mutation.
Materials and Methods
Library Construction and Screening
Wocko and wild-type (C57BL/J6) genomic libraries were commercially prepared in Lamda Fix II (Stratagene). Twenty 150 mm NZY plates were prepared and plated with 50,000 plaque forming units, preadsorbed to 600 mL of a competent strain of E coli. Duplicate plaque lifts were performed on nitrocellulose, and screened using standard methodology with a 32P-labeled probe prepared by random priming from the transgene sequence.25,26
Southern Blot Analysis
For evaluation of probes on genomic DNA, 10-μg samples of DNA were endonuclease digested to completion in a final volume of 50 μL. The samples were then size separated by overnight electrophoresis at 25 V on an agarose gel. After electrophoresis the gel was photographed under ultraviolet illumination. After denaturation and washing the DNA was transferred to nitrocellulose and linked to the membrane by baking.27 Prior to hybridization, the membranes were soaked in 5× SSC, 1% SDS, 20 mmol/L NaPO4, and 50% formamide at 42°C for 1 hour. DNA probes were labeled by the random priming method.26 Hybridizations were performed using 106 cpm/mL of fresh hybridization buffer. The blots were washed to a final stringency of 0.1× SSC/0.1% SDS at 65°C for 1 to 2 hours.
Pulse Field Gel Electrophoresis
Approximately 10-μg samples of high-molecular-weight genomic DNA prepared in low melt agarose were subjected to endonuclease digestion for 12 hours at the appropriate temperature. The samples were treated with 0.5 mol/L EDTA, and the agarose plugs were loaded onto a 1% agarose gel. The gel was then subjected to pulse field electrophoresis with the following parameters: 1) a ramped pulse time with a beginning pulse duration of eight seconds and a final pulse duration of 45 seconds, 2) electrophoresis at 190 W, and 3) a run time of 16 hours at 16°C.
After electrophoresis each gel was depurinated by bathing in 0.25N HCl for 15 minutes. The gel was denatured in a 0.5N NaOH/1.5 mol/L NaCl solution for 1 hour. The gel was neutralized in a 1 mol/L Tris-HCl pH 7.4/1.5N NaCl solution for 1 hour, and blotted onto a nylon membrane in 10× SSC for 48 hours. The DNA was cross-linked to the membrane after transfer by baking at 80°C for 2 hours.
Identification of Single-Copy Probes
Cloned fragments of DNA were digested with a variety of restriction endonucleases, size separated by electrophoresis and transferred to a nylon membrane as described. Twenty-five nanograms of mouse genomic DNA was radiolabeled by random priming and hybridized to the blot. All fragments not displaying hybridization were purified on low-melt agarose and extracted using standard protocols. The fragments were subsequently used as probes for hybridization to a mouse genomic Southern blot, and those fragments demonstrating single copy hybridization were used for genomic cloning.
Northern Blot Analysis
Total RNA from wild-type tissues was prepared by the acid guanidinium thiocyanate-phenol-chloroform extraction method.28 The RNA was poly(A)+ selected, subjected to electrophoresis, and transferred to nylon membranes by standard methods. After cross-linking, the membranes were prehybridized in a solution containing 6× SSC, 5× Denhardt's solution, 50 mg/mL sheared and denatured salmon sperm DNA, and 0.5% SDS for 1 hour at 65°C. Radiolabeled DNA probe was added to a final concentration of 1 to 2 × 106 cpm/mL in fresh solution and incubated at 65°C for 18 hours. After hybridization the blots were subjected to a series of washes of gradually increasing stringency as follows: 1) incubation in 2× SSC for 15 minutes at 65°C, 2) repeated incubation with 2× SSC and 0.1% SDS for 30 minutes at 65°C, and 3) a high stringency wash using 0.1× SSC for 10 minutes at 65°C followed by autoradiography.
Chromosomal Localization
Interspecific backcross progeny were generated by mating (C57BL/6JxM spretus)F1 females and C57BL/6J males as described.29 A total of 205 N2 mice were used to map two Wocko transgene flanking probes. Southern blot analysis was performed as described. All blots were prepared on HybondN+ membrane (Amersham). The probes were labeled with [α32P]dCTP using a nick translation (Boehringer Mannheim) or random priming (Amersham) kit; washing was done to a final stringency of 0.5× SSCP, 0.1% SDS, 65°C. The probe for D1Afr1 was a 1-kb Pst1 fragment derived from the genomic region flanking the 5′ end of the transgene. The probe for D1Afr2 was a 230-bp fragment derived from the genomic region flanking the 3′ end of the transgene. For D1Afr1, a 7.2-kb fragment was detected in Hinc-II digested C57BL/6J DNA and a 6.1-kb fragment in M spretus DNA. For D1Afr2, a 5.2-kb fragment was detected in EcoRI-digested C57BL/6J DNA and a 6.5-kb fragment in M spretus DNA. The presence or absence of the two M spretus fragments was followed in the backcross mice.
A description of the probes and RFLPs for the loci linked to the D1Afr1 and D1Afr2 flanking markers, including bullous pemphigoid antigen 1 (Bpag1), interleukin 1 receptor type 1 (Il1r1) and cytotoxic T-lymphocyte-associated protein-4 (Ctla4) has been reported previously.30 Recombination distances were calculated as described using the computer program SPRETUS MADNESS.31 Gene order was determined by minimizing the number of recombination events required to explain the allele distribution patterns.
Results
Cloning of Regions Flanking Transgene Insertion
Nine clones containing at least a portion of the transgene sequences were identified. Digestion with NotI revealed each clone to possess a separate and distinct pattern of fragments, indicating that each clone contained a unique portion of the integration site (Fig. 1). Southern blotting indicated that each clone also contained both the 5′ and 3′ ends of the transgene (Fig. 2).
Fig. 1.
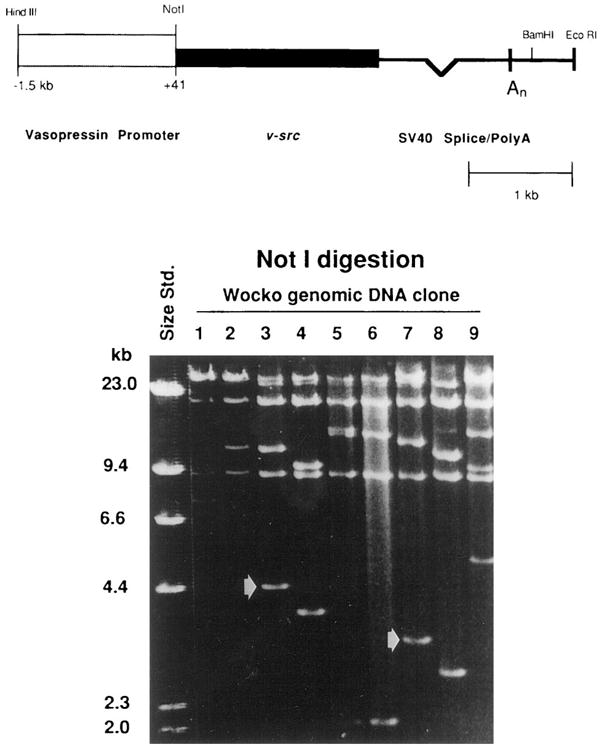
Top. Diagram of the transgene, showing the construct and an internal NotI site. Bottom. Size separation of nine clones isolated from a Wocko genomic library using the transgene as a probe. Each of the nine clones, after digestion to completion with NotI, possessed two insert bands of different lengths, indicating that each is an independent clone containing transgene sequences. The absence of a 5-kb band in all of the clones confirms the integration of a single transgene, without tandem repeats. The arrows indicate bands in clones 3 and 7 that were subsequently found to contain mouse genomic sequences 3′ and 5′ to the insert, respectively.
Fig. 2.
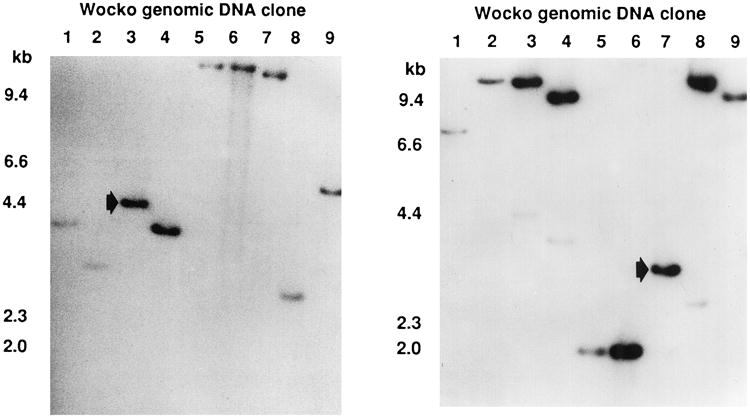
Southern blots of the gel represented in Figure 1. Left. Hybridization with a probe derived from transgene sequences 5′ to the internal NotI site. All clones showed hybridization, indicating that all contained 5′ transgene sequences. The arrow indicates a 3-kb fragment in clone 7 containing 1.5 kb of transgene sequence and 1.5 kb of flanking genomic sequence. Right. Hybridization with a probe derived from transgene sequences 3′ to the internal NotI site. Again, all clones showed hybridization, indicating that all contained 3′ transgene sequences. The arrow indicates a 4.2-kb fragment in clone 3 containing 3.5 kb of transgene sequence and 0.8 kb of flanking genomic sequence.
The mechanisms by which microinjected DNA integrates into the mouse genome are poorly understood. Frequently the event occurs via the insertion, at a single site, of multiple copies of the transgene in a tandem head-to-tail array.32 The transgene used in Wocko contains a single restriction site for NotI, and of course there are additional NotI restriction sites within the sequences of normal mouse DNA flanking the transgene. A single copy integration would therefore yield two bands that hybridize to the transgene Q1probe in the genomic Southern blot, corresponding to the 5′ and 3′ fragments of the transgene and the adjacent flanking DNA. In contrast, multiple tandemly arranged copies of the transgene at the insertion site would be likely to yield three hybridizing bands; a 5′ flank, a 3′ flank, and a band corresponding to the length of the transgene (5 kb) produced by the tandemly arranged NotI sites. Two bands were seen in all nine clones, suggesting the integration of a single transgene.
A 3-kb Not I fragment from clone 7, which hybridized with a probe from the 5′ region of the transgene and a 4.2-kb Not I fragment from clone 3, which hybridized with a 32P-labeled probe made from the 3′ end of the transgene (Fig. 2), was subcloned into pBluescript II KS- (Strat-agene).
Approximately 400 base pairs from each end of the two subclones were sequenced.33,34 Sequences from one end of each subclone proved to be the expected transgene sequences. The opposite ends yielded new sequences suggestive of normal genomic DNA flanking the transgene. These flanking sequences were used as probes for identification of non-mutant clones from a wild-type mouse genomic library.
Physical Mapping of Integration Site
Utilizing a number of unique genomic fragments from the DNA flanking the transgene, several lambda phage clones from the integration site were identified from both the mutant and wild-type genomic libraries. These clones were mapped by endonuclease digestion, and used in a standard walking protocol to isolate overlapping genomic clones. These clones provided extensive physical maps of the integration site, which encompassed more than 40 kb of normal mouse DNA (Fig. 3A). The data were inconsistent with a simple insertion of the transgene, because the maps of normal mouse DNA produced from the two flanks did not appear to overlap.
Fig. 3.
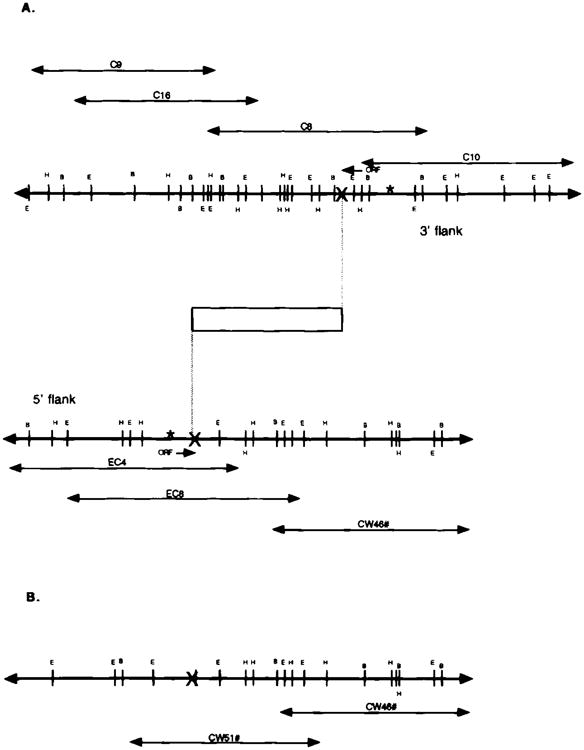
A. Physical map of the Wocko integration site. Open box = transgene; X = breakpoints; C*, C9, C10, and C16 = wild-type genomic clones from the breakpoint region adjacent to the 3′ end of the transgene; EC4 and EC8 = wild-type clones from the breakpoint region adjacent to the 5′ end of the transgene; ORF = open reading frame. B. Physical map of another breakpoint region in Wocko. CW46 and CW51 = mutant genomic clones from the third breakpoint region. The map to the right of the breakpoint corresponds to the right-side portion of the wild-type map isolated with probes from the 5′ flank in Wocko.
Pulse field gel electrophoresis was used to test for the presence of deletion. High-molecular-weight mutant and wild-type genomic DNA was digested with rare cutting restriction endonucleases (BssHII and SfiI) and size separated on a 1% agarose gel by pulse field electrophoresis. After Southern transfer, the membrane was hybridized with the unique 1-kb PstI fragment adjacent to the 5′ end of the transgene. The BssHII lane revealed a large polymorphism in Wocko. Additionally, the SfiI lane demonstrated a polymorphic fragment in the mutant lane approximately 50 kb smaller than the corresponding wild-type band (Fig. 4). These data confirm the presence of a genomic rearrangement at the integration site, and suggest the possibility of an approximately, 50-kb deletion.
Fig. 4.
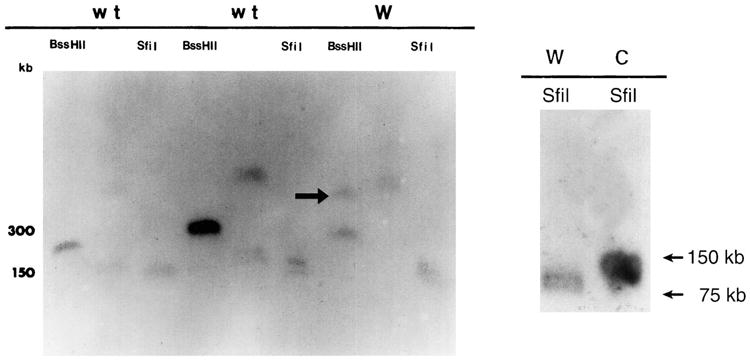
Left. Southern blot generated by pulse field electrophoresis of high-molecular-weight samples of wild-type (wt) and Wocko (W) genomic DNA. The blot was hybridized with a probe from the flank adjacent to the 5′ end of the transgene. An additional band (arrow) can be seen, well above the 300-kb wild-type band in the BssHII digested Wocko DNA. Right. Pulse field Southern blot produced with SfiI digestion of wild-type (C) and Wocko (W) DNA. The Wocko bands appear to be approximately 50 kb shorter than the wild-type bands.
Genetic Mapping of Integration Site
The possibility of a larger deletion or rearrangement was evaluated by chromosomal mapping of the DNA flanking the transgene. The results of backcross mapping of probes from the 5′ and 3′ flanks of the transgene are illustrated in Figure 5. The mapping results indicate that D1Afr1 (5′ flank probe) and D1Afr2 (3′ flank probe) are located in the proximal region of mouse chromosome 1, linked to Bpag1, Il1r1 and Ctla4. Although 129 mice were analyzed for every marker and are shown in the segregation analysis, up to 198 mice were typed for some pairs of markers. Each locus was analyzed in pairwise combinations for recombination frequencies using the additional data. The ratios of the total number of mice exhibiting recombinant chromosomes to the total number of mice tested for each pair of loci and recombinations frequencies between the loci are shown in Figure 5. The distance between D1Afr1 and D1Afr2 is 6.3 ± 1.8 cM, suggesting that the transgene has substantially disrupted the region between the two flanking markers. Il1r1, Il1r2, St2, and Otf-8 are genes that map between the two flanking probes in the normal mouse. None of these genes appear to be candidates for the Wocko mutation.35–37
Fig. 5.
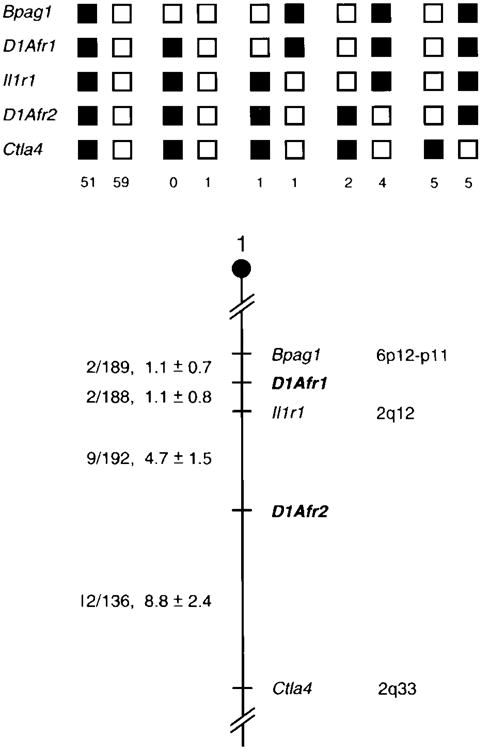
Chromosomal location of the D1Afr1 and D1Afr2 transgene flanking probes in the mouse genome. The probes were mapped by interspecific backcross analysis. The segregation patterns of D1Afr1 and D1Afr2 and flanking genes are shown (top). Each column represents the chromosome identified in the backcross progeny that was inherited from the (C57BL/6J × Mus spretus)F1 parent. The shaded boxes represent the presence of a C57BL/6J allele and white boxes represent the presence of a M spretus allele. The number of offspring inheriting each type of chromosome is listed at the bottom of each column. A partial chromosome 1 linkage map showing the location of D1Afr1 and D1Afr2 in relation to linked genes is shown (bottom). The number of recombinant2 animals over the total number of N2 animals typed plus the recombination frequencies (expressed as genetic distance in cM [±1 SE]) are shown for each pair of loci on the left side of the chromosome map. The positions of loci in human chromosomes, where known, are shown on the right side of the chromosome maps. References for the human map positions of loci cited in this study can be obtained from GDB (Genome Database), a computerized database of human linkage information maintained by The William H. Welch Medical Library of The Johns Hopkins University (Baltimore, MD).
Characterization of Rearrangement
Any one of a number of potential genomic rearrangements may have occurred at the integration site in Wocko. A DNA deletion, inversion, translocation, or a complex combination of these events may have occurred during transgene insertion. Two lines of evidence argue against a translocation. First, each of the flanking probes utilized for interspecific backcrosses were mapped to chromosome 1. Second, a translocation of this size would likely display semidominant lethality, a phenotype not identified in Wocko.
The first task was to identify the possibility of a large, simple genomic deletion of the region between D1Afr1 and D1Afr2. Probes from inside the putative deletion were identified. A 0.8-kb XbaI/EcoRI fragment, lying immediately adjacent to the 5′ breakpoint and between D1Afr1 and D1Afr2 was hybridized to a Southern blot containing genomic DNA from Wocko and its two parent strains, C57BL/6J and DBA. As Wocko is hemizygous, it was hypothesized that this probe, located within the rearrangement and not present at the integration site in Wocko, would be displayed as a single copy hybridization, half intensity compared to the wild-type, if a simple deletion had occurred. In each digest, an additional band could be identified in the Wocko lane that hybridized to this 0.8-kb probe. A 6-kb EcoRI fragment, a 10-kb BamHI fragment, a 7-kb SstI fragment, and a 6.9-kb PstI fragment were seen in addition to the wild-type bands in each lane, indicating that the region is not deleted in Wocko. A similar analysis using a 350-base-pair PstI/XbaI fragment located between D1Afr1 and D1Afr2 and adjacent to the 3′ breakpoint also identified a normal and a mutant fragment in this region (Fig. 6).
Fig. 6.
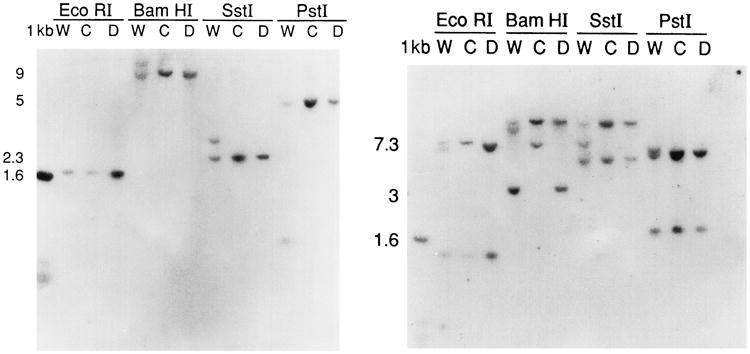
Southern blots of Wocko (W) and parental strains (C and D) genomic DNA digested with a variety of restriction enzymes and hybridized with probes located a few kilobases inside of the two breakpoints that in Wocko flank the insert and that, in the wild-type mouse, are separated by 6.3 cM. Left. Hybridization with a probe adjacent to the 5′ breakpoint. Each lane containing Wocko DNA contains an additional band, indicating re-arrangement and not deletion of the sequence. Right. Hybridization with a probe adjacent to the 3′ breakpoint. Each band containing Wocko DNA, except in the case of EcoRI, contains an additional band. As with the 5′ probe, this indicates rearrangement and not deletion of the sequence.
As the data suggest something other than a simple deletion occurred at the integration site, the next task was to consider the possibility of a simple, 6.3 cM DNA inversion. To investigate this further, it was necessary to identify another breakpoint within the Wocko genome. The unique 0.8-kb XbaI/EcoRI probe described above was used to screen the Wocko genomic library. Several clones were identified that contained the wild-type 7.3-kb fragment from the normal chromosome, and one (CW58) was found that contained the 6-kb EcoRI fragment that was unique to the Wocko genome (Fig. 3B). A restriction endonuclease map of this clone revealed extensive overlap with the genomic region containing the 0.8-kb XbaI/EcoRI fragment in the normal mouse, but displayed no similarity to the map of the region flanking the 5′ end of the transgene, as would be expected if a simple inversion had occurred. This was confirmed by Southern blotting using a probe from the 5′ flanking region, which failed to hybridize to clone CW58.
Identification of Open Reading Frames in Breakpoint Region
Many of the unique probes used for lambda walking were sequenced and analyzed for the presence of homology to known genes and for the probability of protein encoding potential. Two separate fragments, one adjacent to the breakpoint on the 7.3-kb EcoRI genomic fragment and one adjacent to the breakpoint on the 3.5-kb EcoRI genomic fragment (Fig. 3A), were each found to contain approximately 200 base pairs of open reading frame (ORF).
The possibility of messenger RNA expression homologous to either of these two ORF containing fragments was explored by probing Northern blots of poly (A)+ RNA isolated from various adult wild-type tissues and total RNA from wild-type embryos at several times surrounding embryonic day 16. Neither ORF probe displayed specific hybridization to the Northern blots.
Discussion
Although inner ear mutants represent a large proportion of the naturally occurring mouse mutations, only recently, and in a few cases, has the molecular basis for some of these mutations been elucidated. The advent of transgenic technology has brought with it the induction of several insertional developmental mutations, a few that affect the inner ear. The isolation of inner ear specific genes may be facilitated by the study of these insertional mutants. Of the greater than 40 insertional mutations that have been described in the past decade, relatively few have led to the isolation of the specific gene defect. Complex genomic rearrangements may remove some or all of the exon sequences, making gene isolation difficult. Additionally, the presence of highly repetitive sequences around the insertion site can make isolation of single copy flanking sequences difficult.38
In this manuscript we describe a dominant insertional mutation resulting in a waltzing phenotype. The anatomic features associated with this mutation include narrowing of the lumina of the semicircular canals beginning on embryonic day 16 and early postnatal endolymphatic hydrops, followed by collapse of the membranous utricle and semicircular canals. These phenotypic abnormalities segregate with the transgene sequences. The integration site contains a single copy of the 5-kb transgene on proximal chromosome 1 and is associated with a complex genomic rearrangement spanning 6.3 cM. The rearrangement includes at least four breakpoints, but no deletions of DNA have yet been detected. The complexity of the integration is similar to observations in other insertional mutations as described above, and has likewise made the identification of the mutated gene or genes difficult.
The Wocko behavioral phenotype is similar to other mice in the shaker/waltzer class of mutations. Shaker/waltzer syndrome is a name given to a group of mouse mutants sharing a behavioral phenotype characterized by bidirectional circling, vertical head bobbing, hyperactivity, and abnormal responses to changes in position. This phenotype can be inherited as autosomal recessive or dominant with varying degrees of penetrance. This diverse group of mouse mutants are represented by more than 40 loci distributed throughout the genome.39 The inner ears of all mice in the shaker/waltzer class are abnormal.
Although none of the ORFs identified in the region of the break-point yielded sequences suggestive of candidate genes, we recently undertook a positional/candidate cloning approach to identify a gene involved in a spontaneous inner ear mutation in an inbred strain (C3H/HeJ) arising at a map distance of 10 cM on mouse chromosome 1, in the vicinity of one of the Wocko breakpoints. These mutant mice displayed absent cochleas and variable degrees of kidney developmental anomalies, inherited in an autosomal recessive manner. We have identified a mutation in Eya1, the mouse homologue of the gene involved in human branchio-oto-renal syndrome, in this mutant mouse. The mutation resulted from the insertion of an IAP transposon into intron 7 creating cryptic splice donor sites and reducing the wild type mRNA level by approximately 50%.40 Although the mode of inheritance differs between this mutant and Wocko, the Eya1 mutant is not a null and this gene still serves as a candidate for the gene(s) disrupted in Wocko.
An interesting feature of this insertional mutation is the appearance of endolymphatic hydrops in the postnatal period that is followed by degeneration of the vestibular labyrinth. Although the pathophysiology of endolymphatic hydrops is poorly understood, it is generally accepted that it can arise from either overproduction or impaired absorption of endolymph. Experimentally induced obstruction of the endolymphatic sacs in guinea pigs led to the production of endolymphatic hydrops.41 The endolymphatic duct and sac develop normally in Wocko, suggesting an abnormality in the production of endolymph. Abnormal endolymphatic homeostasis and pressures may account for the incomplete canalization of the semicircular canals.
The potassium rich endolymph of the pars inferior is secreted by the stria vascularis. It is believed that the endolymph of the pars superior is secreted by the dark cells present in the macular area of the utricle and the cristae ampullaris of the semicircular canals. Wocko displays edema in both of these areas, suggesting defective homeostatic mechanisms. The stria vascularis of the mouse is composed of three cell layers.42 Neural crest derived pigmented cells in the intermediate layer of the stria vascularis and in the dark cell areas of the vestibular labyrinth are essential for development of the stria vascularis and the subsequent generation of the endocochlear potential.43 The association between pigmentation defects and hearing loss in mice is well established.20,44,45 Although Wocko does not display pigmentation abnormalities, the edema of the stria vascularis and dark cell areas may be secondary to abnormalities of these neural crest cells in the mutant inner ears. The secretion of endolymph depends on the activity of ion pumps, including primarily Na,K-ATPase and on selective ion channels in the membranes of cochlear cells.46,47 No genes encoding ion pumps or channels have been localized to the location of the integration. However, any such genes localized to this region in the future would be attractive candidates for the gene mutated in Wocko.
Acknowledgments
We would like to acknowledge the assistance of Karen Avraham, Nancy Jenkins, and Neil Copeland for their assistance with mapping.
Supported by the American Academy of Otolaryngology—Head and Neck Surgery Foundation, the Deafness Research Foundation, the NIH/NIDCD grants DC00019, DC00139, and DC00028, HHMI and the Veterans Administration Research Service.
Footnotes
Presented as a Candidate's Thesis to the American Laryngological, Rhinological and Otological Society, Inc. Recipient of the Fowler Award.
Bibliography
- 1.Deol MS. The development of the inner ear in mice homozygous for Shaker-with-syndactylism. J Embryol Exp Morphol. 1963;11(3):493–512. [PubMed] [Google Scholar]
- 2.Deol MS. The origin of the abnormalities of the inner ear in Dreher mice. J Embryol Exp Morphol. 1964;12(4):727–733. [PubMed] [Google Scholar]
- 3.Deol MS, Lane PW. A new gene affecting the morphogenesis of the vestibular part of the inner ear in the mouse. J Embryol Exp Morphol. 1966;16(3):543–558. [PubMed] [Google Scholar]
- 4.Deol MS, Dickie MM. Rotating: a new gene affecting behavior and the inner ear in the mouse. J Hered. 1976;58:69–72. [Google Scholar]
- 5.Lyon MF. Twirler: a mutant affecting the inner ear of the house mouse. J Embryol Exp Morphol. 1958;6(1):105–116. [PubMed] [Google Scholar]
- 6.Lyon MF. Zigzag: a genetic defect of the horizontal canals in the mouse. Genet Res. 1960;1:189–195. [Google Scholar]
- 7.Stein KF, Huber SA. Morphology and behavior of waltzertype mice. J Morphol. 1960;106:197–203. doi: 10.1002/jmor.1051060206. [DOI] [PubMed] [Google Scholar]
- 8.Anniko M, Wenngren BI, Wroblewski R. Aberrant elemental composition of otoconia in the Dancer mouse mutant with a semidominant gene causing a morphogenetic type of inner ear defect. Acta Otolaryngol (Stockh) 1988;106:208–212. doi: 10.3109/00016488809106427. [DOI] [PubMed] [Google Scholar]
- 9.Steel KP, Brown SD. Genetics of deafness. Curr Opin Neurobiol. 1996;6:520–525. doi: 10.1016/s0959-4388(96)80059-6. [DOI] [PubMed] [Google Scholar]
- 10.Steel KP, Bock GR. Hereditary inner-ear abnormalities in animals. Arch Otolaryngol Head Neck Surg. 1983;109:22–29. doi: 10.1001/archotol.1983.00800150026005. [DOI] [PubMed] [Google Scholar]
- 11.Brown SD, Steel KP. Genetic deafness-progress with mouse models. Hum Mol Genet. 1994;3:1453–1546. doi: 10.1093/hmg/3.suppl_1.1453. [DOI] [PubMed] [Google Scholar]
- 12.Zheng QY, Johnson KR, Erway LC. Assessment of hearing in 80 inbred strains of mice by ABR threshold analyses. Hear Res. 1999;130:94–107. doi: 10.1016/s0378-5955(99)00003-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lyon MF, Rastan S, Brown SDM. Genetic Variants and Strains of the Laboratory Mouse. Oxford, UK: Oxford University Press; 1996. [Google Scholar]
- 14.Steel KP. Inherited hearing defects in mice. Annu Rev Genet. 1995;29:675–701. doi: 10.1146/annurev.ge.29.120195.003331. [DOI] [PubMed] [Google Scholar]
- 15.Gibson F, Walsh J, Mburu VP, et al. A type VII myosin encoded by the mouse deafness gene shaker-1. Nature. 1995;374:62–64. doi: 10.1038/374062a0. [DOI] [PubMed] [Google Scholar]
- 16.Lui XZ, Walsh J, Mburu P, et al. Mutations in the myosin VIIA gene cause non-syndromic recessive deafness. Nature Genet. 1997;16:188–190. doi: 10.1038/ng0697-188. [DOI] [PubMed] [Google Scholar]
- 17.Weil D, Blanchard S, Kaplan J, et al. Defective myosin VIIA gene responsible for Usher syndrome type 1B. Nature. 1995;374:60–61. doi: 10.1038/374060a0. [DOI] [PubMed] [Google Scholar]
- 18.Probst FJ, Fridell RA, Raphael Y, et al. Correction of deafness in shaker-2 mice by an unconventional myosin in a BAC transgene. Science. 1998;280:1444–1447. doi: 10.1126/science.280.5368.1444. [DOI] [PubMed] [Google Scholar]
- 19.Vahava O, Morell R, Lynch ED, et al. Mutation in transcription factor POU4F3 associated with inherited progressive hearing loss in humans. Science. 1998;279:1950–1954. doi: 10.1126/science.279.5358.1950. [DOI] [PubMed] [Google Scholar]
- 20.Epstein DJ, Vekemans M, Gros P. Splotch (Sp2H), a mutation affecting development of the mouse neural tube, shows a deletion within the paired homeodomain of Pax-3. Cell. 1991;67:767–774. doi: 10.1016/0092-8674(91)90071-6. [DOI] [PubMed] [Google Scholar]
- 21.Baldwin CT, Hoth CF, Amos JA. An exonic mutation in the HuP2 paired domain gene causes Waardenburg's syndrome. Nature. 1992;355:637–638. doi: 10.1038/355637a0. [DOI] [PubMed] [Google Scholar]
- 22.Hodgkinson CA, Moore KJ, Nakayama A, et al. Mutations at the mouse microphthalmia locus are associated with defects in a gene encoding a novel basic-loop-helix-zipper protein. Cell. 1993;74:395–404. doi: 10.1016/0092-8674(93)90429-t. [DOI] [PubMed] [Google Scholar]
- 23.Tassabehji M, Read AP, Newton VE, et al. Waardenburg's syndrome patients have mutations in the human homologue of the Pax-3 paired box gene. Nature. 1992;355:635–636. doi: 10.1038/355635a0. [DOI] [PubMed] [Google Scholar]
- 24.Crenshaw EB, Ryan A, Dillon SR, et al. Wocko, a neurological mutant generated in a transgenic mouse pedigree. J Neurosci. 1991;11(6):1524–1530. doi: 10.1523/JNEUROSCI.11-06-01524.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Benton WD, Davis RW. Screening lambda gt recombinant clones by hybridization to single plaques in-situ. Science. 1977;196:637–638. doi: 10.1126/science.322279. [DOI] [PubMed] [Google Scholar]
- 26.Feinberg AP, Vogelstein B. A technique for radiolabeling DNA restriction endonuclease fragments to high specific activity. Anal Biochem. 1984;137:266–267. doi: 10.1016/0003-2697(84)90381-6. [DOI] [PubMed] [Google Scholar]
- 27.Southern EM. Detection of specific sequences among DNA fragments separated by gel electrophoresis. J Mol Biol. 1975;98(3):503–517. doi: 10.1016/s0022-2836(75)80083-0. [DOI] [PubMed] [Google Scholar]
- 28.Chomoczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 29.Copeland NG, Jenkins NA. Development and applications of a molecular genetic linkage map of the mouse genome. Trends Genet. 1991;7(4):113–118. doi: 10.1016/0168-9525(91)90455-y. [DOI] [PubMed] [Google Scholar]
- 30.Copeland NG, Jenkins NA, Gilbert DJ, et al. A genetic linkage map of the mouse: current applications and future prospects. Science. 1993;262:57–66. doi: 10.1126/science.8211130. [DOI] [PubMed] [Google Scholar]
- 31.Green EL. Genetics and Probability in Animal Breeding Experiments. New York: Oxford University Press; 1981. Linkage, recombination and mapping; pp. 77–113. [Google Scholar]
- 32.Palmiter RD, Brinster RL. Germ-line transformation of mice. Annu Rev Genet. 1986;20:465–499. doi: 10.1146/annurev.ge.20.120186.002341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taber S, Richardson CC. DNA sequence analysis with a modified bacteriophage T7 DNA polymerase. Proc Natl Acad Sci U S A. 1987;84:4767–4771. doi: 10.1073/pnas.84.14.4767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tominaga S. A putative protein of a growth specific cDNA from BALB/c-3T3 cells is highly similar to the extracellular portion of mouse interleukin 1 receptor. FEBS Letters. 1989;258:301–304. doi: 10.1016/0014-5793(89)81679-5. [DOI] [PubMed] [Google Scholar]
- 36.Sims JE, March CJ, Cosman D, et al. cDNA expression cloning of the IL-1 receptor, a member of the immunoglobulin superfamily. Science. 1988;241:585–589. doi: 10.1126/science.2969618. [DOI] [PubMed] [Google Scholar]
- 37.Avraham KB, Cho BC, Gilbert D, et al. Murine chromosomal location of four class III POU transcription factors. Genomics. 1993;18:1–3. doi: 10.1006/geno.1993.1436. [DOI] [PubMed] [Google Scholar]
- 38.Meisler MH. Insertional mutation of “classical” and novel genes in transgenic mice. Trends Genet. 1992;8(10):341–344. doi: 10.1016/0168-9525(92)90278-c. [DOI] [PubMed] [Google Scholar]
- 39.Green MC. Catalog of mutant genes and polymorphic loci. In: Lyon MF, Searle AG, editors. Genetic Variants and Strains of the Laboratory Mouse. 2. Oxford, UK: Oxford University Press; 1989. [Google Scholar]
- 40.Johnson K, Cook S, Erway L, et al. Inner ear and kidney anomalies caused by IAP insertion in an intron of the Eya1 gene in a mouse model of BOR syndrome. Hum Mol Genet. 1999;8(4):645–653. doi: 10.1093/hmg/8.4.645. [DOI] [PubMed] [Google Scholar]
- 41.Kimura RS, Schuknecht HF. Membranous hydrops in the inner ear of the guinea pig after obliteration of the endolymphatic sac. Pract Otorhinolaryngol. 1965;27:343–354. [Google Scholar]
- 42.Kikuchi K, Hilding DA. The development of the stria vascularis in the mouse. Acta Otolaryngol. 1966;62:277–291. doi: 10.3109/00016486609119573. [DOI] [PubMed] [Google Scholar]
- 43.Cable J, Steele KP. Identification of two types of melanocyte within the stria vascularis of the mouse inner ear. Pigment Cell Res. 1991;4:87–101. doi: 10.1111/j.1600-0749.1991.tb00320.x. [DOI] [PubMed] [Google Scholar]
- 44.Cable J, Barkway C, Steele KP. Characteristics of stria vascularis melanocytes of viable dominant spotting (Wv/Wv) mouse mutants. Hear Res. 1992;64:6–20. doi: 10.1016/0378-5955(92)90164-i. [DOI] [PubMed] [Google Scholar]
- 45.Cable J, Jackson IJ, Steele KP. Light (Blt), a mutation that causes melanocyte death, affects stria vascularis function in the mouse inner ear. Pigment Cell Res. 1993;6:215–225. doi: 10.1111/j.1600-0749.1993.tb00605.x. [DOI] [PubMed] [Google Scholar]
- 46.Ryan AF, Watts AG. Expression of genes coding for α and β isoforms of Na/K-ATPase in the cochlea of the rat. Cell Mol Neurosci. 1991;2:179–187. doi: 10.1016/1044-7431(91)90011-c. [DOI] [PubMed] [Google Scholar]
- 47.Fina M, Ryan AF. Expression of mRNAs encoding subunit isoforms of the Na, K-ATPase in the vestibular labyrinth of the rat. Cell Mol Neurosci. 1994;5:604–613. doi: 10.1006/mcne.1994.1074. [DOI] [PubMed] [Google Scholar]