

Abstract
Hepato-renal syndrome (HRS) is one of the most detrimental conditions in patients with end stage liver failure. Historically, HRS was considered a terminal disease associated with cirrhosis and was termed “liver-death syndrome”. Furthermore, despite the improved understanding of pathophysiology and the reversibility of renal dysfunction in HRS, mortality remains extremely high especially for type 1 HRS. This review summarizes the recent advances in the pathophysiology, diagnosis and management of HRS and also provides an evolving area of research in the pathophysiologic mechanisms of HRS, which may open the door for new therapeutic approaches.
Keywords: Acute kidney injury, Cirrhosis, Endothelial cell, Inflammation
INTRODUCTION
Acute kidney injury is a well-known and deadly complication of liver or biliary tract disease for over a century [1]. Freriches and Flint first reported that oliguria in the absence of renal histological changes in patients with advanced cirrhosis and ascites in 1861[2]. Almost a century later, Hecker and Sherlock reported rapid and progressive azotemia and oliguria in patients with cirrhosis in 1957 [3]. They noted near normal kidney histology and full functional recovery of kidneys correlated with recovery of hepatic function. Koppel et al also showed that kidneys from patients who succumbed from hepato-renal syndrome (HRS) were functioned normally when transplanted into patients with chronic uremia [4]. These studies suggested that HRS is a functional renal disorder without underlying abnormalities in kidney structure. Schroeder et al measured the para-aminohippurate (PAH) clearance in cirrhosis with renal failure patients, demonstrated that intense renal arterial vasoconstriction together with systemic and splanchnic vasodilation is the hallmark of HRS [5]. However, despite improved understandings of pathophysiology as well as the reversible nature of renal failure in HRS, the prognosis of HRS remains extremely poor (used to be called as “liver-death syndrome” [6]). Currently, the median survival of untreated HRS type 1 is ~2 weeks while that of type 2 is approximately 4 to 6 months [6]. Liver transplant is the only viable treatment but scarcity of donor organs has been a major hindrance and a majority of patients with HRS type 1 die while awaiting transplant. The latest research discovered the new mechanisms of remote organ injury in local sterile inflammation, which may be applicable to the pathogenesis of HRS. Now it is becoming increasingly clear that HRS is multifactorial phenomenon. The goals of this brief review are to summarize the current understanding and management of HRS and provide an evolving area of research in the pathophysiologic mechanism of HRS.
CURRENT DEFINITION OF HEPATO-RENAL SYNDROME
The International Ascites Club initially defined HRS in 1996 [7] and subsequently revised in 2007 [8]. (Table 1) Those criteria were made from the current understanding of liver-kidney interplay as a pathophysiologic mechanism for HRS. HRS has two distinct types of clinical presentations [9]. Type 1 HRS is an acute form of HRS and characterized by rapidly progressive renal failure. Type 1 HRS usually develops after several precipitating events such as gastrointestinal bleeding [10], large volume paracentesis [10], acute alcoholic hepatitis and spontaneous bacterial peritonitis [11]. Type 1 HRS is commonly associated with rapid deterioration of extra-renal organ function including the heart, brain, liver, and adrenal glands. Type 2 HRS is chronic form of HRS and characterized with moderate and slow progressive renal failure associated with diuretics resistant ascites [12]. Although there is a clear distinction of two different types of HRS, renal impairment is often progressive and can be regarded as “continuum” instead of two different entities, such as most patients initially represents type 2 HRS and turn into type 1 HRS after several episodes of precipitating events.
Table 1.
New diagnostic criteria for hepatorenal syndrome*
|
Criteria have been developed by the International Ascites Club. Modified from [Gut, Salerno, F. et al. 56, 1310-1318, © 2007]
CLINICAL SIGNIFICANCE
HRS is a frequent complication in advanced cirrhosis and the prevalence of HRS parallels the progression of liver disease in patients with cirrhosis [13]. HRS occurs in about 10% of patients admitted to hospital with decompensated cirrhosis, with a cumulative probability of 18% at 1 year and 39% at 5 years [10]. Also patients with spontaneous bacterial peritonitis have a 33% chance of developing HRS [14]. HRS is life-threatening complication and type 1 patients has 80% mortality in two weeks and only 10% of patients survive more than 3 months [6]. Because of this detrimental prognosis, most patients with type 1 HRS die while being evaluated for transplant or waiting for transplant. Those prognoses are even worse for patients with apparent precipitating factors. Patients with type 2 HRS also has a median survival of approximately 6 months [6].
Health care costs related to HRS has not been reported but it is not difficult to imagine that it is very high considering the detrimental nature of this condition. Quiros et al estimates with using their Mexican social security institute database that annual health care costs per person for Child-Pugh Score C is $30,249 (seven times higher than patients with Child-Pugh Score of A, $4,269) [15]. Also Henry Ford study, which compared the economic burden for U.S. patients with chronic hepatitis C disease using a large private health insurance claims database from 2003 to 2010, the annual health care costs per person for decompensated cirrhosis was estimated around $59,995, which is triples compared to patients with no cirrhosis ($17,277) [16]. Considering the fact that 400,000 cirrhotic patients in the United States [17], and grossly up to 2% of those patients develop HRS (the incidence of acute kidney failure in cirrhotic patients was reported to be 10% and HRS causes up to 20% of acute kidney failure) [18], annual US health care costs approximates up to $240 million. HRS creates a major economic burden under the circumstances that currently no effective therapy is available. Therefore there is an urgent need for the clearer understanding of pathophysiologic mechanisms and the development of viable treatment options.
PATHOGENESIS OF HEPATORENAL SYNDROME: BASIC MECHANISMS
The pathophysiological mechanism for HRS remains largely unknown but current understanding dictates that decreased glomerular filtration rate due to reduction of effective blood volume due to splanchnic vasodilation (so-called “splanchnic steal syndrome” [19]) and subsequently compensated with activation of renin-angiotensin-aldosterone and sympathetic nervous system, and release of antidiuretic hormone (ADH), which leads to profound renal afferent artery vasoconstriction (primarily affecting the renal cortex) [20, 21]. Patients with HRS have extremely low urinary sodium excretion due to decreased filtered sodium at glomerulus and increased sodium reabsorption in the proximal tubule (“excessive sodium reabsorption”). Diuretics such as furosemide and spironolactone have a little effect due to decreased delivery and low amount of sodium at the effector site (the loop of Henle and distal tubule). (Figure 1) In addition, a small amount of filtered water is absorbed at the distal tubules in response to high ADH activity leading to oliguria or anuria (“non-osmotic hyper secretion of ADH”) [22, 23].
FIGURE 1. Current Understandings of the Pathophysiology of Hepato-Renal Syndrome.
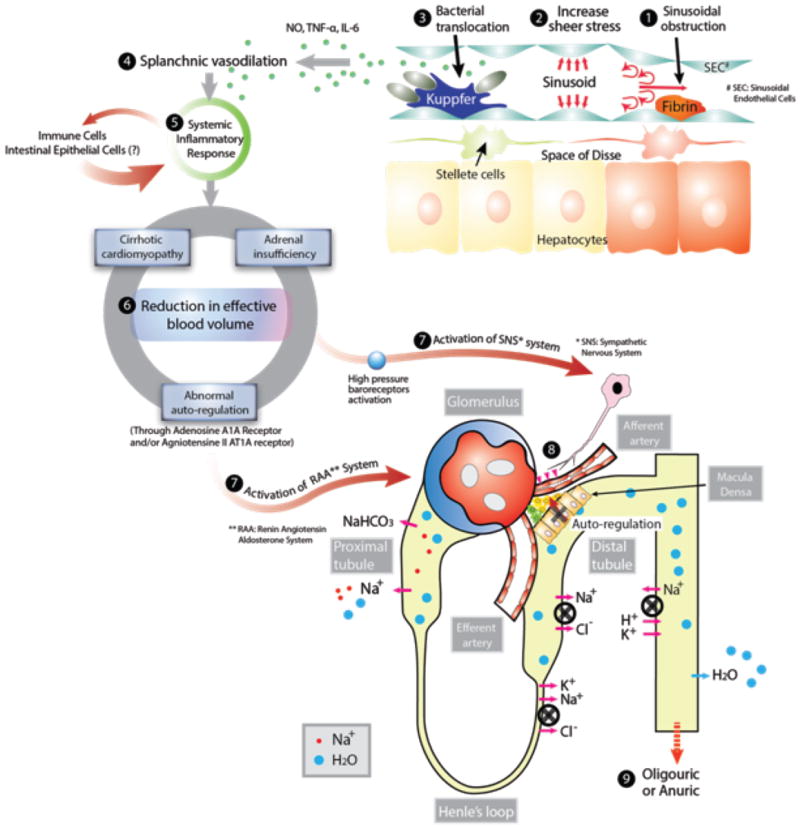
Development of hepato-renal syndrome are thought to occur through the following pathophysiological events; (1) Sinusoidal obstruction due to fibrin formation from stellate cells located in space of Disse following hepatocyte injury, (2) Development of portal hypertension and increased sheer stress to the portal vessel wall leading to increased production of nitric oxide, (3) Bacterial translocation from intestinal flora to the portal circulation activates innate immune system (e.g. mononuclear cell), leading to massive production of cytokines (e.g., TNF-α and IL-6) and nitric oxide leading to (4) Splanchnic vasodilation, (5) These inflammatory mediators causes systemic inflammatory response which further aggregates splanchnic vasodilation and extra-organ damage, (6) Reduction in effective blood volume due to splanchnic dilatation, (7) Activation of systemic vasoconstricting system including sympathetic nervous system and renin-angiotensin-aldosterone system, (8) renal afferent artery vasoconstriction, reduction of glomerular filtration and (8) dramatic reduction in renal function.
Currently splanchnic vasodilation is widely accepted as a key pathophysiologic change for HRS since it can reasonably explain the most of pathophysiologic study data. In early cirrhosis, hepatocyte inflammation activates hepatic stellate cells located in the peri-sinusoid tissue (space of Disse) to secrete collagen into hepatic sinusoids (scar formation) leading to increased portal venous resistance with the progression of liver damage. Increase in portal vascular resistance increases sheer stress on the venous wall of the portal and splanchnic system, and leads to massive production of vasodilators including nitric oxide (NO) from the vascular endothelial cells [24]. Increased sheer stress also causes the formation of the massive collateral network by opening preexisting vessels or by increased angiogenesis due to up-regulation of growth factors such as vascular endothelial growth factor and platelet derived growth factor [25, 26], which further aggregates the reduction of splanchnic vascular resistance [27, 28].
In addition to sheer stress, current evidences have been shown that bacterial translocation from intestinal flora to the portal circulation, which is commonly seen in cirrhotic patients with portal hypertension [29], may activate the innate immune system, leading to massive production of cytokines including tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6) [30, 31]. TNF-α and endotoxins from bacteria further increase the NO production from vascular endothelial cells by up-regulating the endothelial nitric oxide synthase and inducible nitric oxide synthase, and leads to splanchnic vasodilation [32]. In fact, cirrhotic patients with bacterial translocation have lower systemic vascular resistance compared with patients who do not [33]. Also intestinal decontamination with norfloxacin, decreases TNF-α production, alleviate hemodynamic parameters, and renal function [34, 35]. Those studies have indirectly provided the plausibility of bacterial translocation as a mechanism of splanchnic vasodilation in cirrhosis.
It is important to point out that splanchnic vasodilation itself is not sufficient for the development of HRS. Other simultaneous organ dysfunction precipitates in concert to develop HRS. The auto-regulation of renal blood flow in cirrhotic patients is right-shifted due to activation of the renal sympathetic nervous system and other vasoconstrictors [36]. Renal blood flow becomes more dependent on the arterial blood pressure with the progression of liver disease. In patients with advanced cirrhosis, small changes in perfusion pressure will result in a major decrease in renal blood flow. Myocardial pump function, which is required to maintain vital organ perfusion by counteracting the reduction in volume shift to splanchnic circulation, is also impaired, so-called “cirrhotic cardiomyopathy” [37-40]. The impairment to generate adequate cardiac output in response to decrease effective blood volume directly reduces the renal perfusion pressure, contributing to the development of HRS [37, 38]. Hypo-perfusion to adrenal gland may also cause glucocorticoid insufficiency, which leads to circulatory dysfunction and impaired response to vasopressors [41]. Previous study with 101 cirrhotic patients in the intensive care unit showed that adrenal insufficiency was associated with higher mortality with lower mean arterial pressure and higher requirement of vasopressors [42]. Furthermore, hydrocortisone administration rapidly improved systemic hemodynamics, reduced vasopressor requirements and lowered hospital mortality [43]. In summary the pathophysiologic mechanisms of HRS are currently understood that cirrhosis-induced splanchnic vasodilation is primary causative physiologic change and subsequent renal vasoconstriction causes hypo-perfusion of the kidney, which leads to HRS when accompanied by impairment of multiple organ function.
Although splanchnic vasodilation is currently a most plausible causative mechanism of HRS in cirrhosis, there are ample evidences have shown that progressive functional renal failure occurs with a wide variety of liver injury such as trauma, toxic drugs without cirrhosis or splanchnic vasodilation [44, 45]. Therefore it remains possible that liver injury itself directly or indirectly causes HRS. Most up-to date scientific evidences provide that systemic inflammatory response from liver injury (release of inflammatory mediators) causes remote organ injury as well as hemodynamic disturbances which mimics cirrhosis or sepsis. Those evolving new research areas will be discussed later in the new basic science research section.
CURRENT MANAGEMENT OF HEPATORENAL SYNDROME
Unfortunately treatment options for HRS are limited and mostly supportive which are intended to prolong survival of patients awaiting transplant. Those supportive treatments include prevention and immediate treatment for precipitating factors for HRS such as (i) paracentasis with albumin supplement for tense ascites, (ii) antibiotics for spontaneous bacterial peritonitis [9], (iii) avoidance of nephrotoxic medications (e.g. nonsteroidal anti-inflammatory drugs, aminoglycosides [46, 47], radiocontrast media [48], and diuretics), (iii) low-salt diet and free water restriction for patients with hyponatremia [49], (iv) considering cortisone replacement therapy for hypotensive patients [43].
Few pharmacologic agents are currently available to slow the progressive worsening the splanchnic vasodilation and often indicated only for the patients waiting for transplant. Terlipressin is a vasopressin analog with preferential effects on the vasopressin type 1 receptor in splanchnic vasculature, causing greater mesenteric vasoconstriction than in kidney or other organ vascular systems [50]. Currently terlipressin in combination with albumin infusion is the first line vasoconstrictor for HRS type 1 [51, 52] (not currently commercially available in the USA). However, terlipressin is only effective for patients who have mild kidney and liver dysfunction (serum bilirubin < 10mg/dL) [53]. Other vasoconstrictors such as oral midodrine, α-adrenergic receptor agonist, and subcutaneous octreotide, and a long-acting somatostatin analogue, are less effective compared to Terlipressin, and considered to be a second line of treatment. (Only indicated if Terlipressin is contraindicated or not available)
Non-pharmacologic treatments include renal replacement therapy, transjugular intrahepatic portosystemic shunt (TIPS), and liver transplantation. The effectiveness of renal replacement therapy such as hemodialysis or continuous veno-venous hemofiltration remained undetermined for HRS. Therefore renal replacement therapy is only indicated for rescue treatment of transplant candidates with severe hyperkalemia, metabolic acidosis, and volume overload rather as a treatment for HRS [54]. Transjugular intrahepatic portosystemic shunt (TIPS) can temporarily decompress the portal pressure, and decrease sympathetic nervous system and renin-angiotensin-aldosterone activity [55, 56]. However, TIPS acutely increases cardiac output and exaggerates peripheral vasodilatation, and is often indicated only for the patients with stable liver function as either bridge treatment for liver transplant or long-term treatment. Liver transplantation remains the best available treatment for suitable candidates with HRS because it may cure both liver and the renal dysfunction when it performed at an early stage. But this treatment option is limited by the availability of organs. Also patients with cirrhosis and renal failure, especially HRS type 1, are at high risk for death while awaiting transplantation [57]. In 2002, the Model for End-Stage Liver Disease (MELD) score, which incorporates kidney function to calculate the level of sickness of patients waiting for transplant, was introduced to facilitate the systematic organ allocation based on the “sickest first” paradigm for liver transplantation. Use of this scoring system has markedly increased the opportunity of transplantation for patients with renal failure and has aided in reducing mortality among patients awaiting liver transplantation.
NEW BASIC RESEARCH IN HEPATORENAL SYNDROME MECHANISMS
Near 30 years since Schrier and colleagues proposed the “Peripheral Arterial Vasodilatation Hypothesis” as an explanation for the abnormal renal sodium and water retention in patients with cirrhosis in 1988, splanchnic vasodilation has been believed to be a sole causative mechanism for hepato-renal syndrome in cirrhosis [24]. Vaso-constrictive pharmacologic treatments for HRS have been introduced to treat splanchnic vasodilation on this hypothetic ground with partial success. Recently new evidences have provided impetus for other novel mechanisms for HRS including “systemic inflammatory response” hypothesis. After liver ischemia and reperfusion injury, Kuppfer cells release pro-inflammatory mediators. (Figure 1) Significantly higher level of circulating pro-inflammatory cytokines and transcription factors including TNF-α, IL-1α, and IL-6 has been reported after reperfusion of liver [58-60]. Those mediators may promote inflammatory changes in remote organ including lung and kidney [59-61]. In addition, damaged hepatocytes release the intracellular components such as damage associated molecular pattern molecules (DAMPs). DAMPs are molecules released by stressed cells undergoing necrosis that act as endogenous danger signals to promote and exacerbate the inflammatory response. High mobility group box-1 (HMGB1) is a non-histone nuclear protein but functions as DAMPs under stress condition and promotes inflammation [62, 63]. High level of HMGB1 expression was observed in the hepatocytes after ischemia and reperfusion [64]. HMGB1 interacts with the individual members of the Toll-like receptor (TLR) family, TLR2 and TLR4 [65] in the remote organ and innate immune cells, which may also contribute to the HRS. Other inflammatory mediators include, circulating bile acids, uric acids, histones and nuclear DNA and circulating immune complexes may also contribute to the development of acute kidney injury [66]. Those hypotheses were recently supported by the finding that the observation that vascular endothelial cell damage which is shown by the large number of apoptotic cells in the kidney, which is more prominent than the proximal tubule cell apoptosis after liver reperfusion [67]. Pro-inflammatory mediators released into systemic circulation damage renal endothelial cells, and cause a loss of their ability to regulate leukocyte recruitment, leading to disruption of endothelial barrier [68, 69]. Activated neutrophils in response to those inflammatory factors migrate to the injured area guided by endothelial adhesion molecules such as E-selectin, P-selectin, and ICAM-1 in the kidney, which promotes leukocyte recruitment and extravasations to the renal interstitial space[69-72]. Therefore prevention of endothelial damage may improve the survival of renal-endothelial cells after liver ischemia-reperfusion may limit leukocyte infiltration into the kidney parenchyma and improve renal function. Future studies with endothelial stabilizing agents (such as activation of A1 adenosine receptors [73, 74], protein kinase C [75], HSP-27 [76], activated protein C or sphingosine-1-phosphate [77]) will determine whether protecting endothelial integrity will reduce renal injury after liver IR injury.
Another novel discovery of the mechanism of remote organ injury is the involvement of intestine as exacerbating sterile inflammation by releasing internally-stored IL-17A. (Figure 2) Recent spiral of discoveries was started from the initial finding that acute kidney injury in mice causes hepatic injury, which is mediated by inflammatory cytokines (TNF-α, IL-17A, and IL-6). Also portal vein and intestine had higher levels of interleukin 17A than peripheral blood. This finding suggested that IL-17A is derived from small intestine [78]. Contrary to the liver injury following kidney injury, authors found that ischemia-reperfusion injury of the liver cause acute kidney injury, which is also mediated by intestinal-derived IL-17A. High level of IL-17A mRNA was expressed particularly in the Paneth cell. Paneth cells are intestinal epithelial cell, located in the base of intestinal crypts and adjacent to stem cells, secretes bacteriocidal cationic proteins called defensins to protect intestinal epithelial barrier against bacteria. After a liver or kidney ischemia and reperfusion, Paneth cells also secrete large quantities of stored IL-17A, which may be attributed to the aggregation of inflammation at the primary site and also leading to remote organ injury. In fact, the depletion of Paneth cells ameliorates the inflammatory response to primary organ (e.g. liver), but also remote organ damage (e.g. kidney injury after liver ischemia-reperfusion injury). Based on those observations, it is plausible that hepatic and renal injury after hepatic ischemia-reperfusion injury is exacerbated by Paneth-cell derived IL-17A [79]. Exacerbated inflammatory response causes widespread endothelial damage and hemodynamic derangement, which further exacerbate liver dysfunction and also cause extrahepatic organ damage, and eventually draw patients into “downward spiral of inflammation” and multi-organ failure. (Figure 3) This “systemic inflammatory response” hypothesis may provide some clues to answer the question why kidney injury (HRS) occurs in various causes of liver injury without evidence of splanchnic vasodilation.
FIGURE 2. Mechanisms of Sensing of the Hepatocyte Damage and Amplification of Inflammation by Intestinal Epithelial Cells.
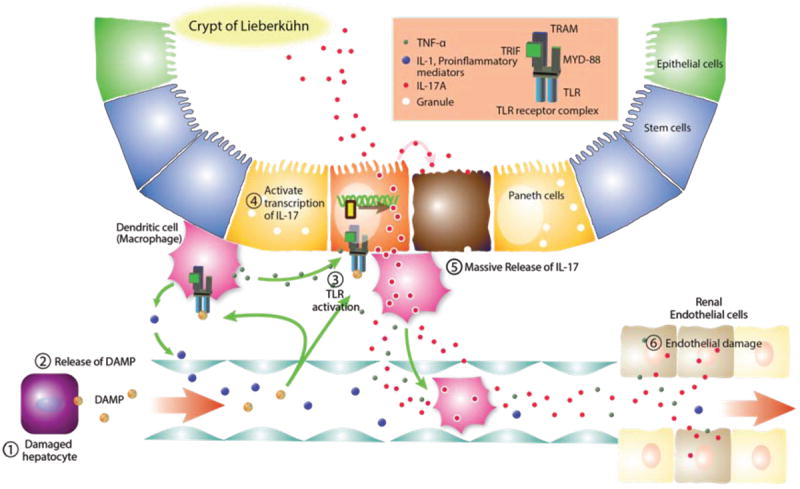
In cirrhosis, chronic ischemia and reperfusion of hepatocytes activates sinusoidal Kuppfer cells, leading to production of pro-inflammatory mediators such as TNF-α, IL-1α, and IL-6. Damaged hepatocytes alert immune system by releasing damage-associated molecular pattern molecules (DAMPs) such as High mobility group box-1 (HMGB1), histones, and uric acids into the systemic circulation. Those pro-inflammatory signals are sensed by intestinal immune system, in which reserve the largest population of immune cells. DAMPs may activate the toll-like receptor (TLR) family in the intestinal epithelial cells (e.g., Paneth cells) and innate immune cells (Dendrite cells). Binding of DMAPs to TLR receptor on the Dendrite cells triggers the production of pro-inflammatory mediators such as TNF-α and IL-6 and IL-1β. Also the binding of DAMPs to TLR receptors in Paneth cells triggers the release of stored IL-17A from Paneth cells. Abbreviations DAMP, Damage Associated Molecular Pattern, IL-1, interleukin-1, IL-17, interleukin-17, MYD-88, Myeloid differentiation primary response gene 88, TNF-α, Tumor Necrosis Factor-α, TLR, Toll Like Receptor, TRAM, TRIF-related adaptor molecule, TRIF, TIR-domain-containing adapter-inducing interferon-β
FIGURE 3. Systemic Inflammatory Response Hypothesis.
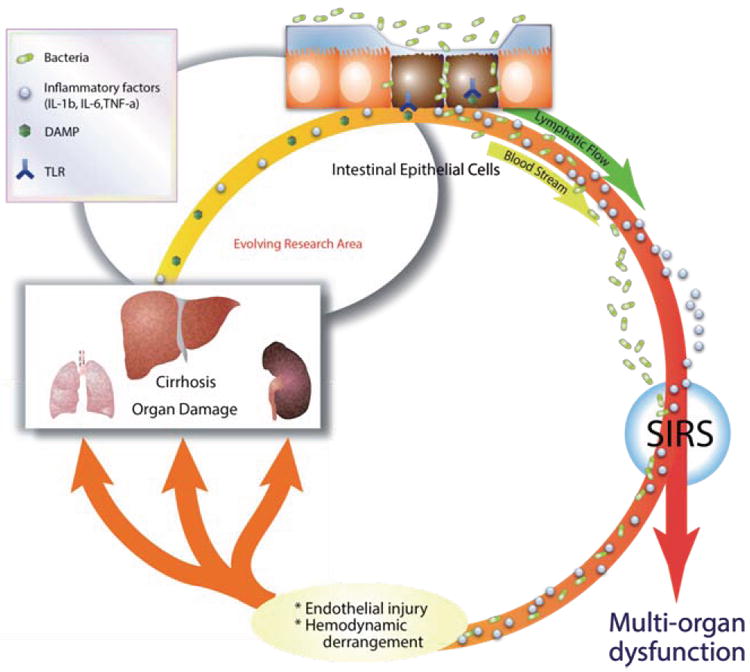
Inflammatory mediators released from cirrhotic liver into systemic circulation reach remote organs including lung, liver and kidney causing endothelial damage and further organ injury. In addition, disruption of intercellular integrity of intestinal epithelial cells associated with cirrhosis causes bacterial invasion into circulation leading to substantial increase in circulating bacterial lipopolysaccharide (LPS). These additional inflammatory mediators elicit endothelial damages in multiple organs (worsening of cirrhosis, development of kidney and lung injury). Damaged organs release additional DAMPs leading to hemodynamic derangements which further exacerbate the systemic inflammatory response, leading to malicious aggregating cycle of inflammation, which ultimately leads to multi-organ failure. Abbreviations: DAMP, Damage Associated Molecular Pattern, IL-1, interleukin-1, TNF-α, Tumor Necrosis Factor-α, SIRS, Systemic Inflammatory Response Syndrome
NEW BIOMARKERS OF HEPATORENAL SYNDROME
Clinical diagnosis of HRS remains difficult and diagnostic criteria warrant further refinement. Current international ascites club criteria requires urine sample to make a diagnosis and therefore cannot establish HRS diagnosis in oligouric or anuric patients. Also they cannot identify HRS superimposed on organic renal disease and in patients with rapidly evolving liver and renal failure[80]. An Italian multicenter study examined the applicability of these diagnostic criteria in a prospective trial[81]. Of the 116 patients diagnosed with HRS only 64% met all diagnostic criteria, whereas the remaining 36% with acute deterioration of serum creatinine to above 1.5 mg/dL could not meet one or more of the diagnostic criteria due to anuria, hematuria, or proteinuria. This is due to a lack of sensitive clinical biomarker to accurately and quickly estimate glomerular filtration rate (GFR)9,123 and to exclude other etiology of kidney failure.
Cystatin C (Cys-C) level is one of the serum markers for GFR and is under investigation for clinical use. Cys-C freely crosses the glomerular membrane and metabolized in the renal proximal tubular cells without extra-renal elimination[82]. Also unlike creatinine, Cys-C levels are independent of muscle mass, age and sex, and are not influenced by inflammatory conditions or malignancy[82]. Serum Cys-C provides a good alternative to serum creatinine for the assessment of glomerular function in cirrhosis.
Another good alternative to creatinine, and has been evaluated for clinical use, is urinary NGAL. NGAL (neutrophil gelatinase-associated lipocalin, lipocalin-2, siderocalin) is expressed by the damaged tubule to induce re-epithelialization and secreted in high levels into the blood and urine within two hours of injury[83]. A recent study examined the urinary NGAL in 118 patients with cirrhosis[84]. 56% of those patients had a kidney failure from a variety of causes consisting chronic kidney disease (12%), prerenal azotemia (14%), HRS (17%) and AKI (13%). Urinary NGAL levels were significantly different amongst AKI (highest), HRS (intermediate), prerenal azotemia, chronic kidney disease, and normal kidney function. Therefore it may be used to differentiate HRS from other causes of renal failure. This research also showed its ability to predict mortality in patients who have cirrhosis and kidney failure[84]. However, interpretation of NGAL is affected by concomitant systemic infection. Also uninary NGAL is difficult to measure in oligouric or anuric patients.
Other urinary biomarkers being investigated for detection of renal function include γ-glutamyl transpeptidase, transaminases, liver-type fatty acid binding protein, IL-18 and hepatitis A virus cellular receptor-1 but these markers warrants further investigation in cirrhotic patients. Markers of the severity of renal afferent artery vasoconstriction have been suggested for the diagnosis of HRS. These markers include sympathetic nervous system (SNS) activity (plasma noradrenaline level) or renin-angiotensin-aldosterone (RAA) activity (plasma renin activity)[10], or renal arterial resistance (“renal artery resistive index” in ultrasonography)[85].
CONCLUSIONS AND FUTURE IMPLICATIONS
In this brief review, we summarized the current understanding of pathophysiological mechanisms, therapeutic approach and evolving pathophysiologic mechanism of HRS. Collecting the currently available evidences together, the pathophysiology of HRS has been recognized as more complex rather than solely due to splanchnic vasodilation. Splanchnic vasodilation, intestinal barrier disruption and bacterial translocation, and exacerbation of systemic inflammatory response may work in concert to develop this detrimental condition, HRS. Understanding of pathophysiological mechanisms will enable more physiology-oriented therapeutic approach (such as pharmacological therapy and gene modulation targeted at these signaling molecules to improve liver/kidney function and delay the development of HRS) other than supportive therapy, which is expected to reduce morbidity and mortality of patients with HRS, and significantly reduce the medical care cost related to HRS. Future research will elucidate the mechanism of development of the systemic inflammatory response syndrome (SIRS) after liver ischemia-reperfusion injury, which includes the type of inflammatory mediators other than mediators discussed in the previous section, and the origin of those mediators (necrotic hepatocytes, Kuppfer cells, endothelial cells, etc.). Although hepatic sinusoidal Kuppfer cells are the largest sessile macrophage in the body, and large number of hepatic endothelial cells exists in the hepatic microcirculation, it is completely unknown whether the mediators from Kuppfer cells or hepatic endothelial cells are enough to produce sustained and powerful systemic inflammatory response to develop HRS. Role of intestine in sterile inflammation is evolving research area and may play an important role in transmitting the local inflammation to systemic inflammation, which leads to the development of remote organ injury including HRS. The mechanisms of renal injury from the systemic inflammatory syndrome, especially the role of endothelial cell needs to be elucidated.
Acknowledgments
DISCLOSURE OF FUNDING:
This work was supported by Department of Anesthesiology, College of Physicians and Surgeons of Columbia University and by the National Institutes of Health Grants RO1-GM-067081 and T32-GM00846.
Footnotes
CONFLICT OF INTEREST:
No relevant conflicts of interest were declared for each author.
References
- 1.Quincke H, Nothnagel H. Specielle Pathologie und Therapie. xviii. Vienna: 1899. p. 63. [Google Scholar]
- 2.F A. Clinical report on hydro-peritoneum, based on analysis of forty-six cases. Am J Med Sci. 1863;45:306–39. [Google Scholar]
- 3.Hecker R, Sherlock S. Electrolyte and circulatory changes in terminal liver failure. Lancet. 1956;271(6953):1121–5. doi: 10.1016/s0140-6736(56)90149-0. [DOI] [PubMed] [Google Scholar]
- 4.Koppel MH, Coburn JW, Mims MM, Goldstein H, Boyle JD, Rubini ME. Transplantation of cadaveric kidneys from patients with hepatorenal syndrome Evidence for the functionalnature of renal failure in advanced liver disease. N Engl J Med. 1969;280(25):1367–71. doi: 10.1056/NEJM196906192802501. [DOI] [PubMed] [Google Scholar]
- 5.Schroeder ET, Shear L, Sancetta SM, Gabuzda GJ. Renal failure in patients with cirrhosis of the liver. 3. Evaluation of intrarenal blood flow by para-aminohippurate extraction and response to angiotensin. Am J Med. 1967;43(6):887–96. doi: 10.1016/0002-9343(67)90247-1. [DOI] [PubMed] [Google Scholar]
- 6.Gines P, Guevara M, Arroyo V, Rodes J. Hepatorenal syndrome. Lancet. 2003;362(9398):1819–27. doi: 10.1016/S0140-6736(03)14903-3. [DOI] [PubMed] [Google Scholar]
- 7.Arroyo V, Gines P, Gerbes AL, Dudley FJ, Gentilini P, Laffi G, et al. Definition and diagnostic criteria of refractory ascites and hepatorenal syndrome in cirrhosis. International Ascites Club. Hepatology. 1996;23(1):164–76. doi: 10.1002/hep.510230122. [DOI] [PubMed] [Google Scholar]
- 8.Salerno F, Gerbes A, Gines P, Wong F, Arroyo V. Diagnosis, prevention and treatment of hepatorenal syndrome in cirrhosis. Gut. 2007;56(9):1310–8. doi: 10.1136/gut.2006.107789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.European Association for the Study of the L. EASL clinical practice guidelines on the management of ascites, spontaneous bacterial peritonitis, and hepatorenal syndrome in cirrhosis. J Hepatol. 2010;53(3):397–417. doi: 10.1016/j.jhep.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 10.Gines A, Escorsell A, Gines P, Salo J, Jimenez W, Inglada L, et al. Incidence, predictive factors, and prognosis of the hepatorenal syndrome in cirrhosis with ascites. Gastroenterology. 1993;105(1):229–36. doi: 10.1016/0016-5085(93)90031-7. [DOI] [PubMed] [Google Scholar]
- 11.Follo A, Llovet JM, Navasa M, Planas R, Forns X, Francitorra A, et al. Renal impairment after spontaneous bacterial peritonitis in cirrhosis: incidence, clinical course, predictive factors and prognosis. Hepatology. 1994;20(6):1495–501. doi: 10.1002/hep.1840200619. [DOI] [PubMed] [Google Scholar]
- 12.Gines P, Schrier RW. Renal failure in cirrhosis. N Engl J Med. 2009;361(13):1279–90. doi: 10.1056/NEJMra0809139. [DOI] [PubMed] [Google Scholar]
- 13.Wadei HM, Gonwa TA. Hepatorenal syndrome in the intensive care unit. J Intensive Care Med. 2013;28(2):79–92. doi: 10.1177/0885066611408692. [DOI] [PubMed] [Google Scholar]
- 14.Sort P, Navasa M, Arroyo V, Aldeguer X, Planas R, Ruiz-del-Arbol L, et al. Effect of intravenous albumin on renal impairment and mortality in patients with cirrhosis and spontaneous bacterial peritonitis. N Engl J Med. 1999;341(6):403–9. doi: 10.1056/NEJM199908053410603. [DOI] [PubMed] [Google Scholar]
- 15.Quiroz ME, Flores YN, Aracena B, Granados-Garcia V, Salmeron J, Perez R, et al. Estimating the cost of treating patients with liver cirrhosis at the Mexican Social Security Institute. Salud Publica Mex. 2010;52(6):493–501. [PubMed] [Google Scholar]
- 16.Levin J. Disease burden in patients with chronic hepatitis C virus (HCV) infection in a United States (US) private health insurance claims database analysis from 2003 to 2010. [2013];2011 Available from: http://www.natap.org/2011/AASLD/AASLD_48.htm.
- 17.NIDDK. Digestive diseases in the United States: Epidemiology and Impact. NIH; Bethesda, MD: 1994. p. 1447. [Google Scholar]
- 18.Moreau R, Durand F, Poynard T, Duhamel C, Cervoni JP, Ichai P, et al. Terlipressin in patients with cirrhosis and type 1 hepatorenal syndrome: a retrospective multicenter study. Gastroenterology. 2002;122(4):923–30. doi: 10.1053/gast.2002.32364. [DOI] [PubMed] [Google Scholar]
- 19.Moller S, Hobolth L, Winkler C, Bendtsen F, Christensen E. Determinants of the hyperdynamic circulation and central hypovolaemia in cirrhosis. Gut. 2011;60(9):1254–9. doi: 10.1136/gut.2010.235473. [DOI] [PubMed] [Google Scholar]
- 20.Sacerdoti D, Bolognesi M, Merkel C, Angeli P, Gatta A. Renal vasoconstriction in cirrhosis evaluated by duplex Doppler ultrasonography. Hepatology. 1993;17(2):219–24. [PubMed] [Google Scholar]
- 21.Maroto A, Gines A, Salo J, Claria J, Gines P, Anibarro L, et al. Diagnosis of functional kidney failure of cirrhosis with Doppler sonography: prognostic value of resistive index. Hepatology. 1994;20(4 Pt 1):839–44. doi: 10.1002/hep.1840200411. [DOI] [PubMed] [Google Scholar]
- 22.Linas SL, Anderson RJ, Guggenheim SJ, Robertson GL, Berl T. Role of vasopressin in impaired water excretion in conscious rats with experimental cirrhosis. Kidney Int. 1981;20(2):173–80. doi: 10.1038/ki.1981.119. [DOI] [PubMed] [Google Scholar]
- 23.Bichet DG, Van Putten VJ, Schrier RW. Potential role of increased sympathetic activity in impaired sodium and water excretion in cirrhosis. N Engl J Med. 1982;307(25):1552–7. doi: 10.1056/NEJM198212163072504. [DOI] [PubMed] [Google Scholar]
- 24.Schrier RW, Arroyo V, Bernardi M, Epstein M, Henriksen JH, Rodes J. Peripheral arterial vasodilation hypothesis: a proposal for the initiation of renal sodium and water retention in cirrhosis. Hepatology. 1988;8(5):1151–7. doi: 10.1002/hep.1840080532. [DOI] [PubMed] [Google Scholar]
- 25.Van Steenkiste C, Geerts A, Vanheule E, Van Vlierberghe H, De Vos F, Olievier K, et al. Role of placental growth factor in mesenteric neoangiogenesis in a mouse model of portal hypertension. Gastroenterology. 2009;137(6):2112–24 e1-6. doi: 10.1053/j.gastro.2009.08.068. [DOI] [PubMed] [Google Scholar]
- 26.Paternostro C, David E, Novo E, Parola M. Hypoxia, angiogenesis and liver fibrogenesis in the progression of chronic liver diseases. World J Gastroenterol. 2010;16(3):281–8. doi: 10.3748/wjg.v16.i3.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Langer DA, Shah VH. Nitric oxide and portal hypertension: interface of vasoreactivity and angiogenesis. J Hepatol. 2006;44(1):209–16. doi: 10.1016/j.jhep.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 28.Sumanovski LT, Battegay E, Stumm M, van der Kooij M, Sieber CC. Increased angiogenesis in portal hypertensive rats: role of nitric oxide. Hepatology. 1999;29(4):1044–9. doi: 10.1002/hep.510290436. [DOI] [PubMed] [Google Scholar]
- 29.Bellot P, Frances R, Such J. Pathological bacterial translocation in cirrhosis: pathophysiology, diagnosis and clinical implications. Liver Int. 2013;33(1):31–9. doi: 10.1111/liv.12021. [DOI] [PubMed] [Google Scholar]
- 30.Sugano S. Endotoxin levels in cirrhotic rats with sterile and infected ascites. Gastroenterol Jpn. 1992;27(3):348–53. doi: 10.1007/BF02777753. [DOI] [PubMed] [Google Scholar]
- 31.Heller J, Sogni P, Barriere E, Tazi KA, Chauvelot-Moachon L, Guimont MC, et al. Effects of lipopolysaccharide on TNF-alpha production, hepatic NOS2 activity, and hepatic toxicity in rats with cirrhosis. J Hepatol. 2000;33(3):376–81. doi: 10.1016/s0168-8278(00)80272-x. [DOI] [PubMed] [Google Scholar]
- 32.Wiest R, Das S, Cadelina G, Garcia-Tsao G, Milstien S, Groszmann RJ. Bacterial translocation in cirrhotic rats stimulates eNOS-derived NO production and impairs mesenteric vascular contractility. J Clin Invest. 1999;104(9):1223–33. doi: 10.1172/JCI7458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frances R, Zapater P, Gonzalez-Navajas JM, Munoz C, Cano R, Moreu R, et al. Bacterial DNA in patients with cirrhosis and noninfected ascites mimics the soluble immune response established in patients with spontaneous bacterial peritonitis. Hepatology. 2008;47(3):978–85. doi: 10.1002/hep.22083. [DOI] [PubMed] [Google Scholar]
- 34.Tazi KA, Moreau R, Herve P, Dauvergne A, Cazals-Hatem D, Bert F, et al. Norfloxacin reduces aortic NO synthases and proinflammatory cytokine up-regulation in cirrhotic rats: role of Akt signaling. Gastroenterology. 2005;129(1):303–14. doi: 10.1053/j.gastro.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 35.Fernandez J, Navasa M, Planas R, Montoliu S, Monfort D, Soriano G, et al. Primary prophylaxis of spontaneous bacterial peritonitis delays hepatorenal syndrome and improves survival in cirrhosis. Gastroenterology. 2007;133(3):818–24. doi: 10.1053/j.gastro.2007.06.065. [DOI] [PubMed] [Google Scholar]
- 36.Stadlbauer V, Wright GA, Banaji M, Mukhopadhya A, Mookerjee RP, Moore K, et al. Relationship between activation of the sympathetic nervous system and renal blood flow autoregulation in cirrhosis. Gastroenterology. 2008;134(1):111–9. doi: 10.1053/j.gastro.2007.10.055. [DOI] [PubMed] [Google Scholar]
- 37.Ruiz-del-Arbol L, Urman J, Fernandez J, Gonzalez M, Navasa M, Monescillo A, et al. Systemic, renal, and hepatic hemodynamic derangement in cirrhotic patients with spontaneous bacterial peritonitis. Hepatology. 2003;38(5):1210–8. doi: 10.1053/jhep.2003.50447. [DOI] [PubMed] [Google Scholar]
- 38.Ruiz-del-Arbol L, Monescillo A, Arocena C, Valer P, Gines P, Moreira V, et al. Circulatory function and hepatorenal syndrome in cirrhosis. Hepatology. 2005;42(2):439–47. doi: 10.1002/hep.20766. [DOI] [PubMed] [Google Scholar]
- 39.Arroyo V, Fernandez J, Gines P. Pathogenesis and treatment of hepatorenal syndrome. Semin Liver Dis. 2008;28(1):81–95. doi: 10.1055/s-2008-1040323. [DOI] [PubMed] [Google Scholar]
- 40.Alqahtani SA, Fouad TR, Lee SS. Cirrhotic cardiomyopathy. Semin Liver Dis. 2008;28(1):59–69. doi: 10.1055/s-2008-1040321. [DOI] [PubMed] [Google Scholar]
- 41.Cooper MS, Stewart PM. Corticosteroid insufficiency in acutely ill patients. N Engl J Med. 2003;348(8):727–34. doi: 10.1056/NEJMra020529. [DOI] [PubMed] [Google Scholar]
- 42.Tsai MH, Peng YS, Chen YC, Liu NJ, Ho YP, Fang JT, et al. Adrenal insufficiency in patients with cirrhosis, severe sepsis and septic shock. Hepatology. 2006;43(4):673–81. doi: 10.1002/hep.21101. [DOI] [PubMed] [Google Scholar]
- 43.Fernandez J, Escorsell A, Zabalza M, Felipe V, Navasa M, Mas A, et al. Adrenal insufficiency in patients with cirrhosis and septic shock: Effect of treatment with hydrocortisone on survival. Hepatology. 2006;44(5):1288–95. doi: 10.1002/hep.21352. [DOI] [PubMed] [Google Scholar]
- 44.Helwig F, Schutz C. A further contribution to the liver-kidney syndrome. J Lab and Clin Med. 1935;21:264. [Google Scholar]
- 45.Wilensky A. Occurrence, Distribution and Pathogenesis of So-Called Liver Death and/or the Hepatorenal Syndrome. Arch Surg. 1939;38:625. [Google Scholar]
- 46.Boyer TD, Zia P, Reynolds TB. Effect of indomethacin and prostaglandin A1 on renal function and plasma renin activity in alcoholic liver disease. Gastroenterology. 1979;77(2):215–22. [PubMed] [Google Scholar]
- 47.Hampel H, Bynum GD, Zamora E, El-Serag HB. Risk factors for the development of renal dysfunction in hospitalized patients with cirrhosis. Am J Gastroenterol. 2001;96(7):2206–10. doi: 10.1111/j.1572-0241.2001.03958.x. [DOI] [PubMed] [Google Scholar]
- 48.Guevara M, Fernandez-Esparrach G, Alessandria C, Torre A, Terra C, Montana X, et al. Effects of contrast media on renal function in patients with cirrhosis: a prospective study. Hepatology. 2004;40(3):646–51. doi: 10.1002/hep.20373. [DOI] [PubMed] [Google Scholar]
- 49.Cardenas A, Gines P. Pathogenesis and treatment of fluid and electrolyte imbalance in cirrhosis. Semin Nephrol. 2001;21(3):308–16. doi: 10.1053/snep.2001.21666. [DOI] [PubMed] [Google Scholar]
- 50.Moller S, Hansen EF, Becker U, Brinch K, Henriksen JH, Bendtsen F. Central and systemic haemodynamic effects of terlipressin in portal hypertensive patients. Liver. 2000;20(1):51–9. doi: 10.1034/j.1600-0676.2000.020001051.x. [DOI] [PubMed] [Google Scholar]
- 51.Davenport A, Ahmad J, Al-Khafaji A, Kellum JA, Genyk YS, Nadim MK. Medical management of hepatorenal syndrome. Nephrol Dial Transplant. 2012;27(1):34–41. doi: 10.1093/ndt/gfr736. [DOI] [PubMed] [Google Scholar]
- 52.Uriz J, Gines P, Cardenas A, Sort P, Jimenez W, Salmeron JM, et al. Terlipressin plus albumin infusion: an effective and safe therapy of hepatorenal syndrome. J Hepatol. 2000;33(1):43–8. doi: 10.1016/s0168-8278(00)80158-0. [DOI] [PubMed] [Google Scholar]
- 53.Nazar A, Pereira GH, Guevara M, Martin-Llahi M, Pepin MN, Marinelli M, et al. Predictors of response to therapy with terlipressin and albumin in patients with cirrhosis and type 1 hepatorenal syndrome. Hepatology. 2010;51(1):219–26. doi: 10.1002/hep.23283. [DOI] [PubMed] [Google Scholar]
- 54.Wadei HM. Hepatorenal syndrome: a critical update. Semin Respir Crit Care Med. 2012;33(1):55–69. doi: 10.1055/s-0032-1301735. [DOI] [PubMed] [Google Scholar]
- 55.Somberg KA, Lake JR, Tomlanovich SJ, LaBerge JM, Feldstein V, Bass NM. Transjugular intrahepatic portosystemic shunts for refractory ascites: assessment of clinical and hormonal response and renal function. Hepatology. 1995;21(3):709–16. [PubMed] [Google Scholar]
- 56.Quiroga J, Sangro B, Nunez M, Bilbao I, Longo J, Garcia-Villarreal L, et al. Transjugular intrahepatic portal-systemic shunt in the treatment of refractory ascites: effect on clinical, renal, humoral, and hemodynamic parameters. Hepatology. 1995;21(4):986–94. [PubMed] [Google Scholar]
- 57.Ng CK, Chan MH, Tai MH, Lam CW. Hepatorenal syndrome. Clin Biochem Rev. 2007;28(1):11–7. [PMC free article] [PubMed] [Google Scholar]
- 58.Wanner GA, Ertel W, Muller P, Hofer Y, Leiderer R, Menger MD, et al. Liver ischemia and reperfusion induces a systemic inflammatory response through Kupffer cell activation. Shock. 1996;5(1):34–40. doi: 10.1097/00024382-199601000-00008. [DOI] [PubMed] [Google Scholar]
- 59.Tsung A, Hoffman RA, Izuishi K, Critchlow ND, Nakao A, Chan MH, et al. Hepatic ischemia/reperfusion injury involves functional TLR4 signaling in nonparenchymal cells. J Immunol. 2005;175(11):7661–8. doi: 10.4049/jimmunol.175.11.7661. [DOI] [PubMed] [Google Scholar]
- 60.Levy RM, Mollen KP, Prince JM, Kaczorowski DJ, Vallabhaneni R, Liu S, et al. Systemic inflammation and remote organ injury following trauma require HMGB1. Am J Physiol Regul Integr Comp Physiol. 2007;293(4):R1538–44. doi: 10.1152/ajpregu.00272.2007. [DOI] [PubMed] [Google Scholar]
- 61.Tanaka Y, Maher JM, Chen C, Klaassen CD. Hepatic ischemia-reperfusion induces renal heme oxygenase-1 via NF-E2-related factor 2 in rats and mice. Mol Pharmacol. 2007;71(3):817–25. doi: 10.1124/mol.106.029033. [DOI] [PubMed] [Google Scholar]
- 62.Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285(5425):248–51. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 63.Andersson U, Wang H, Palmblad K, Aveberger AC, Bloom O, Erlandsson-Harris H, et al. High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J Exp Med. 2000;192(4):565–70. doi: 10.1084/jem.192.4.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT, et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med. 2005;201(7):1135–43. doi: 10.1084/jem.20042614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Ishizaka A, et al. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem. 2004;279(9):7370–7. doi: 10.1074/jbc.M306793200. [DOI] [PubMed] [Google Scholar]
- 66.Davis CL, Gonwa TA, Wilkinson AH. Pathophysiology of renal disease associated with liver disorders: implications for liver transplantation. Part I. Liver Transpl. 2002;8(2):91–109. doi: 10.1053/jlts.2002.31516. [DOI] [PubMed] [Google Scholar]
- 67.Lee HT, Park SW, Kim M, D’Agati VD. Acute kidney injury after hepatic ischemia and reperfusion injury in mice. Lab Invest. 2009;89(2):196–208. doi: 10.1038/labinvest.2008.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Teoh NC, Farrell GC. Hepatic ischemia reperfusion injury: pathogenic mechanisms and basis for hepatoprotection. J Gastroenterol Hepatol. 2003;18(8):891–902. doi: 10.1046/j.1440-1746.2003.03056.x. [DOI] [PubMed] [Google Scholar]
- 69.Sutton TA, Mang HE, Campos SB, Sandoval RM, Yoder MC, Molitoris BA. Injury of the renal microvascular endothelium alters barrier function after ischemia. Am J Physiol Renal Physiol. 2003;285(2):F191–8. doi: 10.1152/ajprenal.00042.2003. [DOI] [PubMed] [Google Scholar]
- 70.Molitoris BA, Sandoval R, Sutton TA. Endothelial injury and dysfunction in ischemic acute renal failure. Crit Care Med. 2002;30(5 Suppl):S235–40. doi: 10.1097/00003246-200205001-00011. [DOI] [PubMed] [Google Scholar]
- 71.Okusa MD. The inflammatory cascade in acute ischemic renal failure. Nephron. 2002;90(2):133–8. doi: 10.1159/000049032. [DOI] [PubMed] [Google Scholar]
- 72.Klausner JM, Paterson IS, Goldman G, Kobzik L, Rodzen C, Lawrence R, et al. Postischemic renal injury is mediated by neutrophils and leukotrienes. Am J Physiol. 1989;256(5 Pt 2):F794–802. doi: 10.1152/ajprenal.1989.256.5.F794. [DOI] [PubMed] [Google Scholar]
- 73.Park SW, Chen SW, Kim M, D’Agati VD, Lee HT. Selective intrarenal human A1 adenosine receptor overexpression reduces acute liver and kidney injury after hepatic ischemia reperfusion in mice. Lab Invest. 2010;90(3):476–95. doi: 10.1038/labinvest.2009.143. [DOI] [PubMed] [Google Scholar]
- 74.Park SW, Chen SW, Kim M, Brown KM, D’Agati VD, Lee HT. Protection against acute kidney injury via A(1) adenosine receptor-mediated Akt activation reduces liver injury after liver ischemia and reperfusion in mice. J Pharmacol Exp Ther. 2010;333(3):736–47. doi: 10.1124/jpet.110.166884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Park SW, Chen SW, Kim M, D’Agati VD, Lee HT. Human activated protein C attenuates both hepatic and renal injury caused by hepatic ischemia and reperfusion injury in mice. Kidney Int. 2009;76(7):739–50. doi: 10.1038/ki.2009.255. [DOI] [PubMed] [Google Scholar]
- 76.Park SW, Chen SW, Kim M, D’Agati VD, Lee HT. Human heat shock protein 27-overexpressing mice are protected against acute kidney injury after hepatic ischemia and reperfusion. Am J Physiol Renal Physiol. 2009;297(4):F885–94. doi: 10.1152/ajprenal.00317.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Park SW, Kim M, Chen SW, Brown KM, D’Agati VD, Lee HT. Sphinganine-1-phosphate protects kidney and liver after hepatic ischemia and reperfusion in mice through S1P1 receptor activation. Lab Invest. 2010;90(8):1209–24. doi: 10.1038/labinvest.2010.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Park SW, Chen SW, Kim M, Brown KM, Kolls JK, D’Agati VD, et al. Cytokines induce small intestine and liver injury after renal ischemia or nephrectomy. Lab Invest. 2011;91(1):63–84. doi: 10.1038/labinvest.2010.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Park SW, Kim M, Brown KM, D’Agati VD, Lee HT. Paneth cell-derived interleukin-17A causes multiorgan dysfunction after hepatic ischemia and reperfusion injury. Hepatology. 2011;53(5):1662–75. doi: 10.1002/hep.24253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wong F, Nadim MK, Kellum JA, Salerno F, Bellomo R, Gerbes A, et al. Working Party proposal for a revised classification system of renal dysfunction in patients with cirrhosis. Gut. 2011;60(5):702–9. doi: 10.1136/gut.2010.236133. [DOI] [PubMed] [Google Scholar]
- 81.Salerno F, Cazzaniga M, Merli M, Spinzi G, Saibeni S, Salmi A, et al. Diagnosis, treatment and survival of patients with hepatorenal syndrome: a survey on daily medical practice. J Hepatol. 2011;55(6):1241–8. doi: 10.1016/j.jhep.2011.03.012. [DOI] [PubMed] [Google Scholar]
- 82.Randers E, Erlandsen EJ. Serum cystatin C as an endogenous marker of the renal function--a review. Clin Chem Lab Med. 1999;37(4):389–95. doi: 10.1515/CCLM.1999.064. [DOI] [PubMed] [Google Scholar]
- 83.Bennett M, Dent CL, Ma Q, Dastrala S, Grenier F, Workman R, et al. Urine NGAL predicts severity of acute kidney injury after cardiac surgery: a prospective study. Clin J Am Soc Nephrol. 2008;3(3):665–73. doi: 10.2215/CJN.04010907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Verna EC, Brown RS, Farrand E, Pichardo EM, Forster CS, Sola-Del Valle DA, et al. Urinary neutrophil gelatinase-associated lipocalin predicts mortality and identifies acute kidney injury in cirrhosis. Dig Dis Sci. 2012;57(9):2362–70. doi: 10.1007/s10620-012-2180-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Platt JF, Ellis JH, Rubin JM, Merion RM, Lucey MR. Renal duplex Doppler ultrasonography: a noninvasive predictor of kidney dysfunction and hepatorenal failure in liver disease. Hepatology. 1994;20(2):362–9. [PubMed] [Google Scholar]