

Abstract
Eukaryotic organelles can interact with each other through stable junctions where the two membranes are kept in close apposition. The junction that connects the endoplasmic reticulum to the plasma membrane (ER-PM junction) is unique in providing a direct communication link between the ER and the PM. In a recently discovered signaling process, STIM (stromal-interacting molecule) proteins sense a drop in ER Ca2+ levels and directly activate Orai PM Ca2+ channels across the junction space. In an inverse process, a voltage-gated PM Ca2+ channel can directly open ER ryanodine-receptor Ca2+ channels in striated-muscle cells. Although ER-PM junctions were first described 50 years ago, their broad importance in Ca2+ signaling, as well as in the regulation of cholesterol and phosphatidylinositol lipid transfer, has only recently been realized. Here, we discuss research from different fields to provide a broad perspective on the structures and unique roles of ER-PM junctions in controlling signaling and metabolic processes.
Keywords: ER-PM junction, plasma-membrane-associated membrane (PAM), triad, dyad, Ca2+, lipid-transfer proteins
INTRODUCTION
The endoplasmic reticulum (ER) was first described in 1945 by K. R. Porter, who discovered in electron microscope images a new structure in the cytoplasm that he called “a lace-like reticulum” (1). It has since been shown that the ER consists of sacs and tubules that extend across the cell; connect to the nuclear envelope; and form a dynamic, constantly reorganizing, continuous membrane that separates the ER lumen from the cytosol (2). The ER is necessary for key cell functions including membrane and secreted-protein synthesis, lipid metabolism, and protein sorting. Most enzymes involved in these processes are exclusively localized in the ER, placing this organelle at the center of a cell’s lipid synthesis and sorting system (3). In addition, the ER also functions in animal cells as the primary storage site for intracellular Ca2+ (4) that can be released by receptor stimulation to generate Ca2+ signals (5).
How is this network of sacs and cisternae organized to perform its functions? Electron microscopy studies defined specialized regions including rough, smooth, and transitional ER. Most of the rough ER is localized around the nucleus as a continuation of the external nuclear membrane. It is the primary place where membrane and secreted proteins are synthesized. The rough microscopic appearance reflects the presence of membrane-associated ribosomes. In contrast, smooth ER is characterized by a much lower density of ribosomes and extends more uniformly across the cytosolic space. Smooth ER can be markedly expanded in cells where lipid synthesis or Ca2+ regulation are prominent, such as cells that synthesize steroid-related hormones or muscle cells. This suggests that smooth ER has important lipid metabolism and Ca2+-storage roles. The transitional ER, also smooth in appearance, localizes close to the Golgi apparatus and its main function is to transfer the proteins synthesized in the ER to the Golgi where they will be modified (6, 7).
Vesicular trafficking has long been believed to be the main mechanism by which the ER communicates with other organelles, but recent studies show that close juxtaposition of ER membranes with mitochondria, plasma membrane (PM), Golgi, and endosomes can facilitate the transfer of lipids or mediate signaling functions (these have been termed membrane contact sites) (8–12). This review focuses on one of the most intriguing of these sites where the ER comes into close contact with the PM. It has only recently been recognized that these contact sites are ubiquitous cellular structures in eukaryotes with important roles in Ca2+ signaling, lipid transfer, and other processes.
Our review begins with an historical discussion and definition of ER-PM junctions and then focuses on specific examples. We particularly discuss the role of ER-PM junctions in STIM (stromal-interacting molecule)-Orai-mediated “inside-out” Ca2+ signaling, in striated-muscle excitation-contraction coupling, in directional ergosterol lipid transfer from the ER to the PM in yeast, in phosphatidylinositol (PtdIns) lipid transfer in Drosophila photoreceptor cells, as well as in the creation of plasmodesmata cell-cell communication structures in plants. Finally, we discuss a conserved targeting mechanism that localizes ER proteins such as STIM1 to ER-PM junctions, summarize key ER-PM junction features, and highlight important open questions about the structure and function of ER-PM junctions.
AN HISTORICAL PERSPECTIVE
The existence of contacts between the ER and the PM was first reported in 1957 in muscle (13) and a few years later in neurons (14). In both cases, the contacts were identified as structures in which the ER and PM were kept in close proximity to generate a small gap filled by electrodense cytosol. Because both muscle cells and neurons are electrically excitable and Ca2+ release from ER stores is of special importance in these cells, many researchers considered these contacts to be unique features of specialized cells.
A more ubiquitous relevance of contacts between the ER and PM in Ca2+ signaling became apparent in 2005 when STIM proteins were discovered in independent Drosophila melanogaster (15) and human (16) siRNA screens by the group of Stauderman/Cahalan and our group that included the Ferrell laboratory, respectively. Both teams showed that STIM1 is necessary for cells to trigger PM Ca2+ influx when ER Ca2+ levels decrease (15, 16) by a mechanism whereby STIM1 is kept in an inactive state by binding of Ca2+ to an EF hand in the lumen of the ER (16, 17). A striking finding in the study from our laboratory was that lowering the level of Ca2+ in the ER induced the translocation of an ER-localized YFP-tagged STIM1 protein to ER sites near the PM (puncta). The translocation of YFP-STIM1 took approximately 40 s, paralleling the induction of Ca2+ influx into cells. Interestingly, no insertion of STIM into the PM was observed using antibodies against a luminal tag and the YFP-STIM1 translocation to puncta could be rapidly reversed by again increasing ER Ca2+ levels. Total internal reflection fluorescence measurements showed that these puncta are very close to the PM (16). This initial study argued that basal high ER Ca2+ levels keep native STIM1 proteins in an inactive state away from the PM and that receptor-mediated lowering of ER Ca2+ triggers the translocation of STIM1 within the ER to PM proximal sites (16). Although this is now widely accepted to be the primary mechanism of how STIM proteins signal when Ca2+ is lowered in the ER, our findings were initially controversial as another early study proposed that STIM1 is instead inserted into the PM as part of the STIM1-activation process (17). The electron microscopy evidence for the accumulation of STIM1 at ER sites in contact with the PM (18) and the discovery of the Ca2+ channel Orai1 as the STIM target in the PM (19–21) helped to resolve this controversy by providing a molecular connection whereby STIM1 can stay in the ER and signal to the PM by directly interacting with and opening the PM-localized Ca2+ channel Orai1 (22, 23). Thus, a unique mechanism exists whereby the ER-localized Ca2+ sensor STIM1 signals to the PM by reversibly translocating within the ER to ER-PM junctions to regulate directly PM-localized Ca2+ channels and possibly other proteins.
In addition to these findings in mammalian cells, independent work in the yeast Saccharomyces cerevisiae and in D. melanogaster photoreceptors showed that similar types of physical contacts between the ER membrane and the PM must exist with important roles in the transfer of ergosterol (24) and PtdIns lipids (25), respectively. A challenge when reading literature in these and other fields is the lack of a common definition regarding what ER-PM contact sites are. To create a framework to better discuss contacts between the ER and the PM, we first integrate a number of findings from different fields to propose a definition.
DEFINITION OF ER-PM JUNCTIONS
Contacts between the ER membranes and the PM have received different names in the literature, including triads, dyads, PM-associated membranes, PM contact sites, peripheral couplings, and ER-PM junctions. We are using the last term, because we believe the word junction, whose etymologic origin is the Latin verb jungere, to join, better represents the idea of a physical protein-mediated association between two separate membranes. How can one best define an ER-PM junction?
On the basis of similarities between contacts in different cell types, ER-PM junctions can be defined as follows: (a) specialized parts of the ER and PM where the two membranes stay in close apposition and create a restricted protein-rich cytoplasmic space, (b) both membranes conserve their identity—fusion has not been observed—and the width of the inter-membrane space ranges from 7 to 30 nm, and (c) the membrane contact sites are stable from minutes to days. Stable contacts differ from more transients contacts between the ER and PM that can result from the continued highly dynamic remodeling of ER structures (26, 27) and those that can bring membranes near each other without stable contacts necessarily being formed. More detailed references about properties of ER-PM junctions in different cell types are discussed below in the various sections.
These properties of the ER-PM junction require the existence of protein bridges that keep the membranes close and may prevent their fusion. Although overexpression of STIM1 (28, 29), junctophilin (30) or junctate (31) generate such bridges and increase the number or extension of ER-PM junctions, it is not yet established what the physiological components are that form native ER-PM junctions in most cell types. Our review focuses first on the roles of ER-PM junctions in Ca2+ signaling and then in subsequent sections on lipid transfer. Even though neuronal ER-PM junctions were among the first to be described in electron microscopy studies (14), they are not further discussed here because their structure, components, or functions are not well understood. Furthermore, although yeast and plants have ER-PM junctions and Ca2+ signaling is essential for their physiology, none have STIM or Orai, and in the case of yeast, the ER does not have a clear function in controlling Ca2+ homeostasis.
MOLECULAR STEPS OF STIM TRANSLOCATION TO ER-PM JUNCTIONS
The ER is in many cell types the primary Ca2+ source for triggering the initial Ca2+ signal when receptor and other stimuli activate phospholipase C (PLC) (32). The classic stimulus-response pathway begins with receptor activation of either trimeric G proteins that couple to PLCbeta, or tyrosine kinases that couple to PLCgamma (32). In turn, both isoforms hydrolyze the headgroup of phosphatidylinositol-4,5-bisphosphate lipids (PIP2) and release inositol-(1,4,5)-trisphosphate (InsP3), which rapidly diffuses and binds to the InsP3-receptor Ca2+ channels in the ER membrane, inducing Ca2+ release. To detect and respond to receptor stimuli, cells tightly control resting Ca2+ levels, maintaining steep gradients between the Ca2+ concentrations outside the cell and in the cytosol and ER lumen, which are approximately 1.5 mM, 50 nM, and 400 μM, respectively (32).
This InsP3-mediated Ca2+-release pathway involves positive and negative feedback that often results in multiple cycles of release (oscillations), followed by reuptake of Ca2+ into the ER as well as extrusion of Ca2+ across the PM. To compensate for the loss of Ca2+ across the PM, to replenish the Ca2+ reservoir in the ER, and to generate persistently elevated Ca2+ signals, a control system is therefore necessary that increases Ca2+ influx across the PM when the ER Ca2+ levels have dropped (33). Although evidence for such an ER-store-regulated Ca2+ influx existed for tens of years, the molecular mechanism remained obscure for a long time.
This hypothesis of store-operated Ca2+ entry (SOCE), arguing that depletion of ER Ca2+ triggers Ca2+ influx across the PM, was first explicitly proposed by J.W. Putney in a review in 1986 (34) (initially termed capacitative Ca2+ entry, CCE). The hypothesis implied that there must exist a Ca2+ sensor capable of detecting the reduction of Ca2+ levels in the ER, starting a signaling pathway that opens a Ca2+ channel in the PM. As mentioned above, the discovery of the STIM1 and STIM2 genes (15, 16) and of the Orai1-3 genes (19–21) provided such an ER Ca2+ sensor and a PM Ca2+ channel. These discoveries had a transforming effect on our understanding of Ca2+ signaling and ER-PM junctions. The current generated by expression of STIM1 and Orai1 matches the characteristics of one of the most thoroughly studied types of Ca2+ flux induced by depletion of intra-cellular Ca2+ stores, i.e., Ca2+-release-activated Ca2+ influx (CRAC) (19–21). Other described SOCE currents that are not directly generated by STIM and Orai have remained more controversial (35–38) and are only touched on in this review. Given some of these controversies, one could argue that it would be clearer to switch to a molecular description and refer to the STIM- and Orai-mediated Ca2+ influx currents as STIM/Orai currents.
We first summarize what we consider to be the most plausible mechanistic steps explaining STIM signal transduction. STIM1 was first described as an integral membrane protein that may alter the proliferation and survival of pre-B cells (39) or function as a recessive tumor suppressor (40, 41). It is now widely assumed that its primary role is that of an ER-luminal Ca2+ sensor that controls SOCE into cells. STIM1 and its homolog STIM2 proteins are single-pass transmembrane proteins in the ER membrane with a combined EF-SAM-domain Ca2+-binding site in their luminal part that inhibits STIM activity in the Ca2+-bound state.
Site-directed mutagenesis studies showed first for STIM1 that Ca2+ binding to its EF hand keeps the protein in a suppressed state (Figure 1a) (16, 17). In addition, structural studies of the EF-SAM domain showed that the EF and SAM subdomains are closely coupled and form a compact structure in the Ca2+-bound state (42), suggesting that Ca2+ dissociation leads to a more open configuration that may mediate the subsequent oligomerization of this luminal domain (43, 44). Inactive STIM1 is likely present in the ER in a monomeric or dimeric form and Ca2+ dissociation from the EF-SAM domain triggers an increase in oligomerization (43, 45). The evidence for basal dimer formation comes from studies where STIM1 has been overexpressed in cells (46–49) but has not yet been confirmed for native STIM1 proteins. A potential concern with the overexpression experiments is that oligomerization of proteins can be driven by either changes in ligand concentration or increases in the protein concentration, possibly shifting the basal equilibrium state toward a partial oligomerization in cells that overexpress STIM1.
Figure 1.

Schematic representation of the STIM1 translocation process to endoplasmic reticulum (ER)–plasma membrane (PM) junctions following a decrease in ER Ca2+ levels. (a) Main regulatory domains of STIM1. Numbers in parenthesis depict domain-boundary residues in human STIM1. (b) Induced coupling of STIM1 and Orai1 at the ER-PM junction: (I) At high basal Ca2+ (blue circles) in the ER, STIM1 is present as a monomer or dimer relatively uniformly distributed in the ER. Orai1, likely a tetramer, is uniformly present in the PM and is inactive when not bound to STIM1. (II and III) When Ca2+ is depleted in the ER, Ca2+ dissociates from the STIM1 EF-SAM domain, changes its conformation, thereby inducing protein oligomerization, generating a bigger cluster of positively charged amino acids (plus sign), which is now sufficient to interact with PIP2 (black; phosphate group represented as an asterisk) and other negatively charged lipids in the PM. (IV) STIM1 localization to the ER-PM junction, together with an oligomerization-mediated conformational change of STIM1, triggers binding and activation of Orai1, further triggering Ca2+ entry across the PM. CAD, CRAC activation domain; CMD, CRAC modulatory domain.
Independent of the starting point, oligomerization is likely the key step in activation, given that forced oligomerization of STIM1 is sufficient for its activation (50). In our view, the best current mechanistic explanation of how STIM1 activates Orai1 is that the observed oligomerization exposes interaction surfaces in STIM1 that can then bind to and activate Orai1 (22, 51, 52). In addition, the same oligomerization generates a second interaction site that allows binding of STIM1 oligomers to PM lipids (Figure 1b). In a third process, oligomerization likely leads to a loss in binding between STIM1 and EB1 (end binding protein 1) proteins as suggested by an observed loss in colocalization between STIM1 and EB1 following ER Ca2+ depletion (53). EB1 proteins are microtubule plus-ends connector proteins that bind STIM1 in cells that have basal high ER Ca2+ levels (53, 54). Both STIM1 and STIM2 have consensus sequences for binding to EBs, and this binding can be observed as a dynamic apparent movement of STIM1 with EB1 along the ER membrane. Nevertheless, the STIM1-localization change appears to be a reversible binding interaction with EB1 and not a processive STIM1-transport process (53) (see below for possible functions of this attachment in STIM distribution).
Fluorescence resonance energy transfer studies between STIM1 proteins showed that oligomerization of STIM is rapid and occurs within 4 s, whereas STIM1 is still in the ER away from ER-PM junctions (Figure 1b) (45). Translocation of STIM1 to ER-PM junctions likely occurs by passive local diffusion over a few micrometers (45) so that the oligomer is captured at nearby ER-PM junctions by its interaction with PM PIP2 and PIP(3,4,5)P3 lipids (55, 56). This lipid interaction requires a C-terminal polybasic region of STIM1 (45). With 8 positive charges in STIM1, this is below the threshold of more than 12 charges needed for cytosolic proteins to bind PIP2 and PIP(3,4,5)P3 lipids directly (57), if the basal STIM1 state is indeed monomeric. Oligomerization may increase the number of charges per STIM1 complex or mediate the exposure of more positive charges to the outside and thereby enhance its binding affinity to PM PIP2 lipids so that STIM1 oligomers are effectively retained at ER-PM junctions (58–60) (for more detail, see the Targeting Proteins to ER-PM Junctions section below).
Nevertheless, PM targeting of STIM1 by lipids alone is not sufficient for ORAI activation and the same oligomerization-induced conformational change of STIM1 is required for Orai recruitment and activation (22). The role of the STIM1 polybasic tail and lipid interaction (and likely of the homologous sequence in STIM2) is therefore likely to enhance the rate of recruitment of STIM1 to ER-PM junctions, and because oligomer formation is involved, the cooperativity of translocation and Orai activation following a drop in ER Ca2+ also is likely to be enhanced (Figure 1b) (50, 61). Both STIM isoforms also have a long coiled-coil domain immediately adjacent to the membrane-spanning domain (split into two parts) that may function as a spacer and is predicted from its length to bridge at least 15 nm (on the basis of a ~0.14 nm/amino acid spacing). This may explain how STIM1 and STIM2 can bridge the relatively wide space of ~17 nm observed in electron micrographs of T cell ER-PM junctions (18), and still bind directly to PM lipids.
Once STIM1 interacts with lipids at the ER-PM junction, a second trapping mechanism occurs whereby Orai1, initially distributed across the PM, is recruited by binding to the ER-PM-junction-localized STIM1 oligomers. In this final binding step, Orai1 is activated (22, 46, 51, 62–66) via a domain of STIM1 (23, 67) that has been termed the CAD (22) or SOAR (52, 68) domain, with the domain likely forming a tetramer (Figure 1b) (22). This direct interaction can be forced by joint overexpression of Orai1 plus a STIM1 construct that lacks the polybasic region, or by overexpressing the CAD domain of STIM1 alone (22). This again supports the idea that the role of the preceding STIM1 lipid binding is likely to enhance the local concentration of native STIM at ER-PM junctions to facilitate Orai1 interaction. Activation of Orai1 Ca2+ channels triggers local Ca2+ influx at ER-PM junctions (69), completing the STIM1-initiated signaling process. Finally, STIM1 also has a CRAC modulatory domain (CMD) (70) that is implicated in the fast Ca2+-dependent inactivation (CDI) of Orai1 in a process that also involves calmodulin (71).
STIM1 AND STIM2 ISOFORMS AND THEIR REGULATION AT ER-PM JUNCTIONS
Most vertebrates have two versions of the STIM gene, and recent studies showed that STIM1 has a higher affinity for luminal Ca2+ (~200 μM) and requires larger reductions in ER Ca2+ to be activated (43, 61). In contrast, STIM2 has an approximately twofold lower Ca2+ affinity (~400 μM), requires a smaller reduction in ER Ca2+, and is already partially active at basal Ca2+ levels (61). This suggests that STIM2 has a dual role as a basal regulator of ER Ca2+ homeostasis and as a Ca2+ sensor that can be more readily triggered when receptor stimuli are weak (61). These potential roles are also supported by the observation that STIM2 is typically present at much lower concentrations compared with STIM1 (72) and also triggers a relative lower amplitude of Ca2+ influx when compared with the same concentration of expressed STIM1 (61, 73). The lower relative activity may be a result of a lower maximal activation of Orai1 by STIM2 when compared with STIM1 (74) but also of additional regulatory mechanism controlling the respective STIM functions (for example, STIM2 has a number of conserved additional phosphorylation sites missing in STIM1). In addition to the affinity and regulatory differences, the polybasic tail of STIM2 also has a higher affinity for PIP2 lipids compared with STIM1, which may enhance its basal activity in cells (60). The distinct features of STIM2 may prevent excessive entry of Ca2+ when the changes in the Ca2+ ER are small and only STIM2 is activated. More generally, the different regulatory properties of STIM1 and STIM2 may enable different cell types to use a wider range of signaling mechanisms to control Ca2+ entry into cells.
Some of these additional control mechanisms for STIM1 regulation include an inactivation during mitosis by CDK phosphorylation (75), calmodulin binding to the polybasic region (76), and STIM1 gluthathionylation following oxidative stress affecting its recruitment to the junctions (77). In addition, an interesting recently reported interaction of STIM1 and Orai1 with CRACR2A initially stabilizes STIM1 in the ER-PM junction and enhances the activation of Orai1. In a second step, this same protein suppresses Orai1 channel activity when Ca2+ increases to a level where Ca2+ binds to the two CRACR2A EF hands (78).
Together, these studies show that STIM2 and STIM1 have distinct signaling roles responding to small and large reductions in ER Ca2+ levels, respectively, and that both isoforms can be regulated by additional processes.
BEYOND ORAI REGULATION: ADDITIONAL FUNCTIONS OF STIM PROTEINS
STIM1 also regulates the Ca2+ conductance of Trp (transient receptor potential) channels; however, this connection from STIM1 to Trp channels is not yet generally accepted (79–81). Other recent studies showed that activated STIM1 can inhibit L-type voltage-gated Ca2+ channels (DHPRs) (82, 83) and that these channels can also localize to ER-PM junctions (82). This is interesting in the context of muscle as discussed below, where STIM1 may have a role in signaling a muscle “fatigue state” when the sarcoplasmic reticulum (ER) level is too low and the STIM-mediated inhibition of DH-PRs may help with the reloading of the Ca2+ stores. In addition to regulating channels other than Orai1, studies also suggested that STIM1 has alternative roles such as regulating adenylate cyclase activity (84). Nevertheless, for both types of channels as well as the regulation of other processes, the physiological relevance of the respective STIM1-regulated processes is not yet well understood.
It has also been proposed that STIM1 and SOCE activities are linked to mitochondria on the basis of an observation that mitochondria can be proximal to the PM and ER-PM junctions (85, 86). Mitochondria may take up some of the Ca2+ entering from the PM and thereby prolong SOCE Ca2+ influx (by preventing inactivation), although the mechanism for the localization to ER-PM junctions is controversial (86–88). Because the consequent Ca2+ increase in the mitochondria is important for its metabolic activity (89), this may also suggest that localized mitochondria near ER-PM junctions enhance a cell’s ATP production in response to Ca2+ influx.
Together, these results suggest that different signaling processes and possibly mitochondria can be localized at or near ER-PM junctions and that STIM proteins regulate other targets in addition to Orai Ca2+ channels.
ER-PM JUNCTIONS AND STIM1 TRANSLOCATION AND POLARIZATION
A time-course analysis showed STIM translocates to pre-existing 17-nm-wide ER-PM junctions (18), suggesting that the junctions are not generated in response to STIM activation (90, 91). The regions of the ER that are part of these pre-existing ER-PM junctions can have different shapes and have been described as tubules and sheets that appear to be rough, even though ribosomes are excluded from the junctions (28, 29). Although these studies were important to demonstrate the existence of ER-PM junctions and characterize their morphology, little is currently known about how ER-PM junctions are distributed and shaped and whether changes in localization of these junctions regulate where Ca2+ influx occurs.
It is interesting in this context that the translocation of STIM1 and Orai1 is polarized in pancreatic acinar, T, and other cells. In pancreatic acinar cells, STIM1 is preferentially targeted to the basolateral membrane, whereas Orai1 is distributed more uniformly. When ER Ca2+ is depleted, both proteins translocate to basolateral membrane puncta (28), inducing the Ca2+ influx described above to take place at the basolateral side (for a review of the subcellular segregation of Ca2+ currents in acinar cells, see Reference 92). Another interesting polarization mechanism was observed in T cells, where activation of the T cell receptor led to preferential STIM1 localization to sites near the immunological synapse (93, 94).
Why is it important to have polarized or local Ca2+ influx? If ER-PM junctions are preferentially localized in one area of the cell, the Ca2+ concentration may increase locally in these specific regions and generate intracellular Ca2+ gradients as long as the Ca2+ pumps are distributed uniformly or localized elsewhere. This could be important to spatially control diverse cellular processes such as higher local Ca2+ signals near the T cell synapse or polar Ca2+ signals in pancreatic acinar cells, and it may also explain that STIM1 regulates migrating breast cancer cells (95) where gradients in Ca2+ concentration are believed to be important.
Given the binding interaction between STIM1 and EB1 discussed above, one can hypothesize that the observed cellular polarization of STIM1 may be mediated not only by a polarization of ER-PM junctions, but also by the interaction between inactive STIM1 and microtubule plus ends (53, 54). When ER Ca2+ levels are reduced, STIM1 proteins polarized by EB1 would then be locally released from EB1 to interact with nearby ER-PM junctions, leading to polarized Orai1 activation. Thus, the interaction with the microtubule plus-end protein EB1 may have two roles: (a) to generate a more peripheral distribution of STIM1 and (b) to make use of the often polar orientation of microtubules to polarize the distribution of STIM1 to make it preferentially available at one end of a cell.
STRIATED MUSCLE: THE CLASSIC ER-PM JUNCTION
The best-characterized ER-PM junction is arguably the connection between the sarcolemma (a muscle specialization of the PM) and the sarcoplasmic reticulum (a muscle ER specialization) in cardiac and skeletal striated muscles. These specialized ER-PM junctions can be readily observed by electron microscopy in specific regions of the sarcomere (the contraction unit of the muscle) and are formed by a PM invagination termed the T tubule and one or two ER terminal cisternae. These structures are known as dyads if there is only one terminal cisterna, as is often observed in cardiac cells, and triads if there are two, as is often observed in skeletal muscle cells. The membranes in these dyad or triad ER-PM junctions are kept tightly connected at a distance of 9 to 12 nm (96, 97).
Triads and dyads are present in skeletal and cardiac muscle at periodic sites to ensure proximity to the actin and myosin contraction machinery, to maximize the contraction strength, and to reduce the response time (Figure 2a) (98, 99). Interestingly, the specific localizations, number, and intermembrane distance of these junctions have a clear correlation with the different types of striated muscle. Muscle fibers with fast contraction, such as skeletal fast twitch muscle, have tighter triad junctions, and slow twitch and cardiac fibers have fewer and looser dyads (100).
Figure 2.

Schematics of the endoplasmic reticulum (ER)-plasma membrane (PM) junction in striated muscle. (a) The sarcomer, the contraction unit of the striated muscle, as observed by electron microscopy. Actin is shown in pink, and myosin in black (with barbed ends). The inset depicts triad and dyad muscle ER-PM junctions in their common localization along the sarcomer. At these junctions, the terminal cisternae of the sarcoplasmic reticulum (the muscle ER) and the T tubule of the sarcolemma (the muscle PM) are closely linked. The small dots with more intense color in the terminal cisternae represent the ryanodine receptor (RyR), clearly observable as small protrusions by electron microscopy (“feet structures”). (b) List of selected proteins localized to triad- or dyad-type ER-PM junctions. DHPR, dihydropyridine receptor; SR, sarcoplasmic reticulum.
The key components of triads and dyads are the PM-localized voltage-dependent Ca2+ channel DHPR (dihydropyridine receptor) (101) and the ER-localized Ca2+ channel RyR (ryanodine receptor) (Figure 2b) (102). These receptors work together to mediate excitation-contraction coupling whereby PM action potentials are converted into Ca2+ release from the ER (sarcoplasmic reticulum). The DHPR channel is composed of five different subunits that constitute a single pore across the PM, and the RyR is an enormous homotetramer (each unit more than 500 kDa) that protrudes into the ER in structures known as feet, which are distinctly visible by electron microscopy (103, 104). In cardiac cells and some skeletal muscle, the cardiac-specific RyR isoform, RyR2, can be activated by external Ca2+ by a Ca2+-induced Ca2+ release process whereby Ca2+ entering through the DHPR directly activates Ca2+ release from the ER by direct binding of Ca2+ to regulatory sites on the RyR channel (105).
In skeletal muscle, where the Ca2+ burst in the sarcoplasm needs to be extremely fast, a specific isoform of RyR, RyR1, is activated in triads by a voltage-triggered conformational change of DHPR that induces the mechanical force to pull on the connected RyR1 to open the channel and release Ca2+ from the ER into the cytosol (voltage-gated Ca2+ release) (106). In some skeletal muscle junctions, the RyR1 is also regulated by Ca2+-induced Ca2+ release (105).
In addition to this primary process, the local ER Ca2+ release also induces SOCE, and STIM1 proteins have been shown to be an essential element for the physiology of the skeletal muscle. Mice in which only one copy of STIM1 is present showed muscle fatigue, in a model where contractions are induced without intervals of relaxation, so that after the first contraction, the Ca2+ reservoir does not refill fast enough to reach the Ca2+ concentration required for subsequent contractions (107). Although the range of action of SOCE in terms of ER concentration has not been measured, the STIM1/Orai1-dependent Ca2+ entry in the muscle cells is faster than in nonexcitable cells (108). One possible explanation for this finding is that STIM1 and Orai1 are already prelocalized to ER-PM junctions and that Orai1 can be more rapidly activated in these cells without the delay caused by the previously described translocation step (109).
In addition to defects in the muscle physiology, the absence of functional STIM1 induces a myopathy that results in premature death (107). It is interesting that during the process of muscle differentiation, from the myoblast to the myotube, STIM1 changes its subcellular distribution simultaneously with the reorganization of the ER to form the characteristic triad ER-PM junction structures where STIM1 localizes (107, 110). The relation between ER and microtubules during the growth and expansion of this organelle in the cell is well known (96), suggesting that the interaction between ER-localized STIM1 and EB1 (53, 54) may add an additional link that enhances the coupling between the ER tubules and microtubules and thereby helps in the formation of the correct geometry between the ER, microtubules, and ER-PM junctions necessary for muscle function.
MOLECULAR COMPONENTS OF TRIAD- AND DYAD-TYPE ER-PM JUNCTIONS
Although both DHPR and RyR are necessary to generate the Ca2+ signal in the context of the triad, they are not required to keep the ER and PM in close apposition (111, 112). In the absence of these channels the ER-PM junctions are still present, suggesting that the bridges that connect the membranes are composed of other proteins.
Many additional proteins have been described as components of the junctions (Figure 2b): These include (a) ER luminal proteins such as the Ca2+-storage protein calsequestrin (113); (b) proteins integral to the ER membrane such as RyR (101), junctin (114), mitsugumin29 (115, 116), triadin (117), junctophilin (30), and JP45 (118); (c) cytosolic proteins such as FKBP12 (119); and (d) integral PM proteins such as DHPR (101). Many of these proteins modulate the activity of the RyR channel (119). The main one believed to have a role in bridging the membranes is junctophilin (120).
The family of junctophilins is composed of three members: JP-1 (skeletal muscle), JP-2 (cardiac muscle), and JP-3 (neurons). All have an ER transmembrane region and several MORN (membrane occupation and recognition nexus) motifs in the sarcoplasmic-exposed region (30). These motifs, best described in junctophilins and in plant PtdIns monophosphate kinases, are necessary to link junctophilins with the PM. According to studies done in plants, these motifs can bind phosphatidic acid, phosphatidylinositol-4-phosphate, and PIP2, thereby providing a possible mechanism for the connection between the ER membranes and the PM (121). Data obtained from JP-2 knockdown mice demonstrated that this protein is absolutely required to constitute peripheral coupling structures (the precursors of ER-PM junctions during myocyte differentiation), but researchers could not definitely determine whether the molecular partners of JP-2 are lipids and/or proteins in the PM (30). Another unsolved problem is that, upon overexpression of JP-1 in nonmuscle cells, junctions of 7.6 nm are observed, a distance similar to the 7 nm separating membranes in the junctions of RyR knockdown mice (30, 112). This raises the question of whether junctophilin can be flexible enough to span the 9- to 12-nm space that characterizes the normal dyad ER-PM junction. Nevertheless, the triad-dyad-type ER-PM junctions represent a remarkable adaptation of specialized striated-muscle cells to generate rapid, high-amplitude, and spatially restricted Ca2+ signals to trigger effective muscle contraction.
Indeed, this relationship between structure and function can also be seen when one compares the striated junctions with the ones in smooth muscle where the contraction mechanism, although Ca2+ dependent (122), does not need to be as rapid as that of striated-muscle contraction. In line with such a different requirement, smooth-muscle ER-PM junctions have different characteristics from those in striated muscle (123–125) in part as a result of their being less numerous with a more random spatial pattern. Although such junctions are likely important for local Ca2+-mediated Ca2+ release in smooth-muscle cells (126), analogous molecular evidences of altered structures of ER-PM junctions similar to the ones found in striated muscle (30, 112) are still needed. Nevertheless, electron microscopy studies of guinea-pig urinary-bladder smooth muscle showed that RyRs and DHPRs can be colocalized at ER-PM-like junctions, arguing that analogous local-release mechanisms exist in smooth cells similar to those found in striated muscle (127). Interestingly, the observed ER-PM junctions are more extended in phasic muscle that undergoes cycles of contraction and relaxation compared with tonic muscle that undergoes slow and sustained contraction where rapid local activation is likely less relevant (126, 128). This argues again for a close connection between the observed morphology and the number of ER-PM junctions and the relative importance of Ca2+-signaling functions in a particular cell.
To summarize this section, in addition to the STIM-mediated inside-out signaling pathway, a number of specialized cells such as muscle cells and possibly the less well-studied neurons make use of analogous ER-PM junctions in a process that is inverse to the STIM pathway whereby local Ca2+ influx or a mechanical link signals from the PM to cause a local Ca2+ release from the ER Ca2+ stores.
YEAST ER-PM JUNCTION AND ERGOSTEROL TRANSFER
Studies on yeast ER-PM junctions have mostly focused on lipid transfer rather than on Ca2+ signaling, which in yeast involves other channels and mostly vacuoles instead of ER Ca2+ stores (129, 130). A main point in our discussion in this section is the role of ER-PM junctions in unidirectional transfer of the cholesterol analog ergosterol from the ER to the PM. The yeast ER structure differs from the one in mammalian cells (6) in that the former has a sheet-like perinuclear ER as well as a sheet-like cortical ER that are readily distinguishable. These two compartments are connected by several peripheral tubules that cross the cytoplasm (Figure 3a) (7). The cortical ER spreads out just beneath the cytosolic surface of the PM, which makes it difficult to distinguish ER-PM junctions (131). However, the presence of junctions has been supported in studies where yeast without a cell wall were exposed to low osmotic pressure. Upon swelling, a high percentage of the ER remained associated with the PM, suggesting the presence of ER-PM junctions (132). Electron microscopy identified more than 1,000 ER-PM junctions per cell (defined by an intermembrane space smaller than 30 nm). Together with the smaller size of yeast, this suggests a significantly higher surface density of ER-PM junctions in yeast compared with most mammalian cells.
Figure 3.

Schematics of endoplasmic reticulum (ER) distribution in yeast. (a) Yeast ER can be separated into sheet-like perinuclear ER and cortical ER structures, connected by ER tubules. The cortical ER is close to the plasma membrane (PM) and forms numerous ER-PM junctions, shown as more intense colored patches. (b) Model of the difference between cortical and perinuclear ER inheritance in yeast: (I) Distribution of cortical and perinuclear ER in cells prior to division. (II) In a first step in yeast division, organelles including vacuoles, the Golgi apparatus, and mitochondria redistribute inside the cell toward the area where the bud will appear. In the case of the ER, one tubule of the cortical ER is pulled by actin (green) and myosin into the future bud to establish contact with the PM, creating a new ER-PM junction. (III) The septin ring forms shortly after this step (purple). As the bud grows, the cortical ER is extended from the founding-bud ER-PM junction. (IV) once the nucleus is divided, the new nucleus brings along its perinuclear ER and is transferred in a microtubule-driven process into the bud. (V) The bud separates from the mother to generate a smaller daughter cell, already with separated cortical and perinuclear ER.
In a biochemical isolation of yeast ER-PM junctions, a lipid composition more similar to the ER than to the PM was observed (enriched in PtdIns and depleted in ergosterol) (132). Further analysis showed an enrichment in enzymes involved in lipid metabolism such as phosphatidylserine synthase, PtdIns synthase, and ergosterol-synthesizing enzymes (132) compared with the bulk of the ER. The proximity to the PM of this lipid-synthesis machinery correlates with the recently demonstrated presence of several ergosterol-binding Osh proteins in the yeast ER-PM junction (24). These observations support the hypothesis of the importance of ER-PM junctions for providing the PM with ergosterol and other characteristic lipids.
Osh proteins are the yeast homologs of the mammalian oxysterol-binding-related proteins (ORPs) (7 genes in S. cerevisiae, 5 in Caenorhabditis elegans, 4 in D. melanogaster, and 12 in Mus musculus and Arabidopsis thaliana). They are peripherally associated with membranes and are characterized by the presence of ORP domains (24, 133). They were initially thought to be oxysterol receptors acting in a feedback loop to limit cholesterol synthesis. However, it has now been demonstrated that they are also essential for nonvesicle traffic of cholesterol/ergosterol, a function mediated by the ORP domain. The crystalization of Osh4p revealed that this domain has a similar structure to other lipid transfer proteins with a central hydrophobic cavity where one molecule of the lipid substrate is bound. Upon lipid binding, the protein changes its conformation to cover the cavity with a flexible lid that will protect the lipid during its transit through the hydrophilic cytosol (134). This transfer probably occurs mainly in the ER-PM junctions as these transfer proteins require the acceptor and the donor membranes to be close for efficient transfer. Indeed, the dimensions of the Osh proteins suggest that these contacts could be very tight given that the ORP-related domain is only 6 nm in diameter and the proteins contain two lipid-interacting surfaces, which may mediate simultaneous contacts with both membranes (24).
Another feature of some of the proteins with ORP domains is the presence of additional domains involved in lipid and protein binding that regulate localization and activity. Osh1p, Osh2p, and Osh3p contain a FFAT motif [diphenylalanine (FF) in an acidic tract] that mediates binding to Scs2p, a VAP (VAMP-associated protein) yeast homolog (135). Scs2p is an ER transmembrane protein, and when overexpressed, it recruits Osh2p and Osh3p exclusively to the cortical ER, where they transfer ergosterol (24). Of note, Osh protein activity not only depends on ergosterol availability, but is also accelerated when the donor membrane contains PtdIns (136). When considering the difference in lipid composition, this regulatory feature likely promotes the directed transfer of de novo synthesized ergosterol from the ER to the PM; the high concentration of PtdIns on the ER (132) side could help to establish the directionality of the transfer. Another explanation for the directional transfer of ergosterol between ER and PM proposes that the free ergosterol is the only one transported and kept in equilibrium, but the total concentration of ergosterol is higher in the PM because it accumulates and tightly associates with sphingolipids-enriched areas of the PM (137).
Scs2p is important not only for localization of FFAT motif–containing proteins, but also for cortical ER inheritance (138). This suggests an additional role of ER-PM junctions beyond lipid transfer. During the yeast-budding process, the perinuclear and cortical ER have different mechanisms for transmission to the new cell (139). The perinuclear ER uses a microtubule-dependent mechanism (140), and the cortical ER relies on myosin-mediated transport along actin cables (141). In addition, the cortical ER appears in the bud as soon as it is formed, whereas the perinuclear ER gets segregated with the nucleus during the last steps of cell division (Figure 3b) (6).
Cortical ER inheritance has been studied using deletion mutants, and several steps in the process have been characterized (142). Initially, some peripheral ER tubules are transported or extended to the tip of the bud and contact the PM (Figure 3b). After this step, the cortical ER spreads out to cover the bud (141). Several proteins, including Scs2p (138), as well as members of the translocon complex, accumulate in the tip of the initial tubule (143). This complex is critical for the biogenesis of transmembrane proteins and may be a candidate for establishing contact between the ER and the PM in the bud tip given its ability to interact with components of the exocyst, a complex that targets vesicles to the PM (144). Deletion mutants of Scs2p and of some of the proteins that compose the translocon or the exocyst show a block in cortical ER inheritance, and this block induces aberrant septin ring formation as well as problems during cell division (138, 143, 144). This introduces the interesting possibility that a physical link exists in the bud tip between ER-PM-junction lipid-transfer sites and the exocyst sites where vesicular transport occurs.
Interestingly, a new ER stress-surveillance (ERSU) pathway that is related to cortical ER inheritance has recently been described (145) and could point to a mechanism that senses defects in lipid transfer at ER-PM junctions. When this pathway senses defects in ER function (such as those caused by drugs that inhibit oxidative protein folding), it blocks cortical ER inheritance by an incompletely understood mechanism that involves modification of septins and induces the same phenotype as the Scs2p, translocon, or exocyst deletion mutants. Could ER-PM-junction lipid transfer be part of the sensing mechanism in this pathway? ER malfunction may create a disruption in the traffic of lipids across the junction in the tip of the peripheral tubule. The resulting lack of ergosterol or other lipids in the actively growing PM could then be sensed by the cell-surface receptor that has been implicated in the ERSU pathway. This suggests that dysfunction of the ER-PM junction may trigger ERSU-mediated cell stress by interfering with lipid transfer between the ER and PM. In sum, this could provide a mechanism that allows cells to sense intact ER-PM junctions.
Together, the findings in yeast argue that the ER-PM junctions play a central role in direct ergosterol and possibly other lipid transfer from the ER to the PM; that cells may have control mechanisms to sense intact ER-PM junctions; and that cells may use ER-PM junctions in processes such as cortical ER inheritance, regulation of PM exocyst complexes, and the induction of the ERSU stress pathway. Evidence for a similar transfer mechanism of PtdIns lipids from the ER to the PM has been found in Drosophila photoreceptor cells and is discussed below.
PHOSPHATIDYLINOSITOL TRANSFER IN DROSOPHILA PHOTORECEPTOR ER-PM JUNCTIONS
The ommatidium, the basic unit of the invertebrate compound eye, is formed by eight photoreceptor cells that pack together to generate a cone whose center is occupied by microvilli. These form the photosensitive structure of the ommatidia where the light sensor rhodopsin accumulates (Figure 4) (146).
Figure 4.
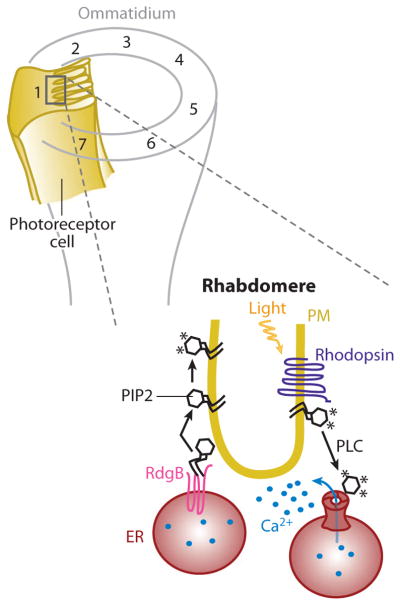
Schematics of rhabdomere endoplasmic reticulum (ER)-plasma membrane (PM) junctions: representation of an ommatidium showing photoreceptor cells (numbered; only number 1 is shown fully) that extend along the cone with the microvilli, constituting the rhabdomere. Rhodopsin (purple) is densely packed in the PM of the rhabdomere and, once activated by light, triggers PLC-dependent PIP2 hydrolysis. The released InsP3 binds to its receptor in the ER and induces Ca2+ release (small blue circles). In the same ER tubules, RdgB (pink) accumulates in the ER-PM junction where it transfers newly synthesized PtdIns to the PM. Once in the PM, PtdIns lipids are phosphorylated to replenish the level of PIP2, enabling a new cycle of rhodopsin activation and PIP2 hydrolysis. Each asterisk represents a phosphate group. Abbreviations: PtdIns, phosphatidylinositol; PIP2, phosphatidylinositol-4, 5-bisphosphate lipid; PLC, phospholipase C; RdgB, retinal degeneration B.
Closer observation of these structures reveals ER tubules that run along the length of the cell in close apposition, 10 to 40 nm, to the base of each microvillus in each rhabdomere (146). Rhodopsins, in response to light, activate PLC that hydrolizes PIP2 to generate diacylglycerol and InsP3 as well as protons. The light-triggered initial photoresponse is mostly mediated by depletion of PIP2 as well as the production of protons (147). The Ca2+ signal generated by the opening of PM Trp channels and the InsP3-mediated release of Ca2+ from these ER tubules (148) is part of a negative-feedback mechanism involved in recovery and adaptation (149, 150). As part of this activation, inactivation, and adaptation process, the hydrolyzed PIP2 in the PM has to be rapidly replenished by a transmembrane PtdIns transfer protein that also contains a FFAT motif (necessary for VAP binding, as explained in the Yeast ER-PM Junction and Ergosterol Transfer section above) (25, 148, 151, 152). This RdgB (retinal degeneration B) is resident in the membrane of the ER and can transfer PtdIns, which can be synthesized only in the ER to the PM, where it is phosphorylated to generate PIP2 (148, 152). Whereas other lipid-transfer proteins are peripheral membrane proteins that may be able to move short distances between membranes, RdgB is a transmembrane protein and requires that the two membranes involved be close, as is the case in ER-PM junctions (153). Thus, this transfer requires that the transfer protein contacts both the donor and the acceptor membrane to avoid the exposure of the transported lipid to the hydrophilic surrounding. This supports the idea that the junction between the PM of the invertebrate rhabdomere and the adjacent ER not only allows rapid spatially restricted Ca2+ signaling, but also allows for the rapid and efficient traffic of PtdIns lipids to replenish PM PIP2 that is degraded by rhodopsin activation of PLC.
Such an intricate architecture and mechanism of this invertebrate photoreceptor ER-PM junction is matched by an equally sophisticated plasmodesmata structure in plants that is also based on an ER-PM junction. These plant ER-PM junctions are discussed in the next section.
PLANT PLASMODESMA: A SPECIALIZED ER-PM JUNCTION NECESSARY FOR CELL-TO-CELL COMMUNICATION
Plants and multicellular algae have, similar to yeast, an extensive cortical ER, though its structure and functions are different in the former. In the case of plants and algae, the cortical ER is formed by interconnected tubules that spread in a polygonal net in which three-way vertices are occupied by lamellar structures that likely anchor the ER to the PM (Figure 5) (154, 155). This morphology is observed by electron microscopy when cells are treated to generate a “footprint” of the inner face of their PM, demonstrating that the cortical ER is firmly attached to the PM (155). The characteristic that makes this cortical ER unique is that it goes through an otherwise impermeable cell wall in structures known as plasmodesmata, facilitating communication with the contiguous adjacent cell (156). In these plasmodesmata, the tunnel penetrating the cell wall is lined by PM and occupied by a furled tubule of the cortical ER (desmotubule) that is separated from the PM by a 13- to 20-nm space (Figure 5).
Figure 5.

Plants’ endoplasmic reticulum (ER)–plasma membrane (PM) junction. The ER tubules close to the PM in plants and multicellular algae form polygonal nets, and in some regions, they go across the PM and cell wall in structures known as plasmodesmata. In these structures, the ER tubule, known as a desmotubule, is kept at different distances from the PM depending on the accumulation of callose in the cell wall or the presence of proteins such as actin in the intermembrane space.
Little is known about how these dual ER-PM tubes are formed, although the actin cytoskeleton plays a role both in maintaining the polygonal arrangement of the ER network and in helping the desmotubule to advance through the perforated cell wall (157). In addition, the presence of filamentous actin and other proteins in the space that separates the desmotubule and PM restricts the size of the particles that can be transferred from cell to cell (158).
When considering the traffic through the plasmodesmata, one has to distinguish four different paths: two concentric tunnel spaces (the inner one is the cortical ER lumen, and the outer is the intermembrane space) and two membranes (the desmotubule ER membrane and the PM). Several studies have demonstrated that the flux through the ER lumen and the ER membrane is likely not restricted. However, the space between the membranes can be remodeled and the PM in this area has a specific lipid and protein composition that facilitates control of the flux rate (159). The plasmodesmata-specific proteins have been characterized as acting in one of two functions, by regulating active transport of proteins and RNA or by degradation of callose, a polysaccharide that accumulates in the cell wall and constricts the tunnel (160). Dynamic regulation of access has been observed during viral infection whereby some plant viruses have evolved systems to control their transport through the plasmodesmata favoring a fast and efficient infection (156).
This distinct use of ER-PM junctions in plants as selective structural barriers that restrict cell-cell transport extends the range of roles of ER-PM junctions beyond their roles discussed above in cell signaling and lipid transfer. As a final point of discussion, we now build on findings from these different fields and focus on molecular-targeting mechanisms that localize proteins to ER-PM junctions.
TARGETING PROTEINS TO ER-PM JUNCTIONS
What can be learned from these examples about how ER-PM junctions are constructed and how proteins are targeted to ER-PM junctions? In principle, there are at least two plausible mechanisms of how ER-PM junctions are created: First, an ER and a PM resident protein have a high binding affinity for each other that stabilizes stochastic encounters between the ER and PM and, once the proteins bind, create stable ER-PM junctions. Examples of binding interactions between ER and PM resident proteins are those between STIM and Orai and between RyR and DHPR, which could, in principle, create ER-PM junctions. Nevertheless, as discussed above, in both examples, other proteins that are primarily responsible for creating the junctions are thought to exist. These proteins seem to be necessary in the case of STIM and Orai in which the interaction is induced only when Ca2+ is depleted in the ER. The ER-PM junctions are already present in the same cells and increase in size primarily as a consequence of Ca2+ depletion in the ER (29).
Second, proteins in the ER that bind to PM-specific lipids could be the source of ER-PM junctions. As discussed above for STIM (45) and junctophilins (30), ER-localized proteins can have targeting sequences or domains that recognize PM-specific lipids and that this can create new ER-PM junctions when such proteins are overexpressed in cells (28, 30). In the case of the junctate, although it is not clear how this ER transmembrane protein can form junctions, the formation of a complex with InsP3 receptor (in the ER) and a TRP channel (in the PM) seems to be required (161).
Although candidate genes responsible for forming the junction exist at least in muscle ER-PM junctions (junctophilin), there is as of yet no clear molecular candidate responsible for the formation of ER-PM junctions in other cell types. In terms of protein-protein versus protein-lipid bridges, both mechanisms appear to be equally plausible explanations for the creation of ER-PM junctions, and additional mechanisms could be envisioned whereby, for example, cytosolic-bridging proteins connect to proteins or lipids in both membranes. In addition to creating new ER-PM junctions, the same mechanisms may also function as reversible targeting mechanisms that localize ER resident proteins to pre-existing ER-PM junctions (such as is the case for STIM proteins).
When considering lipid-mediated targeting to existing ER-PM junctions, one structural feature of the PM is unique compared with the ER and other membranes: The PM is enriched in the negatively charged phospholipids, phosphatidylserine, as well as in PIP2 and PI(3,4,5)P3 (59). Many signaling domains such as the PH and C2 domain make use of the presence of these PM-enriched lipids to selectively target cytosolic proteins to the PM and not to internal membranes (162). A particularly revealing finding was that stretches of positively charged amino acids with adjacent hydrophobic amino acids, which are present, for example, in many Ras family proteins and in proteins such as MARCKS, can be sufficient to selectively target associated proteins to the PM (57). Comparison of dozens of such polybasic sequences showed that the precise sequence of these targeting sequences is less important; however, they must include patches with 3–5 lysine residues (less frequently arginines) in a 5–7 amino acid stretch, often with each patch immediately flanked by a hydrophobic residue such as tryptophan, phenylalanine, valine, or isoleucine (57, 59). These hydrophobic residues likely help to enhance PM binding by inserting into the membrane while the charged residues electrostatically interact with phosphorylated PtdIns and possibly some phosphatidylserine lipids (59). The most effective PM-targeting sequences have two or three closely spaced polybasic patches (~7 amino acid interval) that each adds sufficient countercharges for an additional PIP2 lipid to ultimately generate a high-affinity complex from several low-affinity-binding interactions. The lowest number of total polybasic residues that can target a cytosolic protein to the PM had more than 12 such positive amino acids (57). The PM-selective targeting of such polybasic patches requires the presence of phosphorylated PtdIns lipids in the PM (PIP2 and PtdIns-3,4,5-P3) because induced hydrolysis of phosphorylated PtdIns lipids rapidly dissociated the constructs with polybasic-targeting domains from the PM (57) (indicating that phosphatidylserine is not sufficient).
It is interesting that this targeting mechanism is also conserved in yeast. Ist2p, a yeast ER transmembrane protein, binds PM PIP2 through a cortical sequence rich in polybasic residues (163). In addition, Ist2p localizes to the ER-PM junction when expressed in mammalian cells, and the fusion of this polybasic motif to different transmembrane proteins is sufficient to localize these proteins to ER-PM junctions (60). In addition, a polybasic region from STIM1 with 8 positive charges was not sufficient to target an ER resident protein to ER-PM junctions, whereas the same peptide from STIM2 with 9 charges was sufficient, suggesting that STIM2 has a higher affinity for lipids compared with STIM1, which may help with its higher basal activity (60). This also suggests that ER-localized proteins can be targeted to ER-PM junctions with fewer than the 12 charges needed to target cytosolic proteins to the PM. Together, this suggests that a universal ER-PM lipid-targeting mechanism exists that requires an ER-associated or membrane-spanning protein to have on its cytosolic side patches of polybasic residues flanked by hydrophobic amino acids.
This type of targeting mechanism implies that ER proteins may function analogously to cytosolic proteins, except that the former randomly diffuse along the ER membrane instead of in the cytosol. The translocation is thus mediated by a retention mechanism whereby the diffusion-mediated binding rate is higher than the dissociation rate at which the protein dissociates from ER-PM junctions. Is such a diffusion mechanism effective? ER-resident trans-membrane proteins typically have relatively high diffusion coefficients that can range from 0.02 to 0.5 μm2/s (164, 165); with 0.05 μm2/s, STIM is among the slowest (45), suggesting that its range of action for finding junctions is approximately 2 μm for a time period of 30 s. Together, these considerations suggest that this polybasic-targeting mechanism is effective and ER-resident proteins can be targeted to ER-PM junctions within seconds to minutes.
At the PM, targeting of PM-resident proteins to ER-PM junctions could function by a similar passive trapping mechanism, but without involving lipids, whereby proteins diffusing within the PM membrane bind to already existing ER-PM-junction protein components at a rate faster than they dissociate from these same proteins. This likely explains the recruitment and activation of the Ca2+ channel Orai by STIM proteins as Orai translocates to ER-PM junctions following STIM recruitment to ER-PM junctions (69, 166). It is more difficult to explain how cytosolic proteins, such as lipid-transfer proteins, localize to these same junction sites; however, a similar protein-retention mechanism is also plausible.
As discussed in Yeast ER-PM Junction and Ergosterol Transfer (see section above), cholesterol transfer proteins (ORPs) are involved in ergosterol and cholesterol traffic, and some of their yeast homologues localize to the ER-PM junction (24). No studies have tried to assess their membrane localization in mammalian cells, but mammalian cholesterol traffic is known to be nonvesicle dependent (167, 168). Additionally, some ORPs (133) have FFAT domains that may help to localize these proteins to ER-PM junctions via binding interactions with VAP proteins, which would require their own mechanisms to be targeted to the junctions. Nevertheless, other unknown targeting mechanisms are likely involved as is the case of Osh6p and Osh7p, which localize to the yeast ER-PM junctions and do not contain an FFAT motif (24).
SUMMARY POINTS.
ER-PM junctions are ubiquitous eukaryotic structures that can have structural roles, such as in cell-cell communication in plants or Ca2+-signaling roles in store-operated Ca2+ influx, and excitation-contraction coupling in muscle. These junctions also have lipid transfer roles as discussed for ergosterol transport and PtdIns transport in yeast and Drosophila, respectively.
The structural components of ER-PM junctions are still mostly unknown. The molecularly best-understood junctions and processes are the triads and dyads in striated muscle where junctophilins act as important structural elements of the junctions. In the process of STIM-Orai signaling from the lumen of the ER to the PM as well as for the PM ergosterol transfer in yeast, the components responsible for generating the involved ER-PM junctions are not yet known.
Ca2+ signaling in ER-PM junctions can be bidirectional. Ca2+ signals can either start with the sensing of a drop in ER luminal Ca2+ leading to PM Ca2+ influx (through Orai channels) or start with the depolarization of the PM, which activates DHPR Ca2+ channels that then directly open connected RyR-type Ca2+ channels in the ER either by mechanical coupling or by local Ca2+-induced Ca2+ release. A number of additional Ca2+ channels and transporters also have been proposed to be locally regulated.
Lipid transfer at ER-PM junctions is likely to be unidirectional. In the case of ergosterol transfer, the transfer proteins Osh preferentially pick up ergosterol on the ER side and deliver it to the PM while contacting both membranes. PtdIns-lipid transfer in rhabdomers may use a similar mechanism to direct ER-synthesized PtdIns lipids to the PM, where they get phosphorylated to generate PIP2.
Accurate spacing between ER and PM at different junctions (from 7 to 30 nm) is important so that ER and PM components of different sizes connect to enable effective Ca2+ signaling and lipid transfer.
Targeting of ER-resident proteins to ER-PM junctions is likely in many cases mediated by polybasic/hydrophobic targeting sequences that selectively bind to negatively charged PIP2, PI(3,4,5)P3, and, possibly, phosphatidylserine lipids, enriched in the PM. In STIM proteins, the signal-induced conformational change likely exposes more of these polybasic PM-lipid-binding sites so that STIM proteins can be retained at ER-PM junctions.
FUTURE ISSUES.
Our understanding of the structural components of ER-PM junctions remains very limited. Studies in different organisms and cell types are needed to gain insights into common and specific building blocks of these junctions and into how cells control the number and turnover of ER-PM junctions.
The spacing between the ER and PM membrane has important functional consequences because proteins can either be excluded from the junction by size limitation or fail to bridge across the space owing to limited linker distances. Regulatory mechanisms that can change or maintain the spacing of ER-PM junctions are therefore promising fields of research.
ER-PM junctions are likely polarized or selectively localized in many cells. Mechanisms controlling the localization of ER-PM junctions need to be better understood.
A major role of ER-PM junctions is related to lipid transfer. Very little is currently known about putative lipid-transfer processes at ER-PM junctions in mammalian cells. These may include PtdIns and cholesterol, but potentially also other hydrophobic molecules.
An important class of functions of ER-PM junctions is related to Ca2+ signaling. A more detailed understanding of additional components involved in STIM-Orai, DHPR-RyR junctions, and possibly other Ca2+-signaling processes is still needed.
Additional signaling processes such as cAMP signaling also appear to occur at ER-PM junctions. Research is needed to better understand the relevance of other local signaling mechanisms.
It is of great interest to understand how the vast differences in structures and function of ER-PM junctions in different eukaryotic species evolved and how they use different molecular-targeting mechanisms to direct ER, PM, and cytosolic proteins to existing ER-PM junctions.
Glossary
- ER
endoplasmic reticulum
- PM
plasma membrane
- Membrane contact site
region of close contact between the membranes of two organelles
- Excitation-contraction coupling
the molecular processes that translate the nerve-induced sarcolemma depolarization into contraction of the sarcomere in mammalian muscle
- PtdIns
phosphatidylinositol
- Puncta
light microscope visualization of the ER-PM junction
- Junctophilins
transmembrane proteins expressed in mammalian muscle cells and neurons that contain a MORN (membrane occupation and recognition nexus) motif
- PIP2
phosphatidylinositol-4,5-bisphosphate
- InsP3
inositol-(1,4,5)-trisphosphate
- SOCE
store-operated Ca2+ entry
- EB1
end binding protein 1
- Trp channel
transient receptor potential channel
- Lipid transfer proteins
proteins that transfer one lipid at a time between membranes keeping it protected from the aqueous cytosol in a hydrophobic pocket
- FFAT
diphenylalanine (FF) in an acidic tract
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding or financial holding that might be perceived as affecting the objectivity of this review.
Contributor Information
Silvia Carrasco, Email: silviac2@stanford.edu.
Tobias Meyer, Email: tobias.meyer@stanford.edu.
LITERATURE CITED
- 1.Porter KR, Claude A, Fullam EF. A study of tissue culture cells by electron microscopy: methods and preliminary observations. J Exp Med. 1945;81(3):233–46. doi: 10.1084/jem.81.3.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Park SH, Blackstone C. Further assembly required: construction and dynamics of the endoplasmic reticulum network. EMBO Rep. 2010;11(7):515–21. doi: 10.1038/embor.2010.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Voeltz GK, Rolls MM, Rapoport TA. Structural organization of the endoplasmic reticulum. EMBO Rep. 2002;3(10):944–50. doi: 10.1093/embo-reports/kvf202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Somlyo AP, Bond M, Somlyo AV. Calcium content of mitochondria and endoplasmic reticulum in liver frozen rapidly in vivo. Nature. 1985;314(6012):622–25. doi: 10.1038/314622a0. [DOI] [PubMed] [Google Scholar]
- 5.Berridge MJ. The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium. 2002;32(5–6):235–49. doi: 10.1016/s0143416002001823. [DOI] [PubMed] [Google Scholar]
- 6.Estrada de Martin P, Novick P, Ferro-Novick S. The organization, structure, and inheritance of the ER in higher and lower eukaryotes. Biochem Cell Biol. 2005;83(6):752–61. doi: 10.1139/o05-159. [DOI] [PubMed] [Google Scholar]
- 7.English AR, Zurek N, Voeltz GK. Peripheral ER structure and function. Curr Opin Cell Biol. 2009;21(4):596–602. doi: 10.1016/j.ceb.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Levine T, Loewen C. Inter-organelle membrane contact sites: through a glass, darkly. Curr Opin Cell Biol. 2006;18(4):371–78. doi: 10.1016/j.ceb.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 9.Pan X, Roberts P, Chen Y, Kvam E, Shulga N, et al. Nucleus-vacuole junctions in Saccharomyces cerevisiae are formed through the direct interaction of Vac8p with Nvj1p. Mol Biol Cell. 2000;11(7):2445–57. doi: 10.1091/mbc.11.7.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kornmann B, Walter P. ERMES-mediated ER-mitochondria contacts: molecular hubs for the regulation of mitochondrial biology. J Cell Sci. 2010;123(Part 9):1389–93. doi: 10.1242/jcs.058636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kornmann B, Currie E, Collins SR, Schuldiner M, Nunnari J, et al. An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science. 2009;325(5939):477–81. doi: 10.1126/science.1175088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Brito OM, Scorrano L. An intimate liaison: spatial organization of the endoplasmic reticulum-mitochondria relationship. EMBO J. 2010;29(16):2715–23. doi: 10.1038/emboj.2010.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Porter KR, Palade GE. Studies on the endoplasmic reticulum. III. Its form and distribution in striated muscle cells. J Biophys Biochem Cytol. 1957;3(2):269–300. doi: 10.1083/jcb.3.2.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rosenbluth J. Subsurface cisterns and their relationship to the neuronal plasma membrane. J Cell Biol. 1962;13:405–21. doi: 10.1083/jcb.13.3.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol. 2005;169(3):435–45. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liou J, Kim ML, Heo WD, Jones JT, Myers JW, et al. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol. 2005;15(13):1235–41. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, et al. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005;437(7060):902–5. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu MM, Buchanan J, Luik RM, Lewis RS. Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J Cell Biol. 2006;174(6):803–13. doi: 10.1083/jcb.200604014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang SL, Yeromin AV, Zhang XH, Yu Y, Safrina O, et al. Genome-wide RNAi screen of Ca2+ influx identifies genes that regulate Ca2+ release-activated Ca2+ channel activity. Proc Natl Acad Sci USA. 2006;103(24):9357–62. doi: 10.1073/pnas.0603161103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vig M, Peinelt C, Beck A, Koomoa DL, Rabah D, et al. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science. 2006;312(5777):1220–23. doi: 10.1126/science.1127883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel S, et al. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441(7090):179–85. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- 22.Park CY, Hoover PJ, Mullins FM, Bachhawat P, Covington ED, et al. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell. 2009;136(5):876–90. doi: 10.1016/j.cell.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou Y, Meraner P, Kwon HT, Machnes D, Ohhora M, et al. STIM1 gates the store-operated calcium channel ORAI1 in vitro. Nat Struct Mol Biol. 2010;17(1):112–16. doi: 10.1038/nsmb.1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schulz TA, Choi M, Raychaudhuri S, Mears JA, Ghirlando R, et al. Lipid-regulated sterol transfer between closely apposed membranes by oxysterol-binding protein homologues. J Cell Biol. 2009;187(6):889–903. doi: 10.1083/jcb.200905007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Milligan SC, Alb JG, Elagina RB, Bankaitis VA, Hyde DR. The phosphatidylinositol transfer protein domain of Drosophila retinal degeneration B protein is essential for photoreceptor cell survival and recovery from light stimulation. J Cell Biol. 1997;139(2):351–63. doi: 10.1083/jcb.139.2.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Waterman-Storer CM, Salmon ED. Endoplasmic reticulum membrane tubules are distributed by microtubules in living cells using three distinct mechanisms. Curr Biol. 1998;8(14):798–806. doi: 10.1016/s0960-9822(98)70321-5. [DOI] [PubMed] [Google Scholar]
- 27.Friedman JR, Webster BM, Mastronarde DN, Verhey KJ, Voeltz GK. ER sliding dynamics and ER-mitochondrial contacts occur on acetylated microtubules. J Cell Biol. 2010;190(3):363–75. doi: 10.1083/jcb.200911024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lur G, Haynes LP, Prior IA, Gerasimenko OV, Feske S, et al. Ribosome-free terminals of rough ER allow formation of STIM1 puncta and segregation of STIM1 from IP(3) receptors. Curr Biol. 2009;19(19):1648–53. doi: 10.1016/j.cub.2009.07.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Orci L, Ravazzola M, Le Coadic M, Shen W, Demaurex N, Cosson P. From the cover: STIM1-induced precortical and cortical subdomains of the endoplasmic reticulum. Proc Natl Acad Sci USA. 2009;106(46):19358–62. doi: 10.1073/pnas.0911280106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takeshima H, Komazaki S, Nishi M, Iino M, Kangawa K. Junctophilins: a novel family of junctional membrane complex proteins. Mol Cell. 2000;6(1):11–22. doi: 10.1016/s1097-2765(00)00003-4. [DOI] [PubMed] [Google Scholar]
- 31.Treves S, Franzini-Armstrong C, Moccagatta L, Arnoult C, Grasso C, et al. Junctate is a key element in calcium entry induced by activation of InsP3 receptors and/or calcium store depletion. J Cell Biol. 2004;166(4):537–48. doi: 10.1083/jcb.200404079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clapham DE. Calcium signaling. Cell. 2007;131(6):1047–58. doi: 10.1016/j.cell.2007.11.028. [DOI] [PubMed] [Google Scholar]
- 33.Putney JW. Capacitative calcium entry: sensing the calcium stores. J Cell Biol. 2005;169(3):381–82. doi: 10.1083/jcb.200503161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Putney JW. A model for receptor-regulated calcium entry. Cell Calcium. 1986;7(1):1–12. doi: 10.1016/0143-4160(86)90026-6. [DOI] [PubMed] [Google Scholar]
- 35.Smyth JT, Dehaven WI, Jones BF, Mercer JC, Trebak M, et al. Emerging perspectives in store-operated Ca2+ entry: roles of Orai, Stim and TRP. Biochim Biophys Acta. 2006;1763(11):1147–60. doi: 10.1016/j.bbamcr.2006.08.050. [DOI] [PubMed] [Google Scholar]
- 36.Shuttleworth TJ. Arachidonic acid, ARC channels, and Orai proteins. Cell Calcium. 2009;45(6):602–10. doi: 10.1016/j.ceca.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Salido GM, Sage SO, Rosado JA. Biochemical and functional properties of the store-operated Ca2+ channels. Cell Signal. 2009;21(4):457–61. doi: 10.1016/j.cellsig.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 38.Lee KP, Yuan JP, Hong JH, So I, Worley PF, Muallem S. An endoplasmic reticulum/plasma membrane junction: STIM1/Orai1/TRPCs. FEBS Lett. 2010;584(10):2022–27. doi: 10.1016/j.febslet.2009.11.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oritani K, Kincade PW. Identification of stromal cell products that interact with pre-B cells. J Cell Biol. 1996;134(3):771–82. doi: 10.1083/jcb.134.3.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sabbioni S, Barbanti-Brodano G, Croce CM, Negrini M. GOK: a gene at 11p15 involved in rhabdomyosarcoma and rhabdoid tumor development. Cancer Res. 1997;57(20):4493–97. [PubMed] [Google Scholar]
- 41.Suyama E, Wadhwa R, Kaur K, Miyagishi M, Kaul SC, et al. Identification of metastasis-related genes in a mouse model using a library of randomized ribozymes. J Biol Chem. 2004;279(37):38083–86. doi: 10.1074/jbc.C400313200. [DOI] [PubMed] [Google Scholar]
- 42.Stathopulos PB, Zheng L, Li G, Plevin MJ, Ikura M. Structural and mechanistic insights into STIM1-mediated initiation of store-operated calcium entry. Cell. 2008;135(1):110–22. doi: 10.1016/j.cell.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 43.Stathopulos PB, Li G, Plevin MJ, Ames JB, Ikura M. Stored Ca2+ depletion-induced oligomerization of stromal interaction molecule 1 (STIM1) via the EF-SAM region: an initiation mechanism for capacitive Ca2+ entry. J Biol Chem. 2006;281(47):35855–62. doi: 10.1074/jbc.M608247200. [DOI] [PubMed] [Google Scholar]
- 44.Stathopulos PB, Ikura M. Partial unfolding and oligomerization of stromal interaction molecules as an initiation mechanism of store operated calcium entry. Biochem Cell Biol. 2010;88(2):175–83. doi: 10.1139/o09-125. [DOI] [PubMed] [Google Scholar]
- 45.Liou J, Fivaz M, Inoue T, Meyer T. Live-cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc Natl Acad Sci USA. 2007;104(22):9301–6. doi: 10.1073/pnas.0702866104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Muik M, Frischauf I, Derler I, Fahrner M, Bergsmann J, et al. Dynamic coupling of the putative coiled-coil domain of ORAI1 with STIM1 mediates ORAI1 channel activation. J Biol Chem. 2008;283(12):8014–22. doi: 10.1074/jbc.M708898200. [DOI] [PubMed] [Google Scholar]
- 47.Williams RT, Senior PV, Van Stekelenburg L, Layton JE, Smith PJ, Dziadek MA. Stromal interaction molecule 1 (STIM1), a transmembrane protein with growth suppressor activity, contains an extracellular SAM domain modified by N-linked glycosylation. Biochim Biophys Acta. 2002;1596(1):131–37. doi: 10.1016/s0167-4838(02)00211-x. [DOI] [PubMed] [Google Scholar]
- 48.Baba Y, Hayashi K, Fujii Y, Mizushima A, Watarai H, et al. Coupling of STIM1 to store-operated Ca2+ entry through its constitutive and inducible movement in the endoplasmic reticulum. Proc Natl Acad Sci USA. 2006;103(45):16704–9. doi: 10.1073/pnas.0608358103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Covington ED, Wu MM, Lewis RS. Essential role for the CRAC activation domain in store-dependent oligomerization of STIM1. Mol Biol Cell. 2010;21(11):1897–907. doi: 10.1091/mbc.E10-02-0145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Luik RM, Wang B, Prakriya M, Wu MM, Lewis RS. Oligomerization of STIM1 couples ER calcium depletion to CRAC channel activation. Nature. 2008;454(7203):538–42. doi: 10.1038/nature07065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kawasaki T, Lange I, Feske S. A minimal regulatory domain in the C terminus of STIM1 binds to and activates ORAI1 CRAC channels. Biochem Biophys Res Commun. 2009;385(1):49–54. doi: 10.1016/j.bbrc.2009.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yuan JP, Zeng W, Dorwart MR, Choi Y, Worley PF, Muallem S. SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat Cell Biol. 2009;11(3):337–43. doi: 10.1038/ncb1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grigoriev I, Gouveia SM, Van Der Vaart B, Demmers J, Smyth JT, et al. STIM1 is a MT-plus-end-tracking protein involved in remodeling of the ER. Curr Biol. 2008;18(3):177–82. doi: 10.1016/j.cub.2007.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Honnappa S, Gouveia SM, Weisbrich A, Damberger FF, Bhavesh NS, et al. An EB1-binding motif acts as a microtubule tip localization signal. Cell. 2009;138(2):366–76. doi: 10.1016/j.cell.2009.04.065. [DOI] [PubMed] [Google Scholar]
- 55.Walsh CM, Chvanov M, Haynes LP, Petersen OH, Tepikin AV, Burgoyne RD. Role of phosphoinositides in STIM1 dynamics and store-operated calcium entry. Biochem J. 2010;425(1):159–68. doi: 10.1042/BJ20090884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Korzeniowski MK, Popovic MA, Szentpetery Z, Varnai P, Stojilkovic SS, Balla T. Dependence of STIM1/Orai1-mediated calcium entry on plasma membrane phosphoinositides. J Biol Chem. 2009;284(31):21027–35. doi: 10.1074/jbc.M109.012252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Heo WD, Inoue T, Park WS, Kim ML, Park BO, et al. PI(3,4,5)P3 and PI(4,5)P2 lipids target proteins with polybasic clusters to the plasma membrane. Science. 2006;314(5804):1458–61. doi: 10.1126/science.1134389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huang GN, Zeng W, Kim JY, Yuan JP, Han L, et al. STIM1 carboxyl-terminus activates native SOC, I(crac) and TRPC1 channels. Nat Cell Biol. 2006;8(9):1003–10. doi: 10.1038/ncb1454. [DOI] [PubMed] [Google Scholar]
- 59.McLaughlin S, Murray D. Plasma membrane phosphoinositide organization by protein electrostatics. Nature. 2005;438(7068):605–11. doi: 10.1038/nature04398. [DOI] [PubMed] [Google Scholar]
- 60.Ercan E, Momburg F, Engel U, Temmerman K, Nickel W, Seedorf M. A conserved, lipid-mediated sorting mechanism of yeast Ist2 and mammalian STIM proteins to the peripheral ER. Traffic. 2009;10(12):1802–18. doi: 10.1111/j.1600-0854.2009.00995.x. [DOI] [PubMed] [Google Scholar]
- 61.Brandman O, Liou J, Park WS, Meyer T. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell. 2007;131(7):1327–39. doi: 10.1016/j.cell.2007.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Penna A, Demuro A, Yeromin AV, Zhang SL, Safrina O, et al. The CRAC channel consists of a tetramer formed by Stim-induced dimerization of Orai dimers. Nature. 2008;456(7218):116–20. doi: 10.1038/nature07338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Frischauf I, Muik M, Derler I, Bergsmann J, Fahrner M, et al. Molecular determinants of the coupling between STIM1 and Orai channels: differential activation of Orai1-3 channels by a STIM1 coiled-coil mutant. J Biol Chem. 2009;284(32):21696–706. doi: 10.1074/jbc.M109.018408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhou Y, Mancarella S, Wang Y, Yue C, Ritchie M, et al. The short N-terminal domains of STIM1 and STIM2 control the activation kinetics of Orai1 channels. J Biol Chem. 2009;284(29):19164–68. doi: 10.1074/jbc.C109.010900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang Y, Deng X, Zhou Y, Hendron E, Mancarella S, et al. STIM protein coupling in the activation of Orai channels. Proc Natl Acad Sci USA. 2009;106(18):7391–96. doi: 10.1073/pnas.0900293106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Muik M, Fahrner M, Derler I, Schindl R, Bergsmann J, et al. A cytosolic homomerization and a modulatory domain within STIM1 C terminus determine coupling to ORAI1 channels. J Biol Chem. 2009;284(13):8421–26. doi: 10.1074/jbc.C800229200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Calloway N, Holowka D, Baird B. A basic sequence in STIM1 promotes Ca2+ influx by interacting with the C-terminal acidic coiled coil of Orai1. Biochemistry. 2010;49(6):1067–71. doi: 10.1021/bi901936q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lee KP, Yuan JP, Zeng W, So I, Worley PF, Muallem S. Molecular determinants of fast Ca2+-dependent inactivation and gating of the Orai channels. Proc Natl Acad Sci USA. 2009;106(34):14687–92. doi: 10.1073/pnas.0904664106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Luik RM, Wu MM, Buchanan J, Lewis RS. The elementary unit of store-operated Ca2+ entry: local activation of CRAC channels by STIM1 at ER-plasma membrane junctions. J Cell Biol. 2006;174(6):815–25. doi: 10.1083/jcb.200604015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Derler I, Fahrner M, Muik M, Lackner B, Schindl R, et al. A Ca2+ release-activated Ca2+ (CRAC) modulatory domain (CMD) within STIM1 mediates fast Ca2+-dependent inactivation of ORAI1 channels. J Biol Chem. 2009;284(37):24933–38. doi: 10.1074/jbc.C109.024083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mullins FM, Park CY, Dolmetsch RE, Lewis RS. STIM1 and calmodulin interact with Orai1 to induce Ca2+-dependent inactivation of CRAC channels. Proc Natl Acad Sci USA. 2009;106(36):15495–500. doi: 10.1073/pnas.0906781106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Oh-Hora M, Yamashita M, Hogan PG, Sharma S, Lamperti E, et al. Dual functions for the endoplasmic reticulum calcium sensors STIM1 and STIM2 in T cell activation and tolerance. Nat Immunol. 2008;9(4):432–43. doi: 10.1038/ni1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Parvez S, Beck A, Peinelt C, Soboloff J, Lis A, et al. STIM2 protein mediates distinct store-dependent and store-independent modes of CRAC channel activation. FASEB J. 2008;22(3):752–61. doi: 10.1096/fj.07-9449com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bird GS, Hwang S, Smyth JT, Fukushima M, Boyles RR, Putney JW. STIM1 is a calcium sensor specialized for digital signaling. Curr Biol. 2009;19(20):1724–29. doi: 10.1016/j.cub.2009.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Smyth JT, Petranka JG, Boyles RR, DeHaven WI, Fukushima M, et al. Phosphorylation of STIM1 underlies suppression of store-operated calcium entry during mitosis. Nat Cell Biol. 2009;11(12):1465–72. doi: 10.1038/ncb1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bauer MC, O’Connell D, Cahill DJ, Linse S. Calmodulin binding to the polybasic C-termini of STIM proteins involved in store-operated calcium entry. Biochemistry. 2008;47(23):6089–91. doi: 10.1021/bi800496a. [DOI] [PubMed] [Google Scholar]
- 77.Hawkins BJ, Irrinki KM, Mallilankaraman K, Lien Y, Wang Y, et al. S-glutathionylation activates STIM1 and alters mitochondrial homeostasis. J Cell Biol. 2010;190(3):391–405. doi: 10.1083/jcb.201004152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Srikanth S, Jung H, Kim K, Souda P, Whitelegge J, Gwack Y. A novel EF-hand protein, CRACR2A, is a cytosolic Ca2+ sensor that stabilizes CRAC channels in T cells. Nat Cell Biol. 2010;12(5):436–46. doi: 10.1038/ncb2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.López JJ, Salido GM, Pariente JA, Rosado JA. Interaction of STIM1 with endogenously expressed human canonical TRP1 upon depletion of intracellular Ca2+ stores. J Biol Chem. 2006;281(38):28254–64. doi: 10.1074/jbc.M604272200. [DOI] [PubMed] [Google Scholar]
- 80.Yuan JP, Zeng W, Huang GN, Worley PF, Muallem S. STIM1 heteromultimerizes TRPC channels to determine their function as store-operated channels. Nat Cell Biol. 2007;9(6):636–45. doi: 10.1038/ncb1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zeng W, Yuan JP, Kim MS, Choi YJ, Huang GN, et al. STIM1 gates TRPC channels, but not Orai1, by electrostatic interaction. Mol Cell. 2008;32(3):439–48. doi: 10.1016/j.molcel.2008.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang Y, Deng X, Mancarella S, Hendron E, Eguchi S, et al. The calcium store sensor, STIM1, reciprocally controls Orai and CaV1.2 channels. Science. 2010;330(6000):105–9. doi: 10.1126/science.1191086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Park CY, Shcheglovitov A, Dolmetsch R. The CRAC channel activator STIM1 binds and inhibits L-type voltage-gated calcium channels. Science. 2010;330(6000):101–5. doi: 10.1126/science.1191027. [DOI] [PubMed] [Google Scholar]
- 84.Lefkimmiatis K, Srikanthan M, Maiellaro I, Moyer MP, Curci S, Hofer AM. Store-operated cyclic AMP signalling mediated by STIM1. Nat Cell Biol. 2009;11(4):433–42. doi: 10.1038/ncb1850. [DOI] [PubMed] [Google Scholar]
- 85.Korzeniowski MK, Szanda G, Balla T, Spät A. Store-operated Ca2+ influx and subplasmalemmal mitochondria. Cell Calcium. 2009;46(1):49–55. doi: 10.1016/j.ceca.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Naghdi S, Waldeck-Weiermair M, Fertschai I, Poteser M, Graier WF, Malli R. Mitochondrial Ca2+ uptake and not mitochondrial motility is required for STIM1-Orai1-dependent store-operated Ca2+ entry. J Cell Sci. 2010;123(15):2553–64. doi: 10.1242/jcs.070151. [DOI] [PubMed] [Google Scholar]
- 87.Quintana A, Schwarz EC, Schwindling C, Lipp P, Kaestner L, Hoth M. Sustained activity of calcium release-activated calcium channels requires translocation of mitochondria to the plasma membrane. J Biol Chem. 2006;281(52):40302–9. doi: 10.1074/jbc.M607896200. [DOI] [PubMed] [Google Scholar]
- 88.Hoth M, Fanger CM, Lewis RS. Mitochondrial regulation of store-operated calcium signaling in T lymphocytes. J Cell Biol. 1997;137(3):633–48. doi: 10.1083/jcb.137.3.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Feldman B, Fedida-Metula S, Nita J, Sekler I, Fishman D. Coupling of mitochondria to store-operated Ca2+-signaling sustains constitutive activation of protein kinase B/Akt and augments survival of malignant melanoma cells. Cell Calcium. 2010;47(6):525–37. doi: 10.1016/j.ceca.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 90.Várnai P, Tóth B, Tóth DJ, Hunyady L, Balla T. Visualization and manipulation of plasma membrane-endoplasmic reticulum contact sites indicates the presence of additional molecular components within the STIM1-Orai1 complex. J Biol Chem. 2007;282(40):29678–90. doi: 10.1074/jbc.M704339200. [DOI] [PubMed] [Google Scholar]
- 91.Smyth JT, Dehaven WI, Bird GS, Putney JW. Ca2+-store-dependent and -independent reversal of Stim1 localization and function. J Cell Sci. 2008;121(Part 6):762–72. doi: 10.1242/jcs.023903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Petersen OH, Tepikin AV. Polarized calcium signaling in exocrine gland cells. Annu Rev Physiol. 2008;70:273–99. doi: 10.1146/annurev.physiol.70.113006.100618. [DOI] [PubMed] [Google Scholar]
- 93.Barr VA, Bernot KM, Srikanth S, Gwack Y, Balagopalan L, et al. Dynamic movement of the calcium sensor STIM1 and the calcium channel Orai1 in activated T-cells: puncta and distal caps. Mol Biol Cell. 2008;19(7):2802–17. doi: 10.1091/mbc.E08-02-0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lioudyno MI, Kozak JA, Penna A, Safrina O, Zhang SL, et al. Orai1 and STIM1 move to the immunological synapse and are up-regulated during T cell activation. Proc Natl Acad Sci USA. 2008;105(6):2011–16. doi: 10.1073/pnas.0706122105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yang S, Zhang JJ, Huang X. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell. 2009;15(2):124–34. doi: 10.1016/j.ccr.2008.12.019. [DOI] [PubMed] [Google Scholar]
- 96.Flucher BE. Structural analysis of muscle development: transverse tubules, sarcoplasmic reticulum, and the triad. Dev Biol. 1992;154(2):245–60. doi: 10.1016/0012-1606(92)90065-o. [DOI] [PubMed] [Google Scholar]
- 97.Rossi AE, Dirksen RT. Sarcoplasmic reticulum: the dynamic calcium governor of muscle. Muscle Nerve. 2006;33(6):715–31. doi: 10.1002/mus.20512. [DOI] [PubMed] [Google Scholar]
- 98.Flucher BE. Structural analysis of muscle development: transverse tubules, sarcoplasmic reticulum, and the triad. Dev Biol. 1992;154(2):245–60. doi: 10.1016/0012-1606(92)90065-o. [DOI] [PubMed] [Google Scholar]
- 99.Rossi AE, Dirksen RT. Sarcoplasmic reticulum: the dynamic calcium governor of muscle. Muscle Nerve. 2006;33(6):715–31. doi: 10.1002/mus.20512. [DOI] [PubMed] [Google Scholar]
- 100.Franzini-Armstrong C. Studies of the triad. 3. Structure of the junction in fast-twitch fibers. Tissue Cell. 1972;4(3):469–78. doi: 10.1016/s0040-8166(72)80023-5. [DOI] [PubMed] [Google Scholar]
- 101.Dirksen RT. Bi-directional coupling between dihydropyridine receptors and ryanodine receptors. Front Biosci. 2002;7:d659–70. doi: 10.2741/A802. [DOI] [PubMed] [Google Scholar]
- 102.Fill M, Copello JA. Ryanodine receptor calcium release channels. Physiol Rev. 2002;82(4):893–922. doi: 10.1152/physrev.00013.2002. [DOI] [PubMed] [Google Scholar]
- 103.Block BA, Imagawa T, Campbell KP, Franzini-Armstrong C. Structural evidence for direct interaction between the molecular components of the transverse tubule/sarcoplasmic reticulum junction in skeletal muscle. J Cell Biol. 1988;107(Part 2):2587–600. doi: 10.1083/jcb.107.6.2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hamilton SL, Serysheva II. Ryanodine receptor structure: progress and challenges. J Biol Chem. 2009;284(7):4047–51. doi: 10.1074/jbc.R800054200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Endo M. Calcium-induced calcium release in skeletal muscle. Physiol Rev. 2009;89(4):1153–76. doi: 10.1152/physrev.00040.2008. [DOI] [PubMed] [Google Scholar]
- 106.Orlova EV, Serysheva II, van Heel M, Hamilton SL, Chiu W. Two structural configurations of the skeletal muscle calcium release channel. Nat Struct Biol. 1996;3(6):547–52. doi: 10.1038/nsb0696-547. [DOI] [PubMed] [Google Scholar]
- 107.Stiber J, Hawkins A, Zhang Z, Wang S, Burch J, et al. STIM1 signalling controls store-operated calcium entry required for development and contractile function in skeletal muscle. Nat Cell Biol. 2008;10(6):688–97. doi: 10.1038/ncb1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Edwards JN, Murphy RM, Cully TR, von Wegner F, Friedrich O, Launikonis BS. Ultra-rapid activation and deactivation of store-operated Ca2+ entry in skeletal muscle. Cell Calcium. 2010;47(5):458–67. doi: 10.1016/j.ceca.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 109.Launikonis BS, Murphy RM, Edwards JN. Toward the roles of store-operated Ca2+ entry in skeletal muscle. Pflugers Arch. 2010;460(5):813–23. doi: 10.1007/s00424-010-0856-7. [DOI] [PubMed] [Google Scholar]
- 110.Dirksen RT. Checking your SOCCs and feet: the molecular mechanisms of Ca2+ entry in skeletal muscle. J Physiol. 2009;587(Part 13):3139–47. doi: 10.1113/jphysiol.2009.172148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Franzini-Armstrong C, Pincon-Raymond M, Rieger F. Muscle fibers from dysgenic mouse in vivo lack a surface component of peripheral couplings. Dev Biol. 1991;146(2):364–76. doi: 10.1016/0012-1606(91)90238-x. [DOI] [PubMed] [Google Scholar]
- 112.Ikemoto T, Komazaki S, Takeshima H, Nishi M, Noda T, et al. Functional and morphological features of skeletal muscle from mutant mice lacking both type 1 and type 3 ryanodine receptors. J Physiol (Lond ) 1997;501(Part 2):305–12. doi: 10.1111/j.1469-7793.1997.305bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Beard NA, Wei L, Dulhunty AF. Ca2+ signaling in striated muscle: the elusive roles of triadin, junctin, and calsequestrin. Eur Biophys J. 2009;39(1):27–36. doi: 10.1007/s00249-009-0449-6. [DOI] [PubMed] [Google Scholar]
- 114.Jones LR, Zhang L, Sanborn K, Jorgensen AO, Kelley J. Purification, primary structure, and immunological characterization of the 26-kDa calsequestrin binding protein (junctin) from cardiac junctional sarcoplasmic reticulum. J Biol Chem. 1995;270(51):30787–96. doi: 10.1074/jbc.270.51.30787. [DOI] [PubMed] [Google Scholar]
- 115.Takeshima H, Shimuta M, Komazaki S, Ohmi K, Nishi M, et al. Mitsugumin29, a novel synaptophysin family member from the triad junction in skeletal muscle. Biochem J. 1998;331(Part 1):317–22. doi: 10.1042/bj3310317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pan Z, Yang D, Nagaraj RY, Nosek TA, Nishi M, et al. Dysfunction of store-operated calcium channel in muscle cells lacking mg29. Nat Cell Biol. 2002;4(5):379–83. doi: 10.1038/ncb788. [DOI] [PubMed] [Google Scholar]
- 117.Knudson CM, Stang KK, Moomaw CR, Slaughter CA, Campbell KP. Primary structure and topological analysis of a skeletal muscle-specific junctional sarcoplasmic reticulum glycoprotein (triadin) J Biol Chem. 1993;268(17):12646–54. [PubMed] [Google Scholar]
- 118.Zorzato F, Anderson AA, Ohlendieck K, Froemming G, Guerrini R, Treves S. Identification of a novel 45-kDa protein (JP-45) from rabbit sarcoplasmic-reticulum junctional-face membrane. Biochem J. 2000;351(Part 2):537–43. [PMC free article] [PubMed] [Google Scholar]
- 119.MacKrill JJ. Protein-protein interactions in intracellular Ca2+-release channel function. Biochem J. 1999;337(Pt 3):345–61. [PMC free article] [PubMed] [Google Scholar]
- 120.Komazaki S, Ito K, Takeshima H, Nakamura H. Deficiency of triad formation in developing skeletal muscle cells lacking junctophilin type 1. FEBS Lett. 2002;524(1–3):225–29. doi: 10.1016/s0014-5793(02)03042-9. [DOI] [PubMed] [Google Scholar]
- 121.Ma H, Lou Y, Lin WH, Xue HW. MORN motifs in plant PIPKs are involved in the regulation of subcellular localization and phospholipid binding. Cell Res. 2006;16(5):466–78. doi: 10.1038/sj.cr.7310058. [DOI] [PubMed] [Google Scholar]
- 122.Devine CE, Somlyo AV, Somlyo AP. Sarcoplasmic reticulum and excitation-contraction coupling in mammalian smooth muscles. J Cell Biol. 1972;52(3):690–718. doi: 10.1083/jcb.52.3.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Gabella G. Caveolae intracellulares and sarcoplasmic reticulum in smooth muscle. J Cell Sci. 1971;8(3):601–9. doi: 10.1242/jcs.8.3.601. [DOI] [PubMed] [Google Scholar]
- 124.Isshiki M, Anderson RG. Calcium signal transduction from caveolae. Cell Calcium. 1999;26(5):201–8. doi: 10.1054/ceca.1999.0073. [DOI] [PubMed] [Google Scholar]
- 125.Kuo K, Herrera AM, Seow CY. Ultrastructure of airway smooth muscle. Respir Physiol Neurobiol. 2003;137(2–3):197–208. doi: 10.1016/s1569-9048(03)00147-2. [DOI] [PubMed] [Google Scholar]
- 126.Wray S, Burdyga T. Sarcoplasmic reticulum function in smooth muscle. Physiol Rev. 2010;90(1):113–78. doi: 10.1152/physrev.00018.2008. [DOI] [PubMed] [Google Scholar]
- 127.Moore ED, Voigt T, Kobayashi YM, Isenberg G, Fay FS, et al. Organization of Ca2+ release units in excitable smooth muscle of the guinea-pig urinary bladder. Biophys J. 2004;87(3):1836–47. doi: 10.1529/biophysj.104.044123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Poburko D, Kuo K, Dai J, Lee C, van Breemen C. Organellar junctions promote targeted Ca2+ signaling in smooth muscle: why two membranes are better than one. Trends Pharmacol Sci. 2004;25(1):8–15. doi: 10.1016/j.tips.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 129.Denis V, Cyert MS. Internal Ca2+ release in yeast is triggered by hypertonic shock and mediated by a TRP channel homologue. J Cell Biol. 2002;156(1):29–34. doi: 10.1083/jcb.200111004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Palmer CP, Zhou XL, Lin J, Loukin SH, Kung C, Saimi Y. A TRP homolog in Saccharomyces cerevisiae forms an intracellular Ca2+-permeable channel in the yeast vacuolar membrane. Proc Natl Acad Sci USA. 2001;98(14):7801–5. doi: 10.1073/pnas.141036198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Perktold A, Zechmann B, Daum G, Zellnig G. Organelle association visualized by three-dimensional ultrastructural imaging of the yeast cell. FEMS Yeast Res. 2007;7(4):629–38. doi: 10.1111/j.1567-1364.2007.00226.x. [DOI] [PubMed] [Google Scholar]
- 132.Pichler H, Gaigg B, Hrastnik C, Achleitner G, Kohlwein SD, et al. A subfraction of the yeast endoplasmic reticulum associates with the plasma membrane and has a high capacity to synthesize lipids. Eur J Biochem. 2001;268(8):2351–61. doi: 10.1046/j.1432-1327.2001.02116.x. [DOI] [PubMed] [Google Scholar]
- 133.Olkkonen VM, Levine TP. Oxysterol binding proteins: in more than one place at one time? Biochem Cell Biol. 2004;82(1):87–98. doi: 10.1139/o03-088. [DOI] [PubMed] [Google Scholar]
- 134.Im YJ, Raychaudhuri S, Prinz WA, Hurley JH. Structural mechanism for sterol sensing and transport by OSBP-related proteins. Nature. 2005;437(7055):154–58. doi: 10.1038/nature03923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kaiser SE, Brickner JH, Reilein AR, Fenn TD, Walter P, Brunger AT. Structural basis of FFAT motif-mediated ER targeting. Structure. 2005;13(7):1035–45. doi: 10.1016/j.str.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 136.Raychaudhuri S, Im YJ, Hurley JH, Prinz WA. Nonvesicular sterol movement from plasma membrane to ER requires oxysterol-binding protein-related proteins and phosphoinositides. J Cell Biol. 2006;173(1):107–19. doi: 10.1083/jcb.200510084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Baumann NA, Sullivan DP, Ohvo-Rekilä H, Simonot C, Pottekat A, et al. Transport of newly synthesized sterol to the sterol-enriched plasma membrane occurs via nonvesicular equilibration. Biochemistry. 2005;44(15):5816–26. doi: 10.1021/bi048296z. [DOI] [PubMed] [Google Scholar]
- 138.Loewen CJR, Young BP, Tavassoli S, Levine TP. Inheritance of cortical ER in yeast is required for normal septin organization. J Cell Biol. 2007;179(3):467–83. doi: 10.1083/jcb.200708205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Preuss D, Mulholland J, Franzusoff A, Segev N, Botstein D. Characterization of the Saccharomyces Golgi complex through the cell cycle by immunoelectron microscopy. Mol Biol Cell. 1992;3(7):789–803. doi: 10.1091/mbc.3.7.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Jacobs CW, Adams AE, Szaniszlo PJ, Pringle JR. Functions of microtubules in the Saccharomyces cerevisiae cell cycle. J Cell Biol. 1988;107(4):1409–26. doi: 10.1083/jcb.107.4.1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Estrada P, Kim J, Coleman J, Walker L, Dunn B, et al. Myo4p and She3p are required for cortical ER inheritance in Saccharomyces cerevisiae. J Cell Biol. 2003;163(6):1255–66. doi: 10.1083/jcb.200304030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Prinz WA, Grzyb L, Veenhuis M, Kahana JA, Silver PA, Rapoport TA. Mutants affecting the structure of the cortical endoplasmic reticulum in Saccharomyces cerevisiae. J Cell Biol. 2000;150(3):461–74. doi: 10.1083/jcb.150.3.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Toikkanen JH, Miller KJ, Söderlund H, Jäntti J, Keränen S. The beta subunit of the Sec61p endoplasmic reticulum translocon interacts with the exocyst complex in Saccharomyces cerevisiae. J Biol Chem. 2003;278(23):20946–53. doi: 10.1074/jbc.M213111200. [DOI] [PubMed] [Google Scholar]
- 144.Wiederkehr A, Du Y, Pypaert M, Ferro-Novick S, Novick P. Sec3p is needed for the spatial regulation of secretion and for the inheritance of the cortical endoplasmic reticulum. Mol Biol Cell. 2003;14(12):4770–82. doi: 10.1091/mbc.E03-04-0229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Babour A, Bicknell AA, Tourtellotte J, Niwa M. A surveillance pathway monitors the fitness of the endoplasmic reticulum to control its inheritance. Cell. 2010;142(2):256–69. doi: 10.1016/j.cell.2010.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Walz B, Baumann O. Structure and cellular physiology of Ca2+ stores in invertebrate photoreceptors. Cell Calcium. 1995;18(4):342–51. doi: 10.1016/0143-4160(95)90030-6. [DOI] [PubMed] [Google Scholar]
- 147.Huang J, Liu C, Hughes SA, Postma M, Schwiening CJ, Hardie RC. Activation of TRP channels by protons and phosphoinositide depletion in Drosophila photoreceptors. Curr Biol. 2010;20(3):189–97. doi: 10.1016/j.cub.2009.12.019. [DOI] [PubMed] [Google Scholar]
- 148.Trivedi D, Padinjat R. RdgB proteins: functions in lipid homeostasis and signal transduction. Biochim Biophys Acta. 2007;1771(6):692–99. doi: 10.1016/j.bbalip.2007.04.014. [DOI] [PubMed] [Google Scholar]
- 149.Hardie RC, Raghu P, Moore S, Juusola M, Baines RA, Sweeney ST. Calcium influx via TRP channels is required to maintain PIP2 levels in Drosophila photoreceptors. Neuron. 2001;30(1):149–59. doi: 10.1016/s0896-6273(01)00269-0. [DOI] [PubMed] [Google Scholar]
- 150.Gu Y, Oberwinkler J, Postma M, Hardie RC. Mechanisms of light adaptation in Drosophila photoreceptors. Curr Biol. 2005;15(13):1228–34. doi: 10.1016/j.cub.2005.05.058. [DOI] [PubMed] [Google Scholar]
- 151.Suzuki E, Hirosawa K. Immunolocalization of a Drosophila phosphatidylinositol transfer protein (rdgB) in normal and rdgA mutant photoreceptor cells with special reference to the subrhabdomeric cisternae. J Electron Microsc (Tokyo) 1994;43(4):183–89. [PubMed] [Google Scholar]
- 152.Vihtelic TS, Goebl M, Milligan S, O’Tousa JE, Hyde DR. Localization of Drosophila retinal degeneration B, a membrane-associated phosphatidylinositol transfer protein. J Cell Biol. 1993;122(5):1013–22. doi: 10.1083/jcb.122.5.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Cockcroft S. Phosphatidylinositol transfer proteins couple lipid transport to phosphoinositide synthesis. Semin Cell Dev Biol. 2001;12(2):183–91. doi: 10.1006/scdb.2000.0235. [DOI] [PubMed] [Google Scholar]
- 154.Sparkes IA, Frigerio L, Tolley N, Hawes C. The plant endoplasmic reticulum: a cell-wide web. Biochem J. 2009;423(2):145–55. doi: 10.1042/BJ20091113. [DOI] [PubMed] [Google Scholar]
- 155.Hepler P, Palevitz B, Lancelle S, McCauley M, Lichtschleidl I. Cortical endoplasmic reticulum in plants. J Cell Sci. 1990;96:355–73. [Google Scholar]
- 156.Lucas WJ, Ham B, Kim J. Plasmodesmata—bridging the gap between neighboring plant cells. Trends Cell Biol. 2009;19(10):495–503. doi: 10.1016/j.tcb.2009.07.003. [DOI] [PubMed] [Google Scholar]
- 157.Overall RL, Blackman LM. A model of the macromolecular structure of plasmodesmata. Trends Plant Sci. 1996;1(9):307–11. [Google Scholar]
- 158.Fitzgibbon J, Bell K, King E, Oparka K. Super-resolution imaging of plasmodesmata using 3D-structured illumination microscopy(3D-SIM) Plant Physiol. 2010;153(4):1453–63. doi: 10.1104/pp.110.157941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Heinlein M, Epel BL. Macromolecular transport and signaling through plasmodesmata. Int Rev Cytol. 2004;235:93–164. doi: 10.1016/S0074-7696(04)35003-5. [DOI] [PubMed] [Google Scholar]
- 160.Faulkner CR, Blackman LM, Cordwell SJ, Overall RL. Proteomic identification of putative plasmodesmatal proteins from Chara corallina. Proteomics. 2005;5(11):2866–75. doi: 10.1002/pmic.200401186. [DOI] [PubMed] [Google Scholar]
- 161.Treves S, Vukcevic M, Griesser J, Armstrong C, Zhu MX, Zorzato F. Agonist-activated Ca2+ influx occurs at stable plasma membrane and endoplasmic reticulum junctions. J Cell Sci. 2010;123(Part 23):4170–81. doi: 10.1242/jcs.068387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Hurley JH, Meyer T. Subcellular targeting by membrane lipids. Curr Opin Cell Biol. 2001;13(2):146–52. doi: 10.1016/s0955-0674(00)00191-5. [DOI] [PubMed] [Google Scholar]
- 163.Fischer MA, Temmerman K, Ercan E, Nickel W, Seedorf M. Binding of plasma membrane lipids recruits the yeast integral membrane protein Ist2 to the cortical ER. Traffic. 2009;10(8):1084–97. doi: 10.1111/j.1600-0854.2009.00926.x. [DOI] [PubMed] [Google Scholar]
- 164.Marguet D, Spiliotis ET, Pentcheva T, Lebowitz M, Schneck J, Edidin M. Lateral diffusion of GFP-tagged H2Ld molecules and of GFP-TAP1 reports on the assembly and retention of these molecules in the endoplasmic reticulum. Immunity. 1999;11(2):231–40. doi: 10.1016/s1074-7613(00)80098-9. [DOI] [PubMed] [Google Scholar]
- 165.Snapp EL, Altan N, Lippincott-Schwartz J. Measuring protein mobility by photobleaching GFP chimeras in living cells. Curr Protoc Cell Biol. 2003;Ch. 21(Unit 21.1) doi: 10.1002/0471143030.cb2101s19. [DOI] [PubMed] [Google Scholar]
- 166.Xu P, Lu J, Li Z, Yu X, Chen L, Xu T. Aggregation of STIM1 underneath the plasma membrane induces clustering of Orai1. Biochem Biophys Res Commun. 2006;350(4):969–76. doi: 10.1016/j.bbrc.2006.09.134. [DOI] [PubMed] [Google Scholar]
- 167.van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol. 2008;9(2):112–24. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Yang H. Nonvesicular sterol transport: two protein families and a sterol sensor? Trends Cell Biol. 2006;16(9):427–32. doi: 10.1016/j.tcb.2006.07.002. [DOI] [PubMed] [Google Scholar]