

Abstract
CRACR2A is a newly discovered Ca2+-binding protein that regulates store-operated Ca2+ entry (SOCE). CRACR2A enhances SOCE by promoting the binding of the endoplasmic reticulum Ca2+ sensor STIM to Orai, a Ca2+ channel located in the plasma membrane. As intracellular Ca2+ levels rise, CRACR2A binds Ca2+ and triggers SOCE inactivation by dissociating from the Orai–STIM complex.
Ca2+ is a ubiquitous second messenger that regulates rapid cellular responses such as secretion and cytoskeletal remodelling, and long-term events such as cell proliferation and differentiation1. Cytosolic calcium signalling occurs through an influx of Ca2+ by voltage-, sensory- or ligand-triggered opening of plasma membrane Ca2+ channels and signal-mediated opening of Ca2+ channels in the endoplasmic reticulum (ER). An intriguing, recently discovered process, triggers persistent Ca2+ increases through a sensor that monitors the Ca2+ concentration in the lumen of the ER and directly gates the opening of Ca2+ channels in the plasma membrane when ER Ca2+ levels drop. This process has been termed store-operated Ca2+ entry (SOCE) and is believed to be responsible for T-cell activation and other known functions of long-term Ca2+ signalling in cells2. Although the existence of this SOCE signalling pathway was proposed in 1986 by Putney3, it was not until 2005 and 2006 that two essential components of the molecular machinery responsible for SOCE were discovered: STIM4,5 and Orai6,7,8 (Fig. 1). There are two STIM and three Orai isoforms in many vertebrates, including humans, and one iso-form of each in invertebrates. STIMs are ER transmembrane proteins, with a Ca2+-binding EF-hand domain exposed to the lumen of the ER, which renders the proteins inactive when the ER Ca2+ level is high. Receptor-triggered Ca2+ release from the ER decreases Ca2+ levels and causes Ca2+ dissociation from the EF hand, thereby promoting the relocalization of STIM to a specialized region of the ER close to the plasma membrane that has been termed the ER–plasma membrane junction4 (Fig. 2). At these junctions, STIM interacts with and activates Orai, a four-pass transmembrane protein in the plasma membrane. It is probably a tetramer of Orai proteins that are then assembled to form the Ca2+ channel responsible for Ca2+-release-activated Ca2+ (CRAC) currents9 (often used synonymously with SOCE currents).
Figure 1.

Milestones in the discovery of molecular mechanisms governing SOCE. The figure was provided by Jesus Sanchez (Centro Nacional de Biotecnología, Madrid 28049, Spain).
Figure 2.
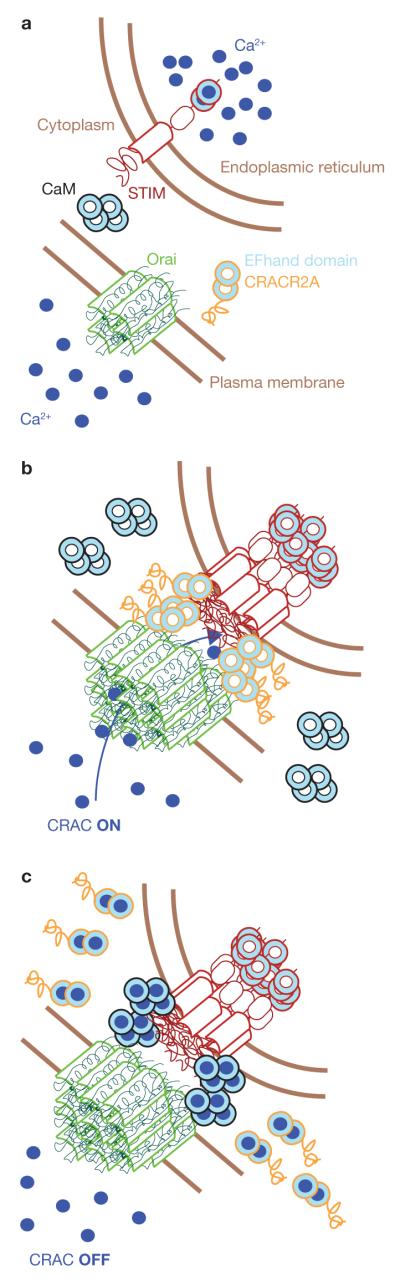
CRACR2A is a novel component of the STIM1–Orai complex that controls SOCE. (a) Cells in resting conditions, with Ca2+ stores filled. (b) Ca2+ depletion in the ER triggers STIM clustering and recruitment to ER–plasma membrane junctions. Orai also accumulates in the junctions, where it interacts with STIM and CRACR2A, inducing channel activation. (c) CRACR2A EF-hand domains bind Ca2+ and trigger the dissociation of CRACR2A from the Orai/STIM complex. This may promote Ca2+/CaM binding to Orai and CRAC inactivation.
Since this initial discovery, studies of STIM1 and Orai1 and their homologues STIM2, Orai2 and Orai3, have mainly provided support for the model that dissociation of Ca2+ from STIM leads to a conformational change, followed by oligomerization and translocation to ER–plasma membrane junctions. Translocation is further mediated by binding of STIM to negatively-charged lipids and to Orai proteins9 (Fig. 2). The same Ca2+-regulated conformational change also allows STIM to become an activator of Orai Ca2+ channels. However, this model does not address the observation that CRAC-mediated Ca2+ influx can be suppressed by an increase in cytosolic Ca2+ concentration, in a process that provides negative feedback regulation when Ca2+ levels stay high for prolonged periods of time. Studies published as early as 1996 showed that calmodulin (CaM) has a function in this Ca2+-dependent inhibition of CRAC channels10, which subsequent experiments have shown is the result of CaM binding to Orai111,12.
The discovery of CRACR2A (CRAC regulator 2A), reported by Srikanth et al. on page 436 in this issue of Nature Cell Biology13, provides a new insight into the regulation of STIM and Orai. The study shows that, at low intracellular Ca2+ levels, CRACR2A enhances the binding of STIM to Orai and thereby promotes SOCE. However, at high Ca2+ levels, CRACR2A dissociates from the channel complex, inhibiting SOCE. The authors also demonstrate that the absence of CRACR2A or expression of a non-activatable version of the protein alter receptor-induced Ca2+ oscillations.
Srikanth et al.13 sought to identify novel Orai1-associated proteins to understand more about how extrinsic components regulate CRAC. Their approach differed from earlier strategies to identify STIM- and Orai-binding proteins by their use of chemical crosslinkers that stabilize potentially weak protein–protein interactions. Orai1 was found to be part of a high-molecular-weight complex that the authors analysed further by mass spectrometry. In cells in which Ca2+ stores were depleted, an uncharacterized 45 kDa protein containing two EF-hands, now called CRACR2A, was identified (in addition to the expected STIM protein). The authors showed that CRACR2A interacts in the absence of Ca2+ with both STIM and Orai. Interestingly, the interaction between CRACR2A and the STIM–Orai complex was attenuated when Ca2+ levels increased. Furthermore, overexpression of a CRACR2A mutant that could not bind Ca2+ markedly enhanced CRAC channel activity, consistent with the authors’ hypothesis that these Ca2+ binding sites are important for dissociation of CRACR2A from STIM and Orai, and subsequent SOCE inactivation.
Functional studies showed that although CRACR2A was not essential for the induction of SOCE, when the protein was depleted using siRNA, Ca2+ entry into cells was significantly reduced and the fraction of STIM and Orai1 accumulating at the ER–plasma membrane junction also decreased. To confirm that the reduced SOCE results in a defect in cellular signalling, the authors used a Jurkat cell line, a classic model for T-cells, in which a persistent Ca2+ response mediates cell activation. In this system, the expression of interleukin-2 receptor was reduced in the absence of CRACR2A.
Based on their binding and functional data, Srikanth et al. propose that CRACR2A is an enhancer of SOCE, which stabilizes the interaction between Orai and STIM in the absence of Ca2+, thereby enhancing CRAC channel activity. However, at a higher cytosolic Ca2+ concentration, CRACR2A dissociates from the complex, precipitating channel inactivation (Fig. 1). These important new findings leave questions unanswered: how is the Orai/STIM/CRACR2A ternary complex formed, and does CRACR2A also participate in the initial steps of STIM oligomerization and STIM/Orai1 clustering? It will be important to determine whether CRACR2A stabilizes dynamic STIM/Orai1 complexes that form independently of CRACR2A, and to assess any potential role for CRACR2A in promoting the translocation of STIM to ER–plasma membrane junctions. Another interesting result from this study is that Orai1 was found to be part of a distinct, larger complex in resting conditions, when stores are filled with Ca2+. This raises the possibility of another step of regulation, whereby Orai1 is recruited from an inactive supramolecular complex to a second complex with STIM and CRACR2A. Finally, it is worth mentioning that the Orai1 residues implicated in CRACR2A binding are the same as those shown previously to bind CaM12, supporting the authors’ hypothesis that Ca2+/CaM and Ca2+-free CRACR2A binding to Orai1 is competitive (Fig. 2). This may indicate the existence of a handover mechanism, whereby the dissociation of CRACR2A allows for binding of Ca2+/CaM to Orai1 resulting in SOCE inactivation, possibly reconciling the earlier studies of Ca2+/CaM-mediated inactivation of Orai1 with these current results.
Although this study focused on CRACR2A, it will be interesting to learn whether its homologue, CRACR2B, has a complementary role in regulating SOCE. However, the apparent differences between the homologues reported in this study, and their relatively low sequence identity, suggests that these proteins have different functions. This raises a number of interesting questions, particularly in regard to dynamic cellular processes such as Ca2+ oscillations, which significantly depend on negative feedback processes. Further studies will be needed to address whether and how differential expression of CRACR2, Orai and STIM isoforms can explain observed differences in cell-type specific dynamic Ca2+ signalling responses.
Thus, although the work of Srikanth et al. enlarges our view of the SOCE field by another small ‘crack’ with addition of a third component to the STIM–Orai complex and a molecular mechanism for the inactivation of SOCE, it raises even more questions about how the newly proposed ternary-active Ca2+ channel complex forms and disassembles within these poorly understood ER-plasma membrane junction sites.
Footnotes
COMPETING FINANCIAL INTERESTS The authors declare no competing financial interests.
References
- 1.Clapham DE. Cell. 2007;131:1047–1058. doi: 10.1016/j.cell.2007.11.028. [DOI] [PubMed] [Google Scholar]
- 2.Parekh AB, Putney JW. Physiol. Rev. 2005;85:757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- 3.Putney JW. Cell Calcium. 1986;7:1–12. doi: 10.1016/0143-4160(86)90026-6. [DOI] [PubMed] [Google Scholar]
- 4.Liou J, et al. Curr. Biol. 2005;15:1235–1241. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roos J, et al. J. Cell Biol. 2005;169:435–445. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vig M, et al. Science. 2006;312:1220–1223. doi: 10.1126/science.1127883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang SL, et al. Proc. Natl Acad. Sci. USA. 2006;103:9357–9362. doi: 10.1073/pnas.0603161103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yeromin AV, et al. Nature. 2006;443:226–229. doi: 10.1038/nature05108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hogan PG, Lewis RS, Rao A. Annu. Rev. Immunol. 2010;28:491–533. doi: 10.1146/annurev.immunol.021908.132550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vaca L. FEBS Lett. 1996;390:289–293. doi: 10.1016/0014-5793(96)00675-8. [DOI] [PubMed] [Google Scholar]
- 11.Parvez S, et al. FASEB J. 2008;22:752–761. doi: 10.1096/fj.07-9449com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mullins FM, Park CY, Dolmetsch RE, Lewis RS. Proc. Natl Acad. Sci. USA. 2009;106:15495–15500. doi: 10.1073/pnas.0906781106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Srikanth S, et al. Nature Cell Biol. 2010;12:436–446. doi: 10.1038/ncb2045. [DOI] [PMC free article] [PubMed] [Google Scholar]