

Abstract
Advanced glycation end products (AGEs) accumulate in patients with diabetes, yet the link between AGEs and the inflammatory and fibrogenic activity in non-alcoholic steatohepatitis (NASH) has not been explored. TNFα converting enzyme (TACE) is at the center of inflammatory processes. As the main natural regulator of TACE activity is the tissue inhibitor of metalloproteinase 3 (Timp3), we hypothesized that AGEs induce TACE through NADPH oxidase 2 (NOX2); and the downregulation of Sirtuin 1 (Sirt1)/Timp3 pathways mediate fibrogenic activity in NASH. The role of NOX2, Sirt1, Timp3 and TACE were evaluated in the choline deficient L-amino acid defined (CDAA) or western diet-fed wild type (wt) and NOX2−/− mice. To restore Timp3, the mice were injected with Ad-Timp3. Sirt1 and Timp3 expressions were studied in livers from NASH patients, and we found that their levels were significantly lower than in healthy controls. In the wt mice on the CDAA or western diet Sirt1 and Timp3 expressions were lower whereas production of reactive oxidative species and TACE activity significantly increased with an increase in active TNFα production, and the induction of fibrogenic transcripts. Ad-Timp3 injection resulted in a significant decline in TACE activity, procollagen α1 (I), αSMA and TGFβ expression. The NOX2−/− mice on the CDAA or western diet had no significant change in Sirt1, Timp3, TACE activity or the fibrosis markers assessed. In vitro, AGEs exposure decreased Sirt1 and Timp3 in hepatic stellate cells by a NOX2-dependent pathway, and TACE was induced after exposure to AGEs.
Conclusion
TACE activation is central to the pathogenesis of NASH, and is mediated by AGEs via NOX2 induction and the downregulation of Sirt1/Timp3 pathways.
Keywords: liver fibrosis, oxidative stress, NADPH oxidase 2, Sirt1, Timp3
Diabetes mellitus is a major risk factor for disease progression with necroinflammation and fibrosis advancing to cirrhotic stage NASH1, 2. The factors implicated in this progression are poorly understood, specifically the effects of diabetes or insulin resistance on fibrogenesis. Advanced glycation end products (AGEs) are produced by a non enzymatic glycation of serum proteins and these modifications significantly influence the structure and function of key protein targets3, 4. AGEs are implicated in diabetic nephropathy, vascular complications and retinopathy5, 6, but their role in inducing inflammatory or fibrogenic changes in the liver have not been adequately explored. Sirtuin 1 (Sirt1) belongs to the class III family of histone deacetylases, and its decreased activity was shown to be linked to the development of NASH7. Accumulating evidence suggests that in NASH hepatic lipid metabolism pathways are affected by Sirt18 and low levels of Sirt1 are implicated in the development of steatosis in animals9. Moreover, in the heterozygous Sirt1 knockout model, when chronically challenged with a 40% fat diet, the mice became obese and insulin resistant, displaying increased serum cytokine levels, and developing hepatomegaly10. Hepatic metabolomic analyses revealed that Sirt1 heterozygous mice had elevated gluconeogenesis and oxidative stress11. Amongst the targets of Sirt1 is the tissue inhibitor of metalloproteinases 3 (Timp3)12, a key regulator and inhibitor of the TNFα converting enzyme (TACE, also called A Metalloprotease and Disintegrin 17, ADAM17) activity. Timp3−/− mice have been described to develop vascular inflammation via increased TNFα13, and hepatic steatosis14.
In this study we showed that exposure to AGEs leads to the downregulation of Sirt 1 and Timp3 in HSC via the activation of NADPH oxidase 2 (NOX2) and the release of reactive oxidative species (ROS). Accordingly, wild type mice fed with the choline deficient L-amino acid defined (CDAA) or western diet showed decreasing Sirt1, Timp3 expression, and an increase in TACE activity, TNFα production and increased fibrogenic activity. Correction of the low TIMP3 levels by injection of Ad-Timp3 into the CDAA or western diet-fed mice reduced TACE activity, TNFα, expression of the receptor of advanced glycation end products (RAGE), and fibrogenic response. NOX2−/− mice on the NASH diets did not develop increased TACE activity or fibrogenic response. In summary, these data suggest an important role of the AGEs in the NOX2-mediated induction of TACE, TNFα and fibrogenic activity during NASH progression.
Experimental Procedures
Liver biopsy samples
The liver biopsy samples were obtained from the UC Davis Cancer Center Biorepository funded by the NCI. Samples from 6 different patients and 6 normal livers were tested. All patients had insulin resistance or diabetes mellitus.
Animal studies
C57BL/6 mice (Jackson Lab) or B6.129S-Cybbtm1Din/J (NOX2−/−, Jackson Lab) were given choline-supplemented L-amino acid-defined (CSAA) or choline-deficient L-amino acid-defined (CDAA) diet (Dyets Inc., Bethlehem, PA) for 10 weeks, or control chow. Both diets contain higher calories, fat and carbohydrates, than standard chow15. The fast food (“western”) diet contains 40% energy as fat (12% SFA, 2% cholesterol (AIN-76, Western diet, Test Diet) and supplemented with high fructose corn syrup (42 g/l)16. To test for the role of Timp3 in TACE regulation, Ad-Timp3 was injected on week 8 via the tail vein (2×107 pfu/200 ul PBS, Applied Biological Materials Inc., Richmond, BC, Canada). As control, Ad-GFP (2×107 pfu/200 ul, Vector Biolabs, Philadelphia PA) was used. In a group of mice gadolinium chloride (GdCl3, 10 mg/kg in saline, Sigma-Aldrich) was injected i.p. every other day throughout the experiment to inhibit macrophages, in both models. The serum and liver tissue were collected and ALT and bilirubin were tested. The tissue was processed for the further assays. The animals were housed in facilities approved by the National Institute of Health. All procedures were reviewed and approved by the Animal Welfare Committee of the University of California Davis.
Cell culture
Primary HSC were isolated either from C57BL/6 mice or Sprague Dawley rats as described previously17. The cells were cultured in medium 199/20% FBS. The primary HSC were used for the experiments within the first 3 days following isolation. The cells were exposed to AGEs (50μg/ml) or BSA in serum free medium for 16 hours and/or transfected with siRNAs. For the siRNA transfection, primary rat HSC were cultured as above for a day then the medium was changed to DMEM, 0.5% FBS. The HSC were transfected with the siRNA to Sirt1 or Timp3 (Santa Cruz Biotechnology Inc., Santa Cruz, CA), or scrambled siRNA using the RiboJuice transfection reagent (EMD Chemicals Inc., Darmstadt, Germany) according to the instructions.
Results
Exposure to AGEs induces Sirt1 and Timp3 downregulation via NOX2 in primary hepatic stellate cells
Activation of NOXs is one of the main sources of ROS in activated HSC. We have previously shown that NOX2 is an important factor in HSC transdifferentiation18 To study the role of AGEs in ROS production, primary cells were transfected with NOX2 or scrambled siRNA and incubated in serum free conditions with glycolaldehyde (GA)-derived AGEs for 16 hours. The superoxide production was significantly increased in control (untransfected) and scrambled siRNA transfected HSC (Figure 1A, 1.74±0.13 fold, p<0.05) whereas this was attenuated in the NOX2 siRNA transfected cells (Figure 1A). To study the effects of AGEs/ROS on Sirt1 and Timp3 expression, real time qPCR was done on the cells from the above experiment. Sirt1 and Timp3 were downregulated in response to AGEs, in a NOX2-dependent manner (Figure 1B, 0.38±0.14-fold, p<0.05; 0.58±0.10-fold, p<0.05 respectively). To test if Sirt1 directly targets Timp3 in primary HSC, the cells were transfected with the Sirt1 siRNA, and the Timp3 expression was found to be downregulated in response to the inhibition of Sirt1 (0.36-fold±0.16, N=3, p<0.05, Figure 1C).
Figure 1. AGEs induce ROS production and downregulate Sirt1 and Timp3 expression via NOX2.

Primary HSC were treated with either scrambled siRNA or NOX2 siRNA for 48hr, followed by GA-derived AGEs exposure or BSA (control) for 16hr. Lucigenin assay showed that superoxide production was significantly increased by AGEs in both untransfected (NT) (*p<0.05) or scrambled siRNA transfected (Scr) HSC (*p<0.05), whereas knock-down of NOX2 attenuated the superoxide production (mean±SED, *p<0.05, N=4) (A). Sirt1 and Timp3 mRNA expression were assessed by real-time qPCR. After treating with AGEs for 16 hrs, Sirt1 and Timp3 mRNA levels decreased in NT and Scr siRNA transfected HSC (shown in fold expression, *p<0.05, N=4), but not in NOX2 siRNA transfected HSC (B). Timp3 mRNA expression decreased in response to siRNA knockdown of Sirt1 (*p<0.05, N=4) as assessed by real-time qPCR (C).
AGEs induce TACE activity in hepatic stellate cells
As Timp3 is the main natural inhibitor of TACE activity, we next examined if TACE could be induced by AGEs. This was studied by two different methods: fluorometry (Figure 2A) and immunoprecipitation with western blotting to assess TACE tyrosine phosphorylation which correlates with the active state of the enzyme (Figure 2B). TACE activity was induced by AGEs in the scrambled siRNA transfected HSC (1.48-fold±0.06,** p<0.01, Figure 2A), and this was significantly attenuated in NOX2 siRNA transfected HSC (1.10±0.09-fold, p<0.05). In addition, inhibiting Sirt1 by siRNA also resulted in an increase in TACE activity (1.43±0.10-fold, p<0.05) which was further increased by AGEs (2.29-fold±0.23,* p<0.05). TACE was phosphorylated following exposure of HSC to AGEs, indicating the activated state of the enzyme (Figure 2B). To discern whether the other major cell types in the liver contribute to the AGEs-induced TACE activation, primary hepatocytes, HSC, and Kupffer cells were incubated either with BSA or AGEs. The TACE activity of hepatocytes was very low and not induced by AGEs (Figure 2C). Only HSC responded with a significant increase (1.73±0.14-fold, p<0.05), and while the baseline activity of Kupffer cells was higher than that of hepatocytes; no induction was seen after incubation with AGEs.
Figure 2. AGEs induce TACE activity in hepatic stellate cells.

After transfection with the scrambled siRNA, NOX2 siRNA or SirT1 siRNA for 48hr, primary HSC were exposed to AGEs or BSA for 16 hrs, and TACE activity studied by fluorometry. TACE activity increased after AGEs treatment in both non-transfected (mean±SED, *p<0.05, N=4) and Scr siRNA transfected (**p<0.01, N=4) groups. This was significantly attenuated in the NOX2 siRNA transfected cells (*p<0.05, N=4). The TACE activity was also induced in response to knockdown of Sirt1 (*p<0.05, N=4) (A). The TACE activity was also analyzed by immunoprecipitation and Western blotting with anti-p-Tyrosine. TACE was phosphorylated after exposure to AGEs (B). TACE activity was tested in isolated primary hepatocytes, HSC and Kupffer cells following incubation with BSA or AGEs. Only HSC TACE activity was significantly induced by AGEs (mean±SED, *p<0.05, N=3). Hepatocytes had a very low TACE activity while Kupffer cells did not exhibit induction after AGEs (C).
Sirt1 and Timp3 are downregulated in the livers of patients with NASH
To study Sirt1 and Timp3 in human livers with NASH, real-time qPCR was performed on liver biopsy samples from different patients with grade 2–3, stage 3–4 NASH, and healthy controls (Figure 3). The patients represented here had either insulin resistance with fasting hyperglycemia or type II DM. The expression of both Sirt1 and Timp3 were significantly decreased in NASH livers compared to normal healthy controls (expressed as fold over control, 0.47-fold±0.11 *p<0.05 and 0.24-fold±0.07, **p<0.01, respectively, N=6, Figure 3). The expression of RAGE was also significantly increased in the patients with NASH (3.93-fold± 0.2, *p<0.05).
Figure 3. Sirt1 and Timp3 are downregulated whereas RAGE expression is induced in NASH patients.
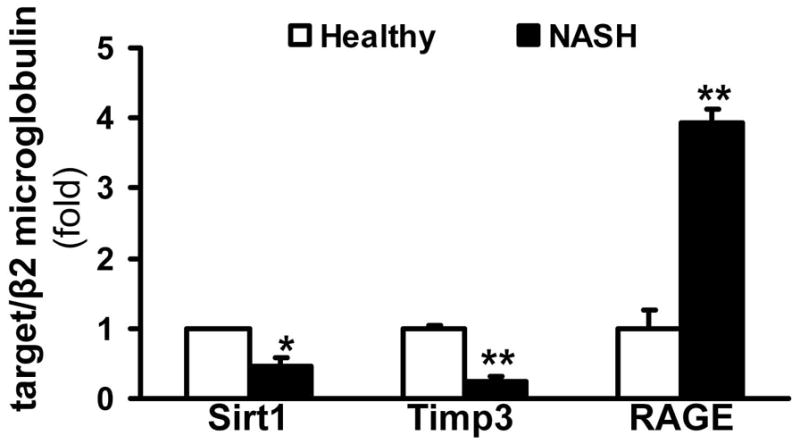
Real-time qPCR was performed on the liver samples from patients with NASH, and healthy controls. Sirt1 and Timp3 were significantly downregulated in livers with NASH (data expressed as fold over values from healthy controls, which was set as 1, mean±SED, *p<0.05, **p<0.01 respectively, N=6). Expression of RAGE was significantly increased in the NASH patients (**p<0.01).
NOX2-dependent ROS production is increased in two different diet models of NASH and is involved in the regulation of Sirt1 and Timp3
To recapitulate our in vitro findings, wild type or NOX2−/− mice were fed either with the CDAA, CSAA, or western diets. The CDAA model was shown to recapitulate the features of human NASH with insulin resistance, inflammatory cell infiltration, hepatocyte death, and liver fibrosis19, 20, whereas mice on the CSAA diet mainly develop steatosis, but no necroinflammation or fibrosis. The advantage of using the CSAA/CDAA model is that we can correlate our findings to a model resulting in simple steatosis or progressive NASH. To confirm our findings we also performed the experiments in mice fed the western diet supplemented with high fructose16. This diet was shown to have higher fidelity to the human pathophysiology with the mice exhibiting significant weight gain, metabolic syndrome, diabetes and histologically developing ballooning, lipoapoptosis, necroinflammation and fibrosis. The body weight has increased similarly both in the wild type and NOX2−/− mice on both NASH diets (Figure 4A). The wild type mice on the CDAA or western diet had significantly increased ALT (**p<0.01) compared to the mice on the CSAA or control chow diet. The mice on the CDAA and western diet had increased glucose levels (216.0mg/dl±36.2, and 239.4mg/dl±12.8, respectively), and these diets were previously shown to induce impaired glucose tolerance or diabetes15, 16. AGEs were increased in both diet models in the wild type but not in the NOX2−/− mice (Supplementary Figure 1). The increase in bilirubin in the wild type CDAA mice may reflect impaired synthetic function, as these mice had more severe steatohepatitis and fibrosis. In the NOX2−/− mice the ALT showed an increasing trend on the NASH diets compared to baseline, albeit not significant, and below the level of liver injury observed in the wild type mice, whilst the bilirubin has not changed. On histology all mice on the CDAA or western diets displayed increased steatosis (Figure 4B).
Figure 4. NOX2-dependent ROS production is increased in two diet models of NASH and is involved in the regulation of Sirt1 and Timp3.


WT and NOX2−/− mice were fed with the chow, CSAA, CDAA or western diet. Compared to the wt mice on the chow or CSAA diet, the wt CDAA and western diet-fed mice displayed an increase in weight; a significant increase of serum ALT (**p<0.01, N=6) and in the CDAA mice in total bilirubin. In the NOX2−/− mice there was no significant increase in ALT or bilirubin. (A). H&E staining showed that the wt and NOX2−/− mice on the CDAA and western diets had increased steatosis (B). The lucigenin assay demonstrated significantly increased superoxide production in the wt mice on the CDAA and western diets compared to those on the chow and CSAA diets (*p<0.05, N=6). No increase was seen in the NOX2−/− mice (*p<0.05, N=6) (C, and D). SirT1 andTimp3 mRNA expression was assessed by real-time qPCR in the wt and NOX2−/− mice. SirT1 and Timp3 expression significantly decreased in the wt CDAA (E) and western diet (F) fed mice (**p<0.01 and *p<0.05, respectively, N=6). In contrast, in NOX2−/− mice Sirt1 andTimp3 expression remained unchanged.
The ROS production has significantly increased in the wt mice on the CDAA or western diet (Figure 4C and D: 1.57-fold±0.16, *p<0.05, and 1.75-fold ± 0.01,* p<0.05) whereas this was attenuated in the NOX2−/− mice in both models (to 0.94-fold±0.11, and 0.89-fold±0.26, *p<0.05). To correlate the data to humans with NASH, quantitative PCR was performed in all experimental conditions (Figure 4E and F). The wild type CDAA or western diet fed mice showed a significant decrease in Sirt1 (to 0.10-fold±0.04, **p<0.01, and 0.11-fold±0.031 *p<0.05, respectively, N=6) and Timp3 expression (to 0.21-fold±0.04, *p<0.05, and 0.43-fold±0.01, *p<0.05, respectively, N=6). In contrast, in the NOX2−/− mice Sirt1 and Timp3 expression was not affected by the CDAA or western diet.
Correction of the low Timp3 expression in wild type mice on the CDAA or western diets by Ad-Timp3 ameliorates TACE and TNFα activity
To confirm the causal link between the low Timp3 expression leading to an unopposed increase in TACE activity and consequent induction of TNFα activity in vivo, we injected Ad-Timp3 or Ad-GFP into wt mice on the CDAA or western diet. Correcting the Timp3 levels resulted in a significant decrease in TACE activity (Figure 5A, B), and TNFα activity (Figure 5C, D) in both diet models. TACE and TNFα activity showed no significant changes in NOX2−/− mice on either diet. As Kupffer cells also express NOX2, a group of mice in both models were also injected by gadolinium chloride (GdCl3) to inhibit the function of these cells and elucidate the respective role of HSC and Kupffer cells in AGEs-induced TACE activation in NASH. Macrophage inhibition did not change the TACE activity in either diet model, but showed a decreasing trend for active TNFα. The triglyceride (TG) content of the liver has significantly increased on both diets in both genotypes, and Ad-Timp3 did not affect the TG content (Figure 5E, and F, p<0.05, N=6 for each group).
Figure 5. Correction of the low Timp3 expression by Ad-Timp3 in the wild type mice on the CDAA and western diet ameliorates TACE and TNFα activity.


The wt mice on the CDAA or western diets were injected with Ad-Timp3 or Ad-GFP, and the TACE activity in the liver was tested. The TACE activity significantly increased in the wt mice on the CDAA diet compared to the CSAA group (mean±SED, *p<0.05, N=6), and in the western diet fed mice (mean±SED, *p<0.05, N=6). This was significantly attenuated in the Ad-Timp3 injected mice in both diets (*p<0.05, N=6, each group). NOX2−/− mice did not exhibit an increase in TACE activity on either diet (*p<0.05, N=6) (A, B). Gadolinium chloride (Gad) was injected into a group of mice on both diets and genotypes to assess the role of Kupffer cells. The TACE activity in the liver was not significantly affected by inhibiting the macrophages. The TNFα protein level was assessed by ELISA in the above conditions. Compared to the mice on the chow and CSAA diets, the wt CDAA or western diet fed mice had significantly higher TNFα production (*p<0.05, N=6) and the Ad-Timp3 prevented this induction (*p<0.05). In the NOX2−/− mice on either diet no increase in the TNFα production was seen (C, D). In the Gad-treated mice there was a trend towards lower TNFα, albeit not significant. The triglyceride content was tested in all groups and both genotypes (E and F). Both diets induced a significant accumulation of TG (*p<0.05, N=6) however; this was not improved by the lack of NOX2; correcting the Timp3 levels, or by Gad injection.
Interestingly, the expression of RAGE showed an increase in the livers of the wt mice on the CDAA diet (2.34-fold±0.17, p<0.05, Suppl. Figure 2), and this was attenuated by Ad-Timp3 (0.86-fold±0.14, p<0.05). No increase in RAGE was seen in the NOX2−/− mice.
CDAA and western diets induce fibrogenic activity in wt mice which is attenuated following Ad-Timp3 injection and in the NOX2−/− mice
Correction of Timp3 may affect fibrogenesis. Therefore, procollagen α1(I), αSMA and TGFβ were tested by real time qPCR and the liver tissues examined following picrosirius red staining and hydoxyproline assay. The procollagen α1(I), αSMA and TGFβ expression were significantly elevated both in the CDAA (5.91-fold±1.85, **p<0.01, 1.41-fold±0.07, **p<0.01; and 3.37- fold±0.76, respectively, p<0.05, N=6, Figure 6A) and western diet-fed mice (1.58-fold±0.12, *p<0.05; 1.75-fold±0.001, *p<0.05; and 1.51-fold±0.13fold, *p<0.05). The expression of these fibrogenic transcripts significantly decreased following Ad-Timp3 injection in both mouse models: in the CDAA diet fed mice procollagen α1(I) to 1.54-fold±0.93, *p<0.05, αSMA to 1.05-fold±0.31,* p<0.05, and TGFβ to 0.96-fold±0.1, p<0.05 In the western diet fed mice following Ad-Timp3 transduction the procollagen α1(I) decreased to 0.76-fold±0.1, *p<0.05; αSMA to 0.62-fold±0.09,* p<0.05, and TGFβ to 0.63-fold±0.1, *p<0.05. These data were confirmed also by the picrosirius red staining and morphometry (Figure 6C, D, and E) and hydroxyproline assay (Figure 7). Injection of the GdCl3 caused a decrease in the expression of αSMA and TGFβ in the CDAA model but not in the western diet. In the NOX2−/− mice on the NASH diets, the fibrosis was less pronounced with an attenuated procollagen α1 (I) and αSMA expression, and a significant decrease in picrosirius red positive area (Figure 6 and 7, *p<0.05) and hydroxyproline quantity (Figure 7,** p<0.01).
Figure 6. The CDAA and western diet-induced fibrogenic activity is attenuated by Ad- Timp3 injection, and also in the NOX2−/− livers.
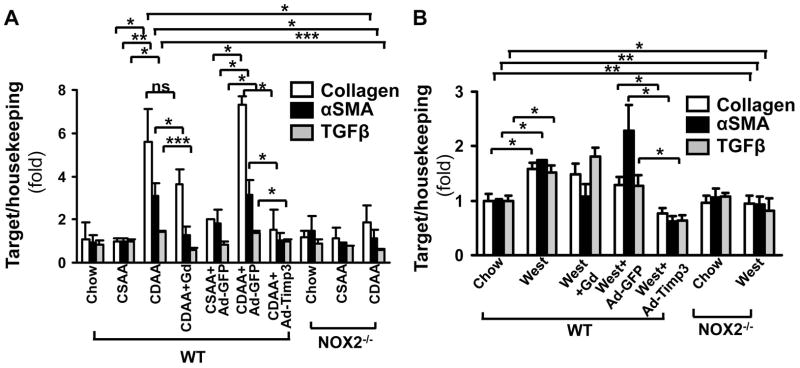

Procollagen α1(I), αSMA and TGFβ expression was analyzed by real-time qPCR. mRNA levels of these transcripts were significantly increased in CDAA and western diet-fed mouse livers injected with the control vector (**p<0.01 and *p<0.05, respectively, N=6), and attenuated by the Ad-Timp3 injection (*p<0.05, N=6). No significant induction of these transcripts was seen in the NOX2−/− group on either diet. Gadolinium injection attenuated the increase in αSMA and TGFβ in the CDAA mice (*p<0.05, ***p<0.001) but not in the western diet fed mice. (A, B). After picrosirius staining (C) and ImageJ analysis (D) in the wt CDAA or western diet-fed and control vector injected mice, the fibrotic area (red, pericellular fibrosis) was significantly increased (*p<0.05, **p<0.01, N=6). In mice injected with Ad-Timp3, the picrosirius positive area decreased (*p<0.05). Also, in the NOX2−/− mice on both diets the fibrotic area was significantly lower than in the wt mice (expressed as fold compared to the samples after chow diet,*p<0.05, **p<0.01 N=6).
Figure 7. The hydroxyl proline content decreased after Ad-Timp3 in the CDAA and western diet fed mice.

OH-proline incorporation assay was performed to assess the amount of collagen in the liver. Compared to the mice on the chow or CSAA diet, the CDAA (A) and western diet (B) fed mice displayed a significant increase in OH-proline incorporation (*p<0.05, N=6). Ad-Timp3 lowered the OH-proline content in the wt mice in both models. In the NOX2−/− mice the OH-proline incorporation was also significantly lower in both models (*p<0.05, N=6).
Discussion
Insulin resistance and diabetes mellitus are major risk factors in progression of NASH. Several important aspects of the pathogenesis are well-defined but how diabetes creates a proinflammatory and profibrogenic milieu, is less clear. Here we have shown that AGEs/NOX2/ROS -induced decrease in Sirt1 expression in HSC culminates in an induction of TACE and consequently TNFα activity. Loss of Sirt1 was shown to be associated with metabolic diseases such as type 2 diabetes, atherosclerosis21, and NASH21. On the other hand, hepatic Sirt1 deficiency was described to induce insulin resistance and an accumulation of ROS was also detected in the liver10. Treating these mice with antioxidants, reversed the phenotype suggesting the key role of ROS in this process. Defining the source of ROS, and analyzing the downstream effects of Sirt1 downregulation are of a great importance.
AGEs are formed at a highly accelerated rate during hyperglycemia by a non enzymatic glycation of serum proteins. The process begins with the conversion of reversible Schiff base adducts to more stable, covalently bound products and eventually the irreversibly bound moieties known as AGEs are formed22, 23. AGE levels in the serum or liver were found to be elevated in patients with steatohepatitis compared to healthy controls or patients with simple steatosis24. The interaction of AGEs with the associated cell surface receptor RAGE has been linked to the induction of oxidative stress in the liver25 and it was postulated that activation of one of the NOXs was the source of ROS production. The specific NOX however, has not been identified. Here we described the key role of NOX2 activation in AGEs-induced ROS production and the downregulation of Sirt1/Timp3 pathways. NOX2 is a phagocytic NADPH oxidase, and we and others have shown that it is highly expressed and enzymatically active in HSC during liver fibrosis18,26. NOX2 directly induces HSC activation, production of collagen I by inducing the promoter activity via H2O2, and NOX2−/− mice develop significantly less fibrosis in the bile duct ligation18 and CCl4 26 models. AGEs have been described to induce ROS formation via NOXs in other systems27, 28, and the postulated mechanism could involve the activation of the PKCα in the kidney28 or PKCδ in the neuronal tissue29 by AGEs. Whether in active HSC other NOXs can elicit a similar response to AGEs could be further investigated in the future. As p47phox knockout HSC had a decrease in ROS production following incubation with AGEs, it is possible that NOX1 also plays a role as a regulator of Sirt125, as p47phox is a common subunit to both NOX1 and 2. In our study demonstrated that in the NOX2−/− mice there was a significant attenuation of fibrosis on both diets. As Kupffer cells also express NOX2, we treated mice with gadolinium to inhibit their function. We found that this did not interfere with TACE activity in the liver. The TACE activity of Kupffer cells was not induced by AGEs in vitro therefore their profibrogenic role during NASH could be attributed to other more dominant mechanisms such as TRL4-mediated induction of HSC30. Steatosis was not affected by the lack of NOX2, corresponding to earlier data from the MCD model31; and it was not influenced by correcting Timp3, either. This suggests that AGEs mainly induced oxidative and inflammatory pathways in these models. The role of Sirt1 downregulation in HSC during NASH progression has not been studied in detail. As the main source of TACE activity in the liver are activated HSC32, and the natural regulator of TACE is Timp3, we sought to determine if low Sirt1 could translate into a downregulation of Timp3, and consequent induction of TACE. First, we confirmed that active HSC are a potent source of TACE production in the liver and that exposure to AGEs directly induces pathways leading to TACE activation whereas hepatocytes and Kupffer cells do not respond to AGE induction. Second, AGEs induced a NOX2-dependent downregulation of Sirt1 and Timp3, and both of these transcripts were downregulated in humans with NASH and also in mice on both diets. Correlating to this, the TACE activity was increased by AGEs and also in the wt mice on the CDAA and western diets, but not on the CSAA diet (steatosis). In contrast, no increase in TACE activity was seen in the NOX2−/− mice on the CDAA and western diets implying that NOX2-derived ROS plays a key a role in TACE activation. To confirm the role of Timp3 in NASH, we injected a group of mice on the CDAA diet with Ad-Timp3. This resulted in a decline in TACE and TNFα activity, and RAGE expression. In addition, Ad-Timp3 transduction also improved fibrosis with the downregulation of profibrogenic transcripts and lowering the hydroxyproline content. As TACE targets multiple other cytokines and chemokines and their receptors33; reducing its activity may have led to a decreased production of fibrogenic mediators. For instance, TACE is known to induce ectodomain shedding of fractalkine (CX3CL1)34 thus decrease in fractalkine could have also contributed to the improvement in fibrogenic activity. Mice deficient in Timp3 demonstrated elevated levels of TNFα and developed insulin resistance and hepatic steatosis, mediated by increased TACE activity10, 21.
In conclusion, we have demonstrated the central role of Sirt1/Timp3/TACE cascade in the AGEs-induced proinflammatory and profibrogenic activity in NASH. Modulation of Timp3 or TACE activity could thus become a successful approach to halt disease progression in NASH.
Supplementary Material
Table 1.
Primer sequences used in the studies
| Rat Timp3 | Forward: 5′-TACCCTGGCTATCAGTCCAAACA-3′ |
| Reverse: 5′-GCTGCAGTAGCCACCCTTCT-3′ | |
| Rat Sirt1 | Forward: 5′-TGAAGCTGTTCGTGGAGATATTTTT-3′ |
| Reverse: 5′-CATGATGGCAAGTGGCTCAT-3′ | |
| Rat Arbp | Forward: 5′-AAGGAGGACCTCACCGAGAT-3′ |
| Reverse: 5′-CCCTCTAGGAAGCGAGTGTG-3′ | |
| Mouse Sirt1 | Forward: 5′-TCCTCACTAATGGCTTTCATTCCT-3′ |
| Reverse: 5′-CGCGGAGTCCAGTCACTAGAG-3′ | |
| Mouse Timp3 | Forward: 5′-GGCCTCAATTACCGCTACCA-3′ |
| Reverse: 5′-CTGATAGCCAGGGTACCCAAAA-3′ | |
| Mouse Collagen IA1 | Forward: 5′-AGAGGCGAAGGCAACAGTCG-3′ |
| Reverse: 5′-GCAGGGCCAATGTCTAGTCC-3′ | |
| Mouse αSMA | Forward: 5′-TCAGCGCCTCCAGTTCCT-3′ |
| Reverse: 5′-AAAAAAAACCACGAGTAACAAATCAA-3′ | |
| Mouse TNF-α | Forward: 5′-TCCCAGGTTCTCTTCAAGGGA-3′ |
| Reverse: 5′-GGTGAGGAGCACGTAGTCGG-3′ | |
| Mouse β-Actin | Forward: 5′-ACGGCCAGGTCATCACTATTG-3′ |
| Reverse: 5′-ATACCCAAGAAGGAAGGCTGGA-3′ | |
| Mouse RAGE | Forward: 5′-GCTGTAGCTGGTGGTCAGAACA-3′ |
| Reverse: 5′-CCCCTTACAGCTTAGCACAAGTG-3 | |
| Mouse TGFβ | Forward: 5′-CATGGAGCTGGTGAAACGG-3′ |
| Reverse: 5′-GCCTTAGTTTGGACAGGATCTGG-3 | |
| Mouse MMP13 | Forward: 5′-TTCTGGTCTTCTGGCACACGCTTT-3′ |
| Reverse: 5′-CCAAGCTCATGGGCAGCAACAATA-3 | |
| Human Sirt1 | Forward: 5′-GCCAGAGTCCAAGTTTAGAAGA-3′ |
| Reverse: 5′-CCATCAGTCCCAAATCCAG-3′ | |
| Human Timp3 | Forward: 5′-CCAGGACGCCTTCTGCAAC-3′ |
| Reverse: 5′-CCCCTCCTTTACCAGCTTCTTC-3′ | |
| Human RAGE | Forward: 5′-GCCAGAAGGTGGAGCAGTAG -3′ |
| Reverse: 5′-CCAGTGGATTTGAGGAGAGG-3′ | |
| Human β2-Microglobulin | Forward: 5′-TTCTGGCCTGGAGGCTATC-3′ |
| Reverse: 5′-TCAGGAAATTTGACTTTCCATTC-3′ |
Acknowledgments
This study was supported by the DK083283 (NJT)
List of Abbreviations
- HSC
hepatic stellate cells
- AGEs
Advanced glycation end products, TNFα, Tumor Necrosis Factor
- CDAA
choline deficient L-amino acid defined
- αSMA
α-smooth muscle actin
- NADPH oxidase
nicotinamide adenine dinucleotide phosphate reduced oxidase
- Timp3
Tissue Inhibitor of Metalloproteinase 3
References
- 1.Chiang DJ, Pritchard MT, Nagy LE. Obesity, diabetes mellitus, and liver fibrosis. Am J Physiol Gastrointest Liver Physiol. 2011;300:G697–702. doi: 10.1152/ajpgi.00426.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Whitlock G, Lewington S, Sherliker P, et al. Body-mass index and cause-specific mortality in 900 000 adults: collaborative analyses of 57 prospective studies. Lancet. 2009;373:1083–96. doi: 10.1016/S0140-6736(09)60318-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vlassara H. Advanced glycation in health and disease: role of the modern environment. Ann N Y Acad Sci. 2005;1043:452–60. doi: 10.1196/annals.1333.051. [DOI] [PubMed] [Google Scholar]
- 4.Vlassara H, Striker GE. AGE restriction in diabetes mellitus: a paradigm shift. Nat Rev Endocrinol. 2011 doi: 10.1038/nrendo.2011.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stitt AW, Jenkins AJ, Cooper ME. Advanced glycation end products and diabetic complications. Expert Opin Investig Drugs. 2002;11:1205–23. doi: 10.1517/13543784.11.9.1205. [DOI] [PubMed] [Google Scholar]
- 6.Zong H, Ward M, Stitt AW. AGEs, RAGE, and diabetic retinopathy. Curr Diab Rep. 2011;11:244–52. doi: 10.1007/s11892-011-0198-7. [DOI] [PubMed] [Google Scholar]
- 7.Herranz D, Serrano M. SIRT1: recent lessons from mouse models. Nat Rev Cancer. 2010;10:819–23. doi: 10.1038/nrc2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tao R, Wei D, Gao H, et al. Hepatic FoxOs regulate lipid metabolism via modulation of expression of the nicotinamide phosphoribosyltransferase gene. J Biol Chem. 2011;286:14681–90. doi: 10.1074/jbc.M110.201061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schug TT, Li X. Sirtuin 1 in lipid metabolism and obesity. Ann Med. 2011;43:198–211. doi: 10.3109/07853890.2010.547211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang RH, Kim HS, Xiao C, et al. Hepatic Sirt1 deficiency in mice impairs mTorc2/Akt signaling and results in hyperglycemia, oxidative damage, and insulin resistance. J Clin Invest. 2011;121:4477–90. doi: 10.1172/JCI46243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Purushotham A, Xu Q, Li X. Systemic SIRT1 insufficiency results in disruption of energy homeostasis and steroid hormone metabolism upon high-fat-diet feeding. Faseb J. 2012;26:656–67. doi: 10.1096/fj.11-195172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cardellini M, Menghini R, Martelli E, et al. TIMP3 is reduced in atherosclerotic plaques from subjects with type 2 diabetes and increased by SirT1. Diabetes. 2009;58:2396–401. doi: 10.2337/db09-0280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Federici M, Hribal ML, Menghini R, et al. Timp3 deficiency in insulin receptor-haploinsufficient mice promotes diabetes and vascular inflammation via increased TNF-alpha. J Clin Invest. 2005;115:3494–505. doi: 10.1172/JCI26052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Menghini R, Menini S, Amoruso R, et al. Tissue inhibitor of metalloproteinase 3 deficiency causes hepatic steatosis and adipose tissue inflammation in mice. Gastroenterology. 2009;136:663–72. e4. doi: 10.1053/j.gastro.2008.10.079. [DOI] [PubMed] [Google Scholar]
- 15.Kodama Y, Kisseleva T, Iwaisako K, et al. c-Jun N-terminal kinase-1 from hematopoietic cells mediates progression from hepatic steatosis to steatohepatitis and fibrosis in mice. Gastroenterology. 2009;137:1467–1477. e5. doi: 10.1053/j.gastro.2009.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Charlton M, Krishnan A, Viker K, et al. Fast food diet mouse: novel small animal model of NASH with ballooning, progressive fibrosis, and high physiological fidelity to the human condition. Am J Physiol Gastrointest Liver Physiol. 2011;301:G825–34. doi: 10.1152/ajpgi.00145.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Geerts A, Niki T, Hellemans K, et al. Purification of rat hepatic stellate cells by side scatter-activated cell sorting. Hepatology. 1998;27:590–8. doi: 10.1002/hep.510270238. [DOI] [PubMed] [Google Scholar]
- 18.Jiang JX, Venugopal S, Serizawa N, et al. Reduced nicotinamide adenine dinucleotide phosphate oxidase 2 plays a key role in stellate cell activation and liver fibrogenesis in vivo. Gastroenterology. 2010;139:1375–84. doi: 10.1053/j.gastro.2010.05.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miura K, Kodama Y, Inokuchi S, et al. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology. 2010;139:323–34. e7. doi: 10.1053/j.gastro.2010.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang B, Majumder S, Nuovo G, et al. Role of microRNA-155 at early stages of hepatocarcinogenesis induced by choline-deficient and amino acid-defined diet in C57BL/6 mice. Hepatology. 2009;50:1152–61. doi: 10.1002/hep.23100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Purushotham A, Schug TT, Xu Q, et al. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 2009;9:327–38. doi: 10.1016/j.cmet.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Glomb MA, Monnier VM. Mechanism of protein modification by glyoxal and glycolaldehyde, reactive intermediates of the Maillard reaction. J Biol Chem. 1995;270:10017–26. doi: 10.1074/jbc.270.17.10017. [DOI] [PubMed] [Google Scholar]
- 23.Bucala R, Cerami A. Advanced glycosylation: chemistry, biology, and implications for diabetes and aging. Adv Pharmacol. 1992;23:1–34. doi: 10.1016/s1054-3589(08)60961-8. [DOI] [PubMed] [Google Scholar]
- 24.Hyogo H, Yamagishi S, Iwamoto K, et al. Elevated levels of serum advanced glycation end products in patients with non-alcoholic steatohepatitis. J Gastroenterol Hepatol. 2007;22:1112–9. doi: 10.1111/j.1440-1746.2007.04943.x. [DOI] [PubMed] [Google Scholar]
- 25.Guimaraes EL, Empsen C, Geerts A, et al. Advanced glycation end products induce production of reactive oxygen species via the activation of NADPH oxidase in murine hepatic stellate cells. J Hepatol. 2010;52:389–97. doi: 10.1016/j.jhep.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 26.Paik YH, Iwaisako K, Seki E, et al. The nicotinamide adenine dinucleotide phosphate oxidase (NOX) homologues NOX1 and NOX2/gp91(phox) mediate hepatic fibrosis in mice. Hepatology. 2011;53:1730–41. doi: 10.1002/hep.24281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wautier MP, Chappey O, Corda S, et al. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am J Physiol Endocrinol Metab. 2001;280:E685–94. doi: 10.1152/ajpendo.2001.280.5.E685. [DOI] [PubMed] [Google Scholar]
- 28.Thallas-Bonke V, Thorpe SR, Coughlan MT, et al. Inhibition of NADPH oxidase prevents advanced glycation end product-mediated damage in diabetic nephropathy through a protein kinase C-alpha-dependent pathway. Diabetes. 2008;57:460–9. doi: 10.2337/db07-1119. [DOI] [PubMed] [Google Scholar]
- 29.Nitti M, Furfaro AL, Traverso N, et al. PKC delta and NADPH oxidase in AGE-induced neuronal death. Neurosci Lett. 2007;416:261–5. doi: 10.1016/j.neulet.2007.02.013. [DOI] [PubMed] [Google Scholar]
- 30.Seki E, De Minicis S, Osterreicher CH, et al. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nature Medicine. 2007;13:1324–1332. doi: 10.1038/nm1663. [DOI] [PubMed] [Google Scholar]
- 31.De Minicis S, Seki E, Paik YH, et al. Role and cellular source of nicotinamide adenine dinucleotide phosphate oxidase in hepatic fibrosis. Hepatology. 2010;52:1420–30. doi: 10.1002/hep.23804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bourd-Boittin K, Basset L, Bonnier D, et al. CX3CL1/fractalkine shedding by human hepatic stellate cells: contribution to chronic inflammation in the liver. J Cell Mol Med. 2009;13:1526–35. doi: 10.1111/j.1582-4934.2009.00787.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cerretti DP. Characterization of the tumour necrosis factor alpha-converting enzyme, TACE/ADAM17. Biochem Soc Trans. 1999;27:219–23. doi: 10.1042/bst0270219. [DOI] [PubMed] [Google Scholar]
- 34.Garton KJ, Gough PJ, Blobel CP, et al. Tumor necrosis factor-alpha-converting enzyme (ADAM17) mediates the cleavage and shedding of fractalkine (CX3CL1) J Biol Chem. 2001;276:37993–8001. doi: 10.1074/jbc.M106434200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.