

Abstract
Stereoselectivity is a hallmark of biomolecular processes from catalysis to self-assembly, which predominantly occur between homochiral species. However, both homochiral and heterochiral complexes of synthetic polypeptides have been observed where stereoselectivity hinges on details of intermolecular interactions. This raises the question whether general rules governing stereoselectivity exist. A geometric ridges-in-grooves model of interacting helices indicates that heterochiral associations should generally be favored in this class of structures. We tested this principle using a simplified molecular screw – a collagen peptide triple- helix composed of either L- or D-proline with a cyclic aliphatic side chain. Calculated stabilities of like- and opposite- handed triple-helical pairings indicated a preference for heterospecific associations. Mixing left and right-handed helices drastically lowered solubility, resulting in micron-scale sheet-like assemblies that are one peptide-length thick as characterized with atomic force microscopy. X-ray scattering measurements of inter-helical spacing in these sheets support a tight ridges-in-grooves packing of left and right-handed triple helices.
In Through the Looking Glass, a fictional account of a mirror- image world, Lewis Carroll presaged our current understanding of biomolecular stereoselectivity suggesting, “Perhaps looking glass milk isn’t good to drink …”. Mirror-image stereoisomers of proteins, sugars and other chiral biomolecules would be useless and possibly poisonous to us, due to the inability of our stereoselective enzymes to metabolize these compounds. The inability of endogenous proteases to degrade non-natural stereoisomers can also be used to our advantage, such as increasing the in vivo half-life of therapeutic peptides by judicious incorporation of D-amino acids.
Although natural proteins tend to prefer self, or homochiral molecular recognition, this is not an absolute rule. Synthetic polypeptides exhibit no consistency in stereoselectivity (Table 1). This lack of consensus suggests that in many cases, detailed aspects of interactions govern recognition.
Table 1.
A list of preferred homo- and heterochiral associations in mixtures of protein sequence enantiomers.
Despite this, general rules that relate shape complementarity to association would be useful in guiding molecular design. One such rule describes interactions between like-versus opposite-handed helical objects. A geometric analysis of the packing of coiled-coils predicted that columnar associations between opposite-handed supercoils would allow for an overall tighter packing density and a greater number of intermolecular contacts than like-handed associations 1. Optimal packing of like-handed threaded rods requires rotation of principle axes of adjacent rods, preventing tight columnar packing 2; this same phenomenon determines helix packing in proteins 3. Molecular simulations of opposite- and like-handed poly-alanine α-helices demonstrate a preference for left-right helical dimers 4. All of these studies support a general rule that supramolecular interactions of opposite-handed helices will be favored over like-handed assemblies. However, it is challenging to develop an appropriate experimental system that evaluates shape complementarity without being strongly influenced by the details of intermolecular interactions. Ridges-in-grooves interactions have been demonstrated on a macroscopic scale between left and right-handed bolts 5. At the molecular scale, a ‘chemically nude’ system is needed where the shape is a primary factor promoting close packing.
The collagen mimetic peptide (PPG)10 (P = proline, G = glycine) is a promising minimal system for evaluating the role of helix handedness on intermolecular association. Collagen is composed of three chains that are supercoiled to form a triple-helix. Except for glycine, all naturally occurring amino acids are levorotatory (L) stereoisomers. Using dextrorotatory (D) stereoisomers results in a mirror-image, opposite- handed triple-helix (Fig. 1(a)). In the collagen mimetic peptide, (PPG)10, proline side chains form the ridges and grooves of the triple-helix. Due to the cyclic aliphatic side chain, one might expect reduced contributions from side chain flexibility, charge-pair interactions or hydrogen bonding that could influence molecular packing specificity. This leaves the complementary shape of the binding interface as the primary determinant for effective packing.
Figure 1.
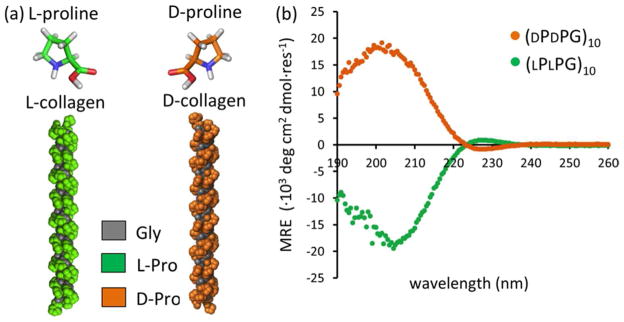
Mirror-image triple helices. (a) Structural models of [(LPLPG)10]3 and [(DPDPG)10]3 triple helices. The [(LPLPG)10]3 model was obtained from a high-resolution (1.30 Å) X-ray crystal structure (PDB: 1K6F) 19. (b) CD spectra at 4 °C. [(LPLPG)10]3 and [(DPDPG)10]3 refer to triple helices comprised of three chains with ten repeating units of PPG.
Supramolecular assembly of collagen is also of interest due to its favorable properties as a biomaterial. Due to pathological and immunological issues with natural collagen 13, bottom- up design of synthetic collagen biomaterials has drawn much attention. Current design strategies utilize non-covalent driving forces such as electrostatics 14,15, hydrophobic interactions 16,17, and metal-chelating interactions 18 to induce supramolecular assembly of short synthetic collagen mimetic peptides. In this work, the additional contributions of stereochemistry and shape complementarity are explored.
Computational Models
(LPLPG)10 and its sequence stereoisomer, (DPDPG)10 both form triple helices with the identical solubility and thermal stability, but opposite helical handedness as detected with circular dichroism (Fig. 1, S2). The (LPLPG)10 peptide forms a right-handed supercoil as shown by its X-ray crystal structure 19. However, when three chains associate, proline side chains form a continuous, left-handed ridge. We therefore use the convention in this study where [(LPLPG)10]3 forms a left-handed helix, and [(DPDPG)10]3 forms a right-handed one. Mixed heterotrimers of (LPLPG)10 and (DPDPG)10 could not readily form as this would require proline residues to adopt highly hindered backbone conformations.
Short-range van der Waals interactions between two like- or opposite-handed triple helices were calculated for a series of structures specified by sampling rigid-body translational and rotational degrees of freedom 20 (for details of the calculations see supplemental methods and Fig. S1). Parallel (Ω= 0°) or antiparallel (Ω= 180°) orientations of the molecular principle axes had optimal contact interfaces as manifested by the lowest interaction scores (Fig. 2(a)). In parallel and antiparallel states, left/right packing was more favorable than left/left packing. Thus, the model predicts that triple helical grooves of opposite-handed molecules interdigitate and interact more tightly, and have a shorter optimal inter-helical distance than like-handed ones (Fig. 2b, c and Table S1). Ridges of two left-handed triple helices were predicted to align by forming edge-to-edge packing, consistent with the lattice packing in X-ray crystal structures of related collagen peptides 21.
Figure 2.

Computational models of opposite (LxD) and like (LxL) packing of collagen triple helices. (a) For each crossing angle, Ω, the packing conformation was optimized and the lowest interaction energy was plotted against Ω. (b, c) Optimal packing conformations of the LxD and LxL.
Association stoichiometry
Previous studies of collagen peptide aggregation 16 showed that (LPLPG)10 was soluble at very high concentrations (14 mg/ml or 5.5 mM). We observed the same behavior, with similar concentrations of both (LPLPG)10 and (DPDPG)10 remaining in solution for several weeks. Mixing the two sequence stereoisomers induced rapid aggregation over the course of a few hours (Fig. 3). This indicates that supramolecular assembly was facilitated by the presence of opposite-handed species.
Figure 3.

Solubilities of like- and opposite-handed collagen mixtures. L = 5.5 mM (LPLPG)10, D = 5.5 mM (DPDPG)10, L:D = 2.75 mM (LPLPG)10 + 2.75 mM (DPDPG)10. Samples prepared in 10 mM phosphate buffer at pH 7.
To establish whether aggregates were composed of both enantiomers or whether one catalyzed condensation of the other, rates and extents of assembly were measured at a series of concentrations of the D-enantiomer while keeping the L-enantiomer concentration fixed. A 1:1 mixture rapidly assembled with significant precipitation exceeding the detection limit of static/dynamic light scattering (SLS/DLS) after ~8 hours (Fig. 4). Reduction of the relative concentration of the (DPDPG)10 species prolonged nucleation times and decreased total scattering intensity indicating less aggregate was made, consistent with a precipitate phase that incorporates both enantiomers.
Figure 4.

Depleting the D-enantiomer slows aggregation and reduces the total amount formed. (a) Total scattering intensity of a fixed concentration, 1 mM (LPLPG)10, combined with 1 mM, 0.2 mM and 0.1 mM (DPDPG)10, at 4 °C was monitored over time by SLS. (b-d) DLS measurement of dominant particle sizes in the mixtures (defined as the size of a particle with the volume percentage > 85%.)
Morphology of the supramolecular assemblies was observed by transmission electron microscopy (TEM) for various mixing ratios of (LPLPG)10:(DPDPG)10. The equimolar mixture formed well-ordered micron-scale sheets (Fig. 5(a), S3). Atomic force microscopy (AFM) measurements of these sheets had an average single layer thickness of ~10 nm (Fig. 5 (b)), which is consistent with the length of a thirty-residue triple-helix 19. These heterochiral triple helices may align to form the sheet-like structure (Figure 5(c)) although higher-resolution data would be needed to confirm this model. This is similar to macroscale assemblies observed in mixtures of millimeter-sized bolts with opposite-handed threads 5. Several sheets were observed to stack in multi-layered structures. Small ~100 nm particles were found in mixtures where the D-proline enantiomer was depleted (Fig. s4).
Figure 5.

Sheet morphology. (a) TEM and (b) AFM of a 1: 1 L to D mixture. Contour heights varied by integer multiples of 10 nm. (c) A proposed model of assembly where triple helices align into a 10 nm thick layer, which further stack. Samples were incubated at 4 °C for ~4 weeks prior to imaging.
Requirement of triple helices
The melting temperatures of (LPLPG)10 and (DPDPG)10 are ~25 °C (Fig. s2). Assembly at temperatures below, at and above the melting temperature was characterized to test the contribution of triple-helix unfolding. Turbidity was observed after incubation at 4, 25 and 40 °C for three days (Fig. 6, 7(a)). At this time point, the 25°C structures were micron sized sheets. Assembly of 4°C structures was slower, with uniform ~100 nm particles observed after 3 days. It took over a month for micronscale sheets to form at 4°C (Fig. 5). Aggregation was also observed at high temperature (40 °C). However, the assembly at 40 °C showed different features such as less turbidity, more irregular morphology, and much smaller sizes in a majority of the particles detected with DLS (Fig. 6, 7(a)). This assembly may be caused by non-specific interactions in the unfolded state although it is interesting to note that similar aggregates were not observed for L or D peptides alone.
Figure 6.

Influence of temperature on sheet morphology. (a) Particle size distribution in 1 mM:1 mM of (LPLPG)10 and (DPDPG)10 mixture detected with DLS at 0 and 3 days incubation at 4, 25, and 40 °C, respectively. (b, c, d) TEM samples prepared under the same condition as in DLS.
Figure 7.
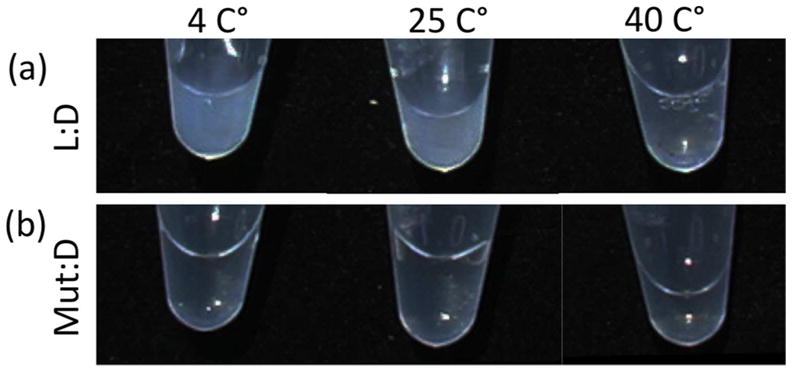
Folding of the triple-helix required for assembly. Photographs of (a) L:D = 1 mM (LPLPG)10 : 1 mM (DPDPG)10. (b) D:Mut = 1 mM (DPDPG)10 : 1 mM mutant-L. The samples were incubated at 4, 25, and 40 °C, respectively, for 3 days.
To further clarify the importance of a folded triple-helix in driving supramolecular assembly, we designed a mutant L-peptide, (LPLPG)4LPLP(LPLPG)5, by removing a conserved glycine in the fifth triplet. This type of mutation has been shown to prevent triple helical formation 22. No triple-helix was observed in the mutant peptide as evidenced by a lack of positive ellipticity at 225 nm in the circular dichroism spectrum (Fig. s5). Mixtures of the mutant L-peptide and (DPDPG)10 remained transparent and soluble regardless of the incubation temperature (Fig. 7(b)). Disrupting triple-helix folding of one of the components was sufficient to prevent sheet formation.
Inter-helical distances
Computational simulations predict close packing of left/right handed triple-helix pairs. The crystalline sheets formed during self-assembly suggest a uniform supramolecular structure. Wide-angle X-ray scattering (WAXS) was used to characterize peptide organization 15,23,24. WAXS profiles confirmed the existence of triple helices, and allowed us to compare inter-helical distances between homo- and heterochiral associations.
The diffraction patterns of the heterochiral sample showed three peaks – two overlapping peaks at ~13Å, and a third peak at ~11 Å. Such peak locations are typical of triple-helix equatorial peaks (13.7, 13.3 and 12.6 Å) found in natural collagen 25 (Also see Table S2 and references therein). The absence of the 2.86 Å meridional reflection 23, which is a strong indication for a triple-helix, is due to the anisotropic nature of the sample.
Clear differences in inter-helical distances of (PPG)10 were observed between hom0chiral versus heterochiral assemblies. Since we were unable to induce the homochiral association under the same concentration as the heterochiral mixture (2 mM), an oversaturated (LPLPG)10 suspension (~80 mM) was prepared to promote homochiral packing. The water content of the homochiral suspension was lower or similar to the heterochiral mixture. Even though drying of the samples may decrease their interhelical distances (Table S2 and references therein), the heterochiral mixture with higher water content still showed tighter packing than the homochiral one. The two inter-helical distances of (LPLPG)10 were both slightly larger than those of the (LPLPG)10:(DPDPG)10 mixture. These experimental differences in the inter-helical distances were consistent with the computationally predicted ones (Table S1).
The mutant L-peptide showed a diffraction pattern lacking any peak characteristic of triple-helix formation within the 10–13 Å region (Fig. 8), consistent with circular dichroism (Fig. s5). The numerous sharp peaks between 2–7 Å in the mutant sample were observed instead of an intense broad peak at 4.5 Å that is typical of native collagen 23. This suggests that the mutant L-peptide is highly crystalline, but does not fold into triple helices.
Figure 8.
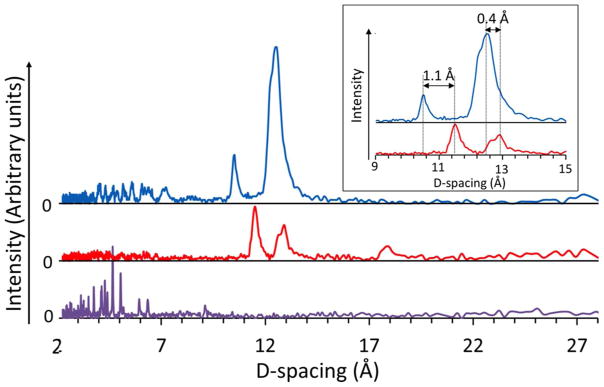
D-spacing measured with wide-angle X-ray scattering of mixture 1 mM (LPLPG)10: 1 mM (DPDPG)10 (upper) compared to oversaturated (LPLPG)10 suspension (middle) and oversaturated mutant L-peptide suspension (bottom). The dspacing differences are shown in the inset.
Conclusion
Using (PPG)10 sequence enantiomers as minimal helical exemplars, the geometric prediction of preferred left/right-handed helical pairings appears to hold true. However, even this system is complicated by chemical detail. In a series of experiments pairing (DPDPG)10 with other right-handed triple-helical peptides such as (POG)10, no interactions were observed. Hydroxylation of the proline side chain may sterically prevent close association of left and right-handed species, or increase the desolvation barrier required to allow tight ridges-in-grooves packing, or even modify the degree of supercoiling of the triple-helix such that different pitch of left and right-handed species could preclude a coherent, columnar interaction. Ironically, this general rule of helix-helix association may best apply in special cases where complete mirror symmetry exists.
Supplementary Material
Acknowledgments
This work was supported by NIH DP2 OD-006478 and NIH R01 GM-089949. We thank Hiroshi Matsui for access to AFM.
Footnotes
The authors declare no competing financial interests.
Details of experimental methods and additional figures and tables are provided in the supplement. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Woodhead-Galloway J. Acta Crystallographica Section B. 1976;32:1880. [Google Scholar]
- 2.Straley JP. Physical Review A. 1976;14:1835. [Google Scholar]
- 3.Chothia C, Levitt M, Richardson D. Journal of molecular biology. 1981;145:215. doi: 10.1016/0022-2836(81)90341-7. [DOI] [PubMed] [Google Scholar]
- 4.Sia SK, Kim PS. Biochemistry. 2001;40:8981. doi: 10.1021/bi010725v. [DOI] [PubMed] [Google Scholar]; Nanda V, DeGrado WF. Journal of the American Chemical Society. 2006;128:809. doi: 10.1021/ja054452t. [DOI] [PubMed] [Google Scholar]
- 5.Boncheva M, Bruzewicz DA, Whitesides GM. Langmuir. 2003;19:6066. [Google Scholar]
- 6.Fuhrhop JH, Krull M, Büdt G. Angewandte Chemie International Edition in English. 1987;26:699. [Google Scholar]
- 7.Saghatelian A, Yokobayashi Y, Soltani K, Ghadiri MR. Nature. 2001;409:797. doi: 10.1038/35057238. [DOI] [PubMed] [Google Scholar]
- 8.Gerber D, Shai Y. Journal of Molecular Biology. 2002;322:491. doi: 10.1016/s0022-2836(02)00807-0. [DOI] [PubMed] [Google Scholar]
- 9.Chung DM, Nowick JS. Journal of the American Chemical Society. 2004;126:3062. doi: 10.1021/ja031632z. [DOI] [PubMed] [Google Scholar]
- 10.Ramagopal UA, Ramakumar S, Mathur P, Joshi R, Chauhan VS. Protein Eng. 2002;15:331. doi: 10.1093/protein/15.4.331. [DOI] [PubMed] [Google Scholar]
- 11.Milton RC, Milton SC, Kent SB. Science. 1992;256:1445. doi: 10.1126/science.1604320. [DOI] [PubMed] [Google Scholar]
- 12.Pentelute BL, Gates ZP, Tereshko V, Dashnau JL, Vanderkooi JM, Kossiakoff AA, Kent SB. Journal of the American Chemical Society. 2008;130:9695. doi: 10.1021/ja8013538. [DOI] [PMC free article] [PubMed] [Google Scholar]; Patterson WR, Anderson DH, DeGrado WF, Cascio D, Eisenberg D. Protein Sci. 1999;8:1410. doi: 10.1110/ps.8.7.1410. [DOI] [PMC free article] [PubMed] [Google Scholar]; Zawadzke LE, Berg JM. Proteins. 1993;16:301. doi: 10.1002/prot.340160308. [DOI] [PubMed] [Google Scholar]
- 13.Lee CH, Singla A, Lee Y. International Journal of Pharmaceutices. 2001;221:1. doi: 10.1016/s0378-5173(01)00691-3. [DOI] [PubMed] [Google Scholar]; O’Grady JE, Bordon DM. Adv Drug Deliv Rev. 2003;55:1699. doi: 10.1016/j.addr.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 14.Rele S, Song Y, Apkarian RP, Qu Z, Conticello VP, Chaikof EL. J Am Chem Soc. 2007;129:14780. doi: 10.1021/ja0758990. [DOI] [PubMed] [Google Scholar]
- 15.O’Leary LER, Fallas JA, Bakota EL, Kang MK, Hartgerink JD. Nat Chem. 2011;3:821. doi: 10.1038/nchem.1123. [DOI] [PubMed] [Google Scholar]
- 16.Kar K, Amin P, Bryan MA, Persikov AV, Mohs A, Wang YH, Brodsky B. J Biol Chem. 2006;281:33283. doi: 10.1074/jbc.M605747200. [DOI] [PubMed] [Google Scholar]
- 17.Cejas MA, Kinney WA, Chen C, Vinter JG, Almond HR, Jr, Balss KM, Maryanoff CA, Schmidt U, Breslav M, Mahan A, Lacy E, Maryanoff BE. Proc Natl Acad Sci U S A. 2008;105:8513. doi: 10.1073/pnas.0800291105. [DOI] [PMC free article] [PubMed] [Google Scholar]; Kar K, Ibrar S, Nanda V, Getz TM, Kunapuli SP, Brodsky B. Biochemistry. 2009;48:7959. doi: 10.1021/bi900496m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Przybyla DE, Chmielewski J. J Am Chem Soc. 2008;130:12610. doi: 10.1021/ja804942w. [DOI] [PubMed] [Google Scholar]
- 19.Berisio R, Vitagliano L, Mazzarella L, Zagari A. Protein Science. 2002;11:262. doi: 10.1110/ps.32602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nanda V, DeGrado WF. Journal of the American Chemical Society. 2005;128:809. doi: 10.1021/ja054452t. [DOI] [PubMed] [Google Scholar]
- 21.Okuyama K, Morimoto T, Narita H, Kawaguchi T, Mizuno K, Bachinger HP, Wu G, Noguchi K. Acta Crystallographica Section D. 2010;66:88. doi: 10.1107/S0907444909046642. [DOI] [PubMed] [Google Scholar]
- 22.Long CG, Braswell E, Zhu D, Apigo J, Baum J, Brodsky B. Biochemistry. 1993;32:11688. doi: 10.1021/bi00094a027. [DOI] [PubMed] [Google Scholar]
- 23.Rich A, Crick FHC. Journal of Molecular Biology. 1961;3:483. doi: 10.1016/s0022-2836(61)80016-8. [DOI] [PubMed] [Google Scholar]
- 24.Okuyama K, Xu X, Iguchi M, Noguchi K. Peptide Science. 2006;84:181. doi: 10.1002/bip.20381. [DOI] [PubMed] [Google Scholar]; Przybyla DE, Rubert Pérez CM, Gleaton J, Nandwana V, Chmielewski J. Journal of the American Chemical Society. 2013;135:3418. doi: 10.1021/ja307651e. [DOI] [PubMed] [Google Scholar]
- 25.Brodsky B, Eikenberry EF. In: Methods Enzymol. Leon W, Cunningham DWF, editors. Vol. 82. Academic Press; 1982. p. 127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.