

Abstract
The TNFAIP3 (tumor necrosis factor alpha–induced protein 3) gene encodes a ubiquitin editing enzyme, A20, that restricts NF-κB–dependent signaling and prevents inflammation. We show that three independent SNPs in the TNFAIP3 region (rs13192841, rs2230926 and rs6922466) are associated with systemic lupus erythematosus (SLE) among individuals of European ancestry. These findings provide critical links between A20 and the etiology of SLE.
Autoimmune diseases are characterized by persistent or recurrent inflammation in the absence of explicit microbial infection. SLE is the prototypic systemic autoimmune disease. Although the disease is genetically complex, substantial work over the past decade has led to the identification of several reproducible genetic risk factors for SLE1.
A20, the product of the TNFAIP3 gene, is an NF-κB–inducible protein expressed in multiple cell types and is required for preventing spontaneous inflammation2. The elimination of A20 from mice leads to severe spontaneous inflammation, cachexia and premature death2. A20 regulates the ubiquitination of key signaling proteins and restricts the duration of both tumor necrosis factor (TNF) and Toll-like-receptor–induced NF-κB signals3–5. Thus, A20 is a potent endogenous anti-inflammatory molecule. As TNFAIP3 is well conserved between humans and mice, and given recent evidence supporting association of this gene with rheumatoid arthritis6,7, we hypothesized that variants in TNFAIP3 might also be associated with SLE.
To examine the potential role of TNFAIP3 in SLE, we used data from a recently published genome-wide association study8. Supplementary Table 1 online shows the number of cases and controls examined before and after quality control filters. In total, 1,239 SLE cases and 1,629 controls were included in this analysis. We initially selected 129 contiguous SNPs from the TNFAIP3 region on chromosome 6, extended with flanking regions approximately 250 kb on either side of the coding sequence (138,000 kb to 138,500 kb). This region also captures the PERP gene, which encodes an apoptosis effector. Because we observed significantly associated SNPs in LD blocks at the boundaries of the initial region, we later extended our analysis to 158 SNPs in the region from 137,975 kb to 138,550 kb.
We carried out additional genotyping for the TNFAIP3 nonsynonymous coding SNP rs2230926 in 393 of the SLE cases, as these individuals were typed on the version 1 Illumina 550K panel, which did not include this SNP. Among controls, 869 individuals were genotyped on the version 1 array and therefore did not have data available for rs2230926. A subgroup analysis of the testing described below using only cases (n = 1,239) and controls (n = 760) that were typed for rs2230926 revealed essentially the same results (data not shown).
A total of 21 SNPs in the region had allelic P ≤ 0.005 (Table 1). At this screening stage, we used a liberal cutoff given the existence of at least ten independent haplotype blocks in the region. rs13192841 had the smallest P value, 5.4 × 10−8 (OR 1.4, 95% CI = 1.2–1.6), and rs2230926 had the highest odds ratio (OR), 2.0 (95% CI = 1.4–3.0, P = 3.0 × 10−4). All of these top 21 SNPs were in the initial 500-kb region covered by 129 SNPs (Fig. 1); the extension to a 575-kb region with 29 additional SNPs did not yield new candidates. Based on data from the HapMap CEU population (r2 = 1), rs6933404 and rs2327832 are perfect proxies for rheumatoid arthritis–associated SNP rs6920220, and rs13192841 and rs12527282 are perfect proxies for another rheumatoid arthritis–associated SNP, rs10499194 (refs. 6,7).
Table 1.
SNPs with allelic P < 0.005 from Haploview
| SNP | Name | Allele | Ratio counts (case, control) | Allele frequencies (case, control) | OR (95% CI) | P value |
|---|---|---|---|---|---|---|
| 2 | rs6933404 | G | 588:1,882, 614:2,640 | 0.24, 0.19 | 1.3 (1.2–1.5) | 5.6 × 10−6 |
| 4 | rs600469 | G | 1,229:1,245, 1,480:1,746 | 0.50, 0.46 | 1.2 (1.05–1.3) | 0.0044 |
| 5 | rs13192841 | G | 1,753:543, 2,208:960 | 0.76, 0.70 | 1.4 (1.2–1.6) | 5.4 × 10−8 |
| 6 | rs12527282 | G | 1,833:629, 2,239:993 | 0.75, 0.69 | 1.3 (1.15–1.5) | 1.8 × 10−5 |
| 8 | rs2327832 | G | 578:1,872, 587:2,613 | 0.24, 0.18 | 1.4 (1.2–1.6) | 1.4 × 10−6 |
| 10 | rs686851 | G | 1,229:1,247, 1,486:1,760 | 0.50, 0.46 | 1.2 (1.1–1.3) | 0.0038 |
| 11 | rs1002658 | G | 2,051:415, 2,583:639 | 0.83, 0.80 | 1.2 (1.1–1.4) | 0.0039 |
| 12 | rs525977 | G | 1,229:1,247, 1,493:1,763 | 0.50, 0.46 | 1.2 (1.05–1.3) | 0.0045 |
| 13 | rs6904167 | G | 1,202:1,214, 1,466:1,728 | 0.50, 0.46 | 1.2 (1.05–1.3) | 0.0042 |
| 17 | rs636393 | A | 1,059:625, 855:663 | 0.63, 0.56 | 1.3 (1.1–1.5) | 2.0 × 10−4 |
| 18 | rs602414 | A | 1,557:917, 1,882:1,374 | 0.63, 0.58 | 1.2 (1.1–1.4) | 8.5 × 10−5 |
| 58 | rs2230926 | C | 114:2,342, 36:1,484 | 0.05, 0.02 | 2.0 (1.4–3.0) | 3.0 × 10−4 |
| 105 | rs2484066 | C | 1,403:1,045, 1,729:1,507 | 0.57, 0.53 | 1.2 (1.1–1.3) | 0.0036 |
| 106 | rs9494941 | A | 1,556:908, 1,908:1,346 | 0.63, 0.59 | 1.2 (1.1–1.3) | 5.0 × 10−4 |
| 108 | rs1931867 | A | 1,534:888, 1,858:1,296 | 0.63, 0.59 | 1.2 (1.1–1.3) | 8.0 × 10−4 |
| 110 | rs6922466 | A | 1,905:531, 2,378:844 | 0.78, 0.74 | 1.3 (1.1–1.4) | 1.0 × 10−4 |
| 111 | rs12660547 | A | 1,853:625, 2,296:962 | 0.75, 0.71 | 1.2 (1.1–1.4) | 3.0 × 10−4 |
| 112 | rs12661926 | A | 1,852:626, 2,293:963 | 0.75, 0.70 | 1.2 (1.1–1.4) | 3.0 × 10−4 |
| 113 | rs7773257 | A | 2,149:329, 2,734:520 | 0.87, 0.84 | 1.2 (1.07–1.4) | 0.0043 |
| 114 | rs6920846 | A | 1,696:782, 2,071:1,183 | 0.68, 0.64 | 1.2 (1.1–1.4) | 2.0 × 10−4 |
| 115 | rs4896318 | G | 1,153:523, 948:560 | 0.69, 0.63 | 1.3 (1.1–1.5) | 4.0 × 10−4 |
SNP number refers to order in Figure 1, which shows 115 SNPs passing quality control in the initial 500-kb region.
Figure 1.
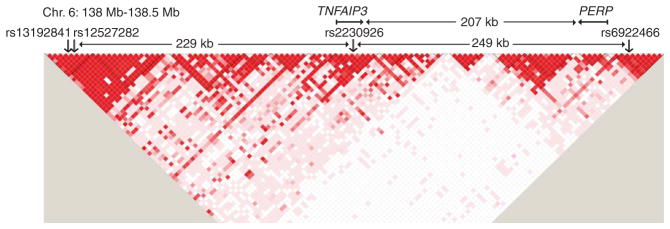
TNFAIP3 region showing D′ for genotypes of all study subjects and location of independently associated SNPs. SNPs shown are those passing quality control in the initial 500-kb region. Independent SNPs based on conditional analysis (Supplementary Table 2) are indicated by rs number. rs13192841 and rs12527282 are collinear; one allele determines the other in >99% of subjects. D′ plot, generated in Haploview10, indicates D′ between pairs of SNPs; darker red indicates higher D′.
Figure 1 also shows the LD pattern among the genotypes in this study. With multiple blocks of high LD in the region, it is clear that the 21 signals are not all independent. Therefore, we performed conditional analysis, starting with the top SNP, to test the additional candidate SNPs for independence (that is, significance when conditioning on the values of the previously confirmed top SNPs). We first confirmed the independence of rs2230926, with P = 0.0014 conditional on rs13192841. We then tested all other candidate SNPs (with allelic P < 0.005) conditional on rs13192841 and rs2230926 (Supplementary Table 2 online). The most significant SNP was rs6922466 (P = 0.00037). Next, we tested all candidate SNPs conditioning on all three independent SNPs and found no strong evidence for additional independent signals (all P ≥ 0.027 in 17 tests). rs12527282 was collinear with rs13192841 in conditional analysis; one allele determined the other in >99% of estimated haplotypes. Finally, we conditioned on rs2230926 within its LD block using all SNPs passing quality control, and did not find any additional significant SNPs (all P < 0.15, data not shown). The final three SNPs are in different LD blocks; each pairwise r2 is <0.01 (Fig. 1).
We further confirmed the three independent signals with multivariate logistic regression using an additive model (Supplementary Table 3 online). This shows protective effects of the rs13192841 minor allele with an OR of 0.72 (95% CI = 0.62–0.83, P = 7.9 × 10−6) and the rs6922466 minor allele with an OR of 0.76 (95% CI = 0.65–0.88, P = 0.00039). In contrast, the minor allele of rs2230926 was associated with an increased risk of SLE with an OR of 1.88 (95% CI = 1.27–2.79, P = 0.0016).
Lastly, we carried out stratified analyses of allelic tests to ensure that the associations were not explained by substructure within the European population. We stratified by (i) whether or not subjects had ≥90% Northern European ancestry and (ii) whether or not subjects were in a genetically homogeneous subset determined by principal components analysis (Supplementary Methods online). Overall, results of these stratified analyses (Supplementary Table 4 online) were consistent with the results summarized above. For rs13192841 and rs6922466, the largest magnitudes of effect (lowest OR for protective SNPs), 0.67 (95% CI = 0.55–0.81, P = 2.9 × 10−5) and 0.72 (95% CI = 0.60–0.88, P = 0.0008), respectively, were in the homogeneous subset of subjects. For the infrequent exonic SNP, rs2230926, the homogeneous subset association was OR = 1.53 (95% CI = 0.84–2.96, P = 0.15); given the number of subjects in this subset, we had only 65% power to detect an OR of 1.53. Combination of the homogeneous and nonhomogeneous strata using the Mantel-Haenszel method produced OR = 1.87 (95% CI = 1.26–2.77, P = 0.0013) with P = 0.34 for the heterogeneity of the stratum-specific associations. We conclude that, although some signal from rs2230926 may be due to intra-European population substructure, there is strong evidence that a signal remains even after controlling for substructure.
The three independently associated SNPs include one coding and two noncoding polymorphisms. The coding SNP, rs2230926, is a nonsynonymous variant resulting in a phenylalanine-to-cysteine change at residue 127 of the A20 protein. To begin to test the biological impact of this SNP, we compared the ability of human A20 proteins encoded by the major (Phe127) and minor (Cys127, risk) allele cDNAs to inhibit TNF-induced NF-κB signaling. These experiments revealed that the minor Cys127 protein is comparably stable relative to the Phe127 protein. Notably, the Cys127 A20 protein is modestly, but consistently, less effective at inhibiting TNF-induced NF-κB activity when similar amounts of the two proteins are expressed (Supplementary Fig. 1 online). This reduced anti-inflammatory activity of A20 may allow excessive cellular responses to TNF. In addition, as A20 is essential for restricting cellular responses triggered by Toll-like receptors, NOD2 and potentially other pro-inflammatory stimuli, it is likely that a hypomorphic A20 protein may contribute to multiple facets of excessive inflammation and auto-immunity in humans bearing this polymorphism4,5.
Our findings show that three independent SNPs in the TNFAIP3 region are associated with SLE. These polymorphisms may cause reduced expression or activity of the anti-inflammatory activity of A20, predisposing individuals to develop SLE. Considered together with recent studies associating TNFAIP3 SNPs with rheumatoid arthritis6,7, it is apparent that TNFAIP3 is a potent regulator of susceptibility to autoimmunity in humans. In an independent study, Graham et al. also observed association to SNPs in TNFAIP3 in their genome-wide association study of SLE9. We identified independent SNPs in the same two LD blocks as Graham et al., plus a third LD block. Future work should attempt to clarify the precise location of the effects seen in these LD blocks through fine-mapping and additional functional experiments, as well as investigating association with specific subphenotypes. As we limited our study to people of European descent, future studies including other ethnic groups are necessary, especially because SLE affects individuals of non-European ancestry at an increased frequency compared to individuals of European ancestry. It is also important to note that our region of interest covers not only TNFAIP3, but also PERP. A missense SNP in PERP, rs648802, was not genotyped in our panel, but a near perfect proxy based on the HapMap CEU data, rs563495 (r2 = 0.966), was genotyped in our cohort and did not have an allelic P value meeting our criteria for significance. Given the recently demonstrated association of human TNFAIP3 SNPs with rheumatoid arthritis and prior functional studies of Tnfaip3-deficient mice, our current genetic and functional experiments support the notion that TNFAIP3 is a causative gene associated with SLE as well as rheumatoid arthritis. Hence, TNFAIP3 may be an important determinant for multiple autoimmune diseases.
Supplementary Material
Acknowledgments
This work was supported by R01 AR44804 (L.A.C.), K24 AR02175 (L.A.C.), N01 AI95386 (P.K.G.), ABCoN contract NO1-AR-1-2256 (P.K.G.), AR050267 (M.F.S.), the Mary Kirkland Center for Lupus Research (L.A.C.), the Rainin Foundation (A.M.), T32 DK07007 (T.T.L.), U19 AI063603 (P.-Y.K.), 2R01 AR4658805 (S.M.), K24 AR02213 (S.M.), R01 HL 54900 (M.I.K.) and R01 HL 74165 (M.I.K.). These studies were performed in part in the General Clinical Research Center, Moffitt Hospital, University of California, San Francisco, with funds provided by the National Center for Research Resources, 5 M01 RR-00079, US Public Health Service. We also thank the individuals and physicians who contributed DNA samples and clinical data for this study.
Footnotes
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions/
AUTHOR CONTRIBUTIONS
S.L.M., K.E.T. and A.M. wrote the manuscript with input from the coauthors. K.E.T., J.N. and M.F.S. performed the analyses. S.L.M., R.C.F. and W.O. performed genotyping of rs2230926. N.S. generated the human cDNA constructs and T.T.L. conducted the functional studies under the supervision of A.M. M.A.P., M.I.K., S.M., P.K.G. and L.A.C. provided samples for this project. T.W.B. and P.K.G. led the project from which the primary SNP data was generated, and A.M., P.-Y.K. and L.A.C. directed the work described in this manuscript.
COMPETING INTERESTS STATEMENT
The authors declare competing financial interests: details accompany the full-text HTML version of the paper at http://www.nature.com/naturegenetics/.
Note: Supplementary information is available on the Nature Genetics website.
References
- 1.Crow MK. N Engl J Med. 2008;358:956–961. doi: 10.1056/NEJMe0800096. [DOI] [PubMed] [Google Scholar]
- 2.Lee EG, et al. Science. 2000;289:2350–2354. doi: 10.1126/science.289.5488.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wertz IE, et al. Nature. 2004;430:694–699. doi: 10.1038/nature02794. [DOI] [PubMed] [Google Scholar]
- 4.Boone DL, et al. Nat Immunol. 2004;5:1052–1060. doi: 10.1038/ni1110. [DOI] [PubMed] [Google Scholar]
- 5.Hitotsumatsu O, et al. Immunity. 2008;28:381–390. doi: 10.1016/j.immuni.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Plenge RM, et al. Nat Genet. 2007;39:1477–1482. doi: 10.1038/ng.2007.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thomson W, et al. Nat Genet. 2007;39:1431–1433. doi: 10.1038/ng.2007.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hom G, et al. N Engl J Med. 2008;358:900–909. doi: 10.1056/NEJMoa0707865. [DOI] [PubMed] [Google Scholar]
- 9.Graham RR, et al. Nat Genet. 2008 Aug 1; doi: 10.1038/ng.200. advance online publication. [DOI] [Google Scholar]
- 10.Barrett JC, Fry B, Maller J, Daly MJ. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.