

SUMMARY
Prostanoids and PGE2 in particular have been long viewed as one of the major mediators of inflammation in arthritis. However, experimental data indicate that PGE2 can serve both pro- and anti-inflammatory functions. We have previously shown (Kojima, F. et al. 2008 J. Immunol. 180, 8361-8368) that microsomal prostaglandin E synthase-1 (mPGES-1) deletion, which regulates PGE2 production, resulted in the suppression of collagen-induced arthritis (CIA) in mice. This suppression was attributable, at least in part, to the impaired generation of type II collagen autoantibodies. In order to examine the function of mPGES-1 and PGE2 in a non-autoimmune form of arthritis, we used the collagen antibody-induced arthritis (CAIA) model in mice deficient in mPGES-1, thereby bypassing the engagement of the adaptive immune response in arthritis development. Here we report that mPGES-1 deletion significantly increased CAIA disease severity. The latter was associated with a significant (~3.6) upregulation of neutrophil, but not macrophage, recruitment to the inflamed joints. The lipidomic analysis of the arthritic mouse paws by quantitative liquid chromatography / tandem mass-spectrometry (LC/MS/MS) revealed a dramatic (~59-fold) reduction of PGE2 at the peak of arthritis. Altogether, this study highlights mPGES-1 and its product PGE2 as important negative regulators of neutrophil-mediated inflammation and suggests that specific mPGES-1 inhibitors may have differential effects on different types of inflammation. Furthermore, neutrophil-mediated diseases could be exacerbated by inhibition of mPGES-1.
Keywords: Nanohybrids, Room-temperature phosphorescence(RTP), Sensor, Rutin
INTRODUCTION
Prostaglandins (PG) are important lipid mediators known to regulate a broad spectrum of physiological functions, including immunity and inflammation (1,2). PG are produced from arachidonic acid (ARA), which is released from membrane phospholipids by phospholipases (3) or synthesized from linoleic acid (LA) (4). The ARA metabolic cascade includes a rate-limiting step of ARA conversion into PGH2 which is controlled by the cyclooxygenase (COX) isoforms 1 and 2 (5). PGH2 is then quickly processed to prostanoids, such as PGF2, PGD2, and PGE2, by their respective PG isomerases (6) (Fig.1).
Fig.1.

Eicosanoid biosynthetic pathway. The plain symbols depict proteins and biosynthetic pathways. The boxed symbols identify ARA and LA as well as their metabolites that were measured in the current study.
Prostanoids and PGE2 in particular, have been shown to play an important role in as mediators of symptoms of inflammatory arthritidies including rheumatoid arthritis (RA) as demonstrated by the widespread use of treatments that inhibit PG production (7–10). However, the current strategies to inhibit PGs by using non-steroidal anti-inflammatory drugs (NSAID) and selective COX-2 inhibitors are often inadequate in providing symptomatic relief of pain, swelling, and stiffness (11,12). Setting aside the incomplete blocking efficiency of NSAID and COX-2 selective inhibitors in vivo, the limited ability of these agents to curb inflammation could be explained, at least in part, by two-faceted and opposing roles of PG, especially PGE2, in the regulation of the inflammatory process. Indeed, the lipidomic analysis of animals with Lyme arthritis revealed that among major PG, only PGE2 production in arthritic joints was undergoing a cyclic change and was rising to a similar level during both the initiation and the resolution stage of the inflammatory disease (13). This would be indicative of a dual role of PGE2 in regulation of inflammation: it could mediate inflammation at the initiation stage while inhibiting inflammation during resolution or by triggering a resolution program. The requirement of PGE2 for the resolution of inflammation was confirmed in the recent studies where mice at the chronic stage of the autoimmune arthritis, when treated with the specific COX-2 inhibitor, displayed an increased disease severity which was corrected downwards upon repletion with a synthetic PGE2 analog (14). A growing body of evidence indicates that PGE2 apparently exerts its antiinflammatory functions at both a cellular and molecular levels. The cellular mode of PGE2 antiinflammatory functions includes suppression of neutrophil functions (15), reprogramming the classically activated macrophages (M1) into the alternatively activated macrophages (M2) (16), and skewing monocyte-to-macrophage differentiation toward M2 (17). At the molecular level, PGE2 has been shown to negatively regulate inflammation by inhibiting CCL5 expression in activated macrophages (18), suppressing macrophage and synovial fibroblast (SF) TNF-α expression induced by T cell-derived IL-17 (19), blocking NF-κB activation and NF-κB/DNA binding in response to LPS stimulation (20), as well as by differentially regulating nuclear translocation of p65 and p50 NF-κB subunits following SF stimulation with IL-1β and TNF-α. In the latter case, PGE2 stimulated p50 nuclear accumulation and inhibited that of p65 thereby promoting formation of p50p50 homodimer and reducing p50p65 heterodimer formation thus efficiently blocking expression of the inflammatory genes (21). It should be noted that sustained p50p50 homodimer nuclear accumulation was shown to play a central role in the resolution of inflammation (22–24). Finally, PGE2 can exert its antiinflammatory function by switching ARA metabolism toward production of anti-inflammatory and pro-resolution lipids, including lipoxin A4 (LXA4) as well as E- and D-series resolvins and protectins (25).
PGE2 production during acute and chronic inflammation is dependent on the enzyme mPGES-1, an inducible enzyme whose expression is upregulated strongly under inflammatory conditions. mPGES-1 functions in the ARA metabolic cascade downstream from COX-2 by catalyzing the isomerization of the endoperoxide PGH2 into PGE2 (26,27) (Fig.1). mPGES-1 works alongside two other PGH2 isomerases, cytosolic PGES (cPGES) and mPGES-2, but the latter two enzymes are constitutively expressed and are mostly responsible for basal production of PGE2 (26). Studies on the deletion of mPGES-1 gene in mice revealed its essential role in female fertility, pain, atherosclerosis, tumorigenesis, and inflammation which made this enzyme an attractive therapeutic target (28).
Our group (29) and others (30) by using CIA model have demonstrated a significant reduction in the incidence and the severity of this autoimmune arthritis in mPGES-1 null mice. The CIA model is dependent on developing cellular and humoral immune responses to type II collagen. We found that the mPGES-1 null mice had a profound defect in their ability to generate collagen-II autoantibodies (29). Therefore, we reasoned that an antiinflammatory role of PGE2 might be masked by a strong effect of PGE2 in the regulation of humoral immunity. We now test this hypothesis by using microsomal PGE synthase-1 (mPGES-1) KO mice. In order to bypass the autoantibody production step in arthritis development and correctly ascertain a role for PGE2 in the regulation of systemic and local inflammation, we chose to induce arthritis in mPGES-1 KO mice through direct injection of collagen-II antibody, i.e. utilizing the well established CAIA model (31,32). In this model, the inflammatory response is triggered after the ligation of Fcγ receptors (FcγR) on polymorphonuclear cells and macrophages by immune complexes (31,33).
In the present study we confirmed our hypothesis and demonstrated that mPGES-1, and consequently, PGE2 deficiency, significantly increased the incidence and severity of CAIA in mice. Furthermore, analysis of specific inflammatory cell markers in the arthritic paws pointed toward a major involvement of neutrophils, and not macrophages, in the maintenance of local inflammation in the mPGES-1 KO animals. Lipidomic profiling of the inflamed joints of arthritic mice revealed changes in the ARA metabolic pathway that were not limited to COX-dependent PGE2 production and could include all branches of the ARA metabolic pathway.
Altogether, our studies further emphasize the pleiotropic activities of mPGES-1-PGE2 in inflammatory arthritis. The development of mPGES-1 inhibitors for treatment of inflammation and pain will need to account for diversity in the mediators of different forms of arthritis that are likely to influence efficacy of these agents.
MATERIALS AND METHODS
Animals
DBA1/lac J mPGES-1 +/− mice were obtained from Pfizer and were described elsewhere (29). mPGES-1 −/− mice were obtained by mating mPGES-1 +/− animals. Wild type littermates were used as control. Mice were housed in microisolator cages in a pathogen-free barrier facility, and all experiments were performed under the Institutional Animal Care and Use Committee guidelines as set forth by the University of Kentucky, Lexington KY.
Arthritis model
CAIA was induced in 8–12 week old, randomly selected male and female mice using mouse monoclonal anti-type II collagen 5 clone antibody cocktail kit with lipopolysaccharide (LPS) from E. coli 0111:B4 (Chondrex) following the manufacturer's instructions. Mice were immunized on day 0 by intraperitoneal (IP) injection of 150 μg of the antibody cocktail and boosted with 25 μg of LPS on day 3. The CAIA severity was assessed using Chondrex mouse arthritis scoring system and was shown to reach its peak on day 7 (http://www.chondrex.com/animal-models/athrogen-cia-arthritogenic-monoclonal-antibody-cocktail) (32).
Real-time RT-PCR
Total RNA was obtained from the synovial capsules that were isolated on day 7 from the front paws of arthritic mice with an arthritis score of ≥ 3. The RNA isolation was performed with TRIzol reagent (Ambion) following the manufacturer's instructions. Total RNA was quantitatively converted into the single stranded cDNA by using High Capacity cDNA Archive Kit (Applied Biosystems). Particular genes were detected using the respective TaqMan Gene Expression Assays (Applied Biosystems) on a 7300 Real-Time RT-PCR system from the same manufacturer by relative quantitation employing glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as the reference gene.
Lipid extraction
Lipids were extracted from pulverized front mouse paws as described (13) with the following modifications. After pulverization, the respective frozen powder was immediately placed into the Pyrex 10 ml screw cap analytical tube with the 3 ml of ice-cold 50% ethanol and weighted. The sample wet weight was later used for the normalization of eicosanoid levels. 10 μl of antioxidant mixture (0.2 mg/ml butylated hydroxytoluene and 0.2 mg/ml EDTA in 2:1:1 methanol:ethanol:H2O solution) was added to each tube and samples were kept at −80°C for a few days prior to their overnight shipment on dry ice to the lipid analytical facility.
Analysis of eicosanoids by LC/MS/MS
This was carried out as previously described in detail (34). Briefly, an Agilent 1200 SL liquid chromatography series (Agilent Corporation, Palo Alto, CA) with an Agilent Eclipse Plus C18 2.1 × 150 mm, 1.8 μm column were used for the eicosanoid separation. The mobile phase A was water with 0.1% acetic acid while the mobile phase B was composed by acetonitrile/methanol (80/15, v/v) and 0.1% acetic acid. Gradient elution was performed at a flow rate of 250 μL/min. The injection volume was 10 μL and the samples were kept at 4 °C in the auto sampler. Analytes were detected by negative MRM mode using a 4000 QTrap tandem mass spectrometer (Applied Biosystems) equipped with an electrospray ionization source (Turbo V). Calibration curves were generated by 10 μL injections of seven standards containing each analyte, internal standard I (d4-PGE2, d11–14,15-DiHETrE, d8-5-HETE, and d11-11(12)-EpETrE), and internal standard II (1-cyclohexyl-dodecanoic acid urea, CUDA) for quantification purpose.
RESULTS
CAIA is exacerbated in mPGES-1KO mice
Using the described protocol, CAIA in mice is a highly reproducible experimental arthritis model the inflammation reaches a maximum at D7 and persists for two weeks (32). Because we were interested in examining the effect of mPGES-1 deficiency on inflammation when the latter reaches its maximum, we did not extend our analysis of experimental animals with CAIA beyond D7 (Fig.2). As shown in this Figure, two major indicators of arthritis, its incidence and clinical score, were significantly higher in mPGES-1 KO compared to WT mice on D7 (Fig.2 A&B). It is worth noting that arthritis was numerically though not statistically more severe in mPGES-1 KO mice throughout the course of the experiment (Fig.2 A&B). The animal body weights, however, were not different comparing mPGES-1 null and WT mice (Fig.2C). The data would be consistent with the requirement of mPGES-1, and consequently PGE2, primarily for the suppressive effect on local, and not systemic, inflammation in the CAIA model of inflammatory arthritis.
Fig.2.

Increased severity of inflammatory arthritis in mPGES-1 KO mice. WT and mPGES-1 KO mice were injected with mouse monoclonal anti-type II collagen 5 clone antibody cocktail kit with lipopolysaccharide (LPS) from E. coli 0111:B4 on D0 and boosted with LPS on D3. The arthritis severity was assessed by arthritis score (A) and by the number of inflamed paws (B). The animal body weight (C) was used as a surrogate, whole-body inflammation marker. Data shown are mean ± SE. (*) depicts P ≤ 0.05 as compared to control (Student t-test, n=9). (**) depicts P ≤ 0.05 as compared to control (Mann-Whitney rank sum test, n=9).
Increased neutrophil infiltration into the inflamed joints of mPGES-1 KO mice with CAIA
It was previously shown that CAIA was driven by the recruitment of neutrophils and macrophages to the inflamed joints (31,33). Therefore, in order to elucidate a mechanism which is responsible for the observed increased CAIA severity in mPGES-1 mice (Fig.2), we assessed the infiltration of neutrophils and macrophages into the arthritic joints of WT and mPGES-1 KO mice with CAIA at its peak by measuring the expression of the respective specific markers on D7 by real-time RT-PCR. We used myeloperoxidase (MPO) as the neutrophil specific marker (35,36) as well as IL-6 (37,38) and PTGS2 (COX-2) (39,40) as the dual neutrophil/macrophage inflammatory markers. We detailed further the phenotype of activated macrophages in the arthritic joints by probing the expression of the classically activated (M1, inflammatory) and alternatively activated (M2, antiinflammatory) specific markers. We used CCL3 and NOS2 to detect M1 macrophages as well as CD163 and CD206 for the M2 macrophage identification (38,41). The respective data are presented in the Figs.3–5. As it is seen from the Figure 3, CAIA in mPGES-1 KO mice at its peak on D7 is characterized by significantly increased MPO (Fig.3A, ~ 3.6-fold) and PTGS2 (Fig.3C, ~ 2.5-fold) expression as compared to the WT mice. Expression of IL-6 demonstrated a trend toward upregulation (Fig.3B, ~ 2.5-fold), although this change did not reach statistical significance. Given that PTGS2 could be expressed by both neutrophils and macrophages, the above data while being clearly indicative of the increased infiltration of neutrophils into the arthritic joints of mPGES-1 KO mice with CAIA, leave in question the respective macrophage infiltration. We addressed the latter by measuring expression of the M1 and M2 macrophage specific markers. (Figs.4&5). As in these Figures, neither M1 (Fig.4) nor M2 (Fig.5) macrophage markers demonstrated any change, thereby efficiently ruling out upregulated macrophage infiltration as a dominant force for the increased severity of the inflammatory arthritis in mPGES-1 KO mice.
Fig.3.
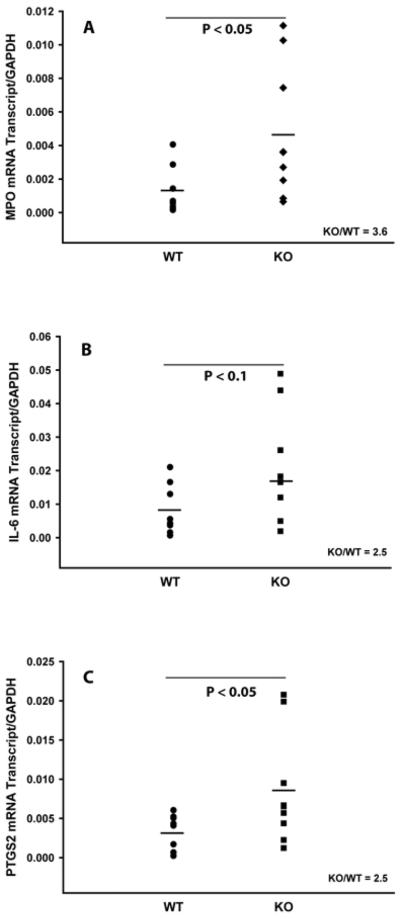
Analysis of inflammatory markers in arthritic paws of WT and mPGES-1 KO animals by real-time RT-PCR. Total RNA was obtained from a synovial capsule isolated from the front arthritic paw on D7. Relative mRNA expression of MPO (A), IL-6 (B), and PTGS2 (C) was measured using the respective TaqMan Gene Expression Assays from Applied Biosystems. Each datum point represents a single animal. Two animal groups were compared by Student t-test with the P ≤ 0.05 considered to be significant (n=9).
Fig.5.

Analysis of the alternatively activated (M2) macrophage markers in arthritic paws of WT and mPGES-1 KO animals by real-time RT-PCR. Total RNA was obtained from a synovial capsule isolated from the front arthritic paw (arthritis score ≥ 3) on D7. Relative mRNA expression of CD163 (A) and CD206 (B) was measured using the respective TaqMan Gene Expression Assays from Applied Biosystems. Each datum point represents a single animal. Two animal groups were compared by Student t-test with the P ≤ 0.05 considered to be significant (n=9).
Fig.4.

Analysis of the classically activated (M1) macrophage markers in arthritic paws of WT and mPGES-1 KO animals by real-time RT-PCR. Total RNA was obtained from a synovial capsule isolated from the front arthritic paw (arthritis score ≥ 3) on D7. Relative mRNA expression of CCL3 (A) and NOS2 (B) was measured using the respective TaqMan Gene Expression Assays from Applied Biosystems. Each data point represents a single animal. Two animal groups were compared by Student t-test with the P ≤ 0.05 considered to be significant (n=9).
Lipidomic profiling of CAIA in mPGES-1 KO mice
Although a role of ARA metabolites, such as lipoxins, resolvins, and protectins in resolution of inflammation is well established (25), very little is known regarding the function of eicosanoids as antiinflammatory agents in arthritis (13). In order to gain more insight into the role of ARA metabolites, and particularly PGE2, in inflammatory arthritis, we performed lipidomic analysis of front paws from the unelicited (non-immunized) WT and mPGES-1 KO mice as well as from the mice with CAIA when the latter reached its peak on D7. The results are presented in the Figs.6&7 where the ARA metabolites are grouped according to their respective biosynthetic pathways (Fig.1),
Fig.6.

Quantitative eicosanoid profiling of arthritic paws from WT and mPGES-1 KO animals by LC/MS/MS. Lipids were extracted from the front paws of WT control, WT arthritic (arthritis score ≥ 3 on D7), mPGES-1 KO control, mPGES-1 KO arthritic (arthritis score ≥ 3 on D7). The lipid analytes were grouped based on their respective metabolic pathways: COX (A), LOX (B), and CYP450 (C) (Fig.1). Data shown are mean ± SE. (*) depicts P ≤ 0.05 as compared to WT arthritic (n=3–5).
Fig.7.

Quantitative eicosanoid profiling of arthritic paws from WT and mPGES-1 KO animals by LC/MS/MS. Lipids were extracted from the front paws of WT control, WT arthritic (arthritis score ≥ 3 on D7), mPGES-1 KO control, mPGES-1 KO arthritic (arthritis score ≥ 3 on D7). The lipid analytes were grouped based on their respective metabolic pathways: Non-Enzymatic (A) and sEH (B) (Fig.1). Data shown are mean ± SE (n=3–5).
COX-dependent pathway
As seen in the Figure 6A, genetic deletion of mPGES-1 did not result in the statistically significant differences in the basal production of COX-dependent ARA metabolites. At the same time, there was a trend toward ~ 2.2-fold reduction in the basal PGE2 production with the concomitant trends toward ~2.7-fold upregulation of PGD2 and ~ 2-fold reduction of 15-deoxy-PGJ2 (Fig. 6A). More importantly, in the setting of inflammatory arthritis, mPGES-1 KO as compared to WT animals displayed a dramatic (~59-fold) and statistically significant reduction in PGE2 production, whereas the sizable downregulation of PGD2 (~3.2-fold), 6-keto-PGF1α (~2.2-fold), and PGF2α (~19-fold) levels can be only viewed as a trend (Fig.6A). We were unable to measure a change in 15-deoxy-PGJ2 levels during arthritis induction in either the WT or mPGES-1 null mice (Fig.6A). These data are consistent with mPGES-1 being a dominant PGH2 isomerase which is responsible for PGE2 production in both the naïve and arthritic mouse joints.
LOX-dependent pathway
There was no significant difference in 5-HETE, 12-HETE, and 15-HETE levels between naïve WT and mPGES-1 KO mice, whereas their production in arthritic mPGES-1 KO mice vs WT mice with arthritis showed a trend toward reduction by respectively ~3.4, 3.8, and 2.6-fold (Fig.6B).
CYP450-dependent pathway
This data set revealed that only ARA metabolites, 14(15)-EET and 11(12)-EET, and not the LA derivatives, 12,13DiHOME, 9,10-DiHOME, 9(10)-EpOME, and 12(13)-EpOME could have been affected by mPGES-1 deletion in arthritic animals. As compared to the WT mice with arthritis, the respective 14(15)-EET and 11(12)-EET displayed a tendency toward reduction in arthritic mPGES-1 KO mice by ~2.9 and ~2.0-fold (Fig.6C).
Non-enzymatic pathway
While the 8-HETE level displayed no difference when it was measured in WT and mPGES-1 under the basal and arthritic conditions, 11-HETE production in the naïve mPGES-1 KO vs WT mice showed a trend toward reduction by ~1.7-fold and this disparity had reached ~5.6-fold difference when the average 11-HETE levels were compared between arthritic WT and mPGES-1 KO animals (Fig.7A).
sEH pathway
The strong antiinflammatory properties of EETs are well-established and their physiological levels are mainly regulated by their conversion into inactive or less active DHETs which is catalyzed by sEH (42). We have already noticed that m-PGES-1 KO mice with arthritis were characterized by a significantly lower average levels of 11.12-EET and 14,15-EET as compared to their WT counterparts (Fig.6C). Since this apparent reduction could be attributable to the upregulated sEH activity, we have measured the levels of the respective EET metabolites, 11,12-DHET and 14,15-DHET (Fig.7B). The data in the Figure 7B show a tendency toward reduction of 11,12-DHET and 14,15-DHET levels in the arthritic mPGES-1 KO mice vs WT mice with arthritis by respectively ~2.0 and 3.4-fold. The calculated ratios of 11,12-DHET/11,12-EET for the arthritic WT and mPGES-1 KO were quite similar (~0.41 and ~0.44) so were the respective 14,15-DHET/14,15-EET values (~0.55 and ~0.55). This supports the hypothesis that sEH activity remains intact in the arthritic mPGES-1 KO mice.
DISCUSSION AND CONCLUSIONS
Prostanoids and particularly PGE2 have often been viewed as simply proinflammatory mediators in arthritis (7–10). This is somewhat surprising because an important role of PGE2 in the resolution of inflammation where it switches ARA metabolism toward production of anti-inflammatory and pro-resolving lipoxins, resolvins, and protectins is well established (25). Moreover, exogenous PGE1 was shown to curb nephritis and adjuvant arthritis in experimental animals (43,44). However, there is a growing body of clinical data (11,12) and experimental studies (13,14) pointing toward a more complicated and sophisticated role of PGE2 in regulation of inflammatory arthritis. A recent study (14) identified an important role for PGE2 in resolving inflammation in the autoimmune (CIA) model of experimental arthritis through regulation of the antiinflammatory lipid, lipoxin A4, thereby serving as pro-resolving lipid molecule. There is a growing understanding of differences between antiinflammatory and pro-resolving modes of action. The former mainly targets neutrophil recruitment to the site of inflammation whereas the latter promotes the removal of apoptotic cells and infectious microorganisms by macrophages (25). Therefore, we designed our study with the aim of examining the antiinflammatory functions of PGE2 using mPGES-1 KO mice and CAIA as a model of inflammatory arthritis.
The current study provides several important insights into the regulation of inflammatory arthritis by mPGES-1 and its product, PGE2. We established that mPGES-1 deficiency increased both the incidence and the severity of CAIA in mice. These results are in the agreement with those of Zurier & Quagliata (44) who demonstrated the antiinflammatory effect of PGE1 in the adjuvant model of inflammatory arthritis. However, our results disagree with those of Kamei et al (45) who reported on the suppression of CAIA in the mPGES-1 KO mice. The above discrepancy could be explained by the different genetic background of mPGES-1 KO animals, pure DBA1/lac J background used in our study and a mixed C57BL/6 × 129/SvJ background used by Kamei et al (45). Yet the different approach for mPGES-1 gene deletion and CAIA induction could also add to the observed discrepancy. The fact that both arthritis score and incidence were increased in the PGES-1 KO animals with arthritis, but the loss of the body weight in arthritic mPGES-1 KO animals was similar to that of arthritic WT mice, would be consistent with local and not systemic inflammation being primarily affected by the deletion of mPGES-1 gene.
The above conclusion is reinforced by our observation that as compared to the WT mice, the peak of CAIA in the mPGES-1 KO animals coincided with significantly (~ 3.6-fold) upregulated neutrophil, and not macrophage infiltration. Therefore, in considering antiinflammatory versus pro-resolving modes of action (25), mPGES-1 and consequently PGE2, appear to exert primarily antiinflammatory effects in the CAIA model of inflammatory arthritis. That said, mPGES-1 deletion results in other changes in eicosanoid metabolism in addition to a dramatic reduction in PGE2 production. PGD2 and 6-keto-PGF1α increased in mPGES-1-deficient arthritic mice compared with control, albeit lower than levels in arthritis WT mice. Nevertheless, the ratio of PGE2 and other PG are dramatically altered by mPGES-1 deficiency and it is certainly plausible that altered relative levels of eicosanoids could account for the increased arthritis severity in this model.
Finally, the involvement of PGE2 in promoting the antiinflammatory action of mPGES-1 is supported by our lipidomics data that demonstrated a staggering (~ 59-fold) PGE2 reduction at the peak of inflammatory arthritis. It should be noted, that mPGES-1 is constitutively expressed at high level in neutrophils and is strongly upregulated by inflammatory stimuli such as LPS (46). More importantly, mPGES-1 expression level correlates strongly with PGE2 production (46). These data provide a strong support to the notion that PGE2 might exert its antiinflammatory effect through the intracrine/autocrine negative regulation of neutrophil recruitment and function, including the suppression of the neutrophil-mediated phagocytosis (15). The antiinflammatory mode of PGE2 could be masked in the autoimmune model of arthritis (CIA) (14,29) by its strong positive regulation of autoantibody formation (29). Indeed, PGE2 may play a critical role at the intersection of innate and acquired immunity.
These conclusions are supported by the work of Serhan and colleagues who demonstrate that early events in an inflammatory response set the resolution program in motion. In this model, specialized pro-resolving mediators, including lipoxins, resolvins, and protectins, are actively generated and act on neutrophils and macrophages to promote resolution of inflammation (25). PG, including PGE2, involved in the initiation phase of inflammation activate the translation of mRNAs encoding enzymes that are necessary for production of these immunoresolvents. Inhibitors of COX-2, in fact, delay the resolution of inflammation (47,48). In summary, all of these data point toward mPGES-1 as a pivotal regulator in the CAIA model of arthritis by inhibition of neutrophil function. When developing and using selective mPGES-1 inhibitors, attention must be paid to the specific mediators of different forms of arthritis in evaluating potential clinical targets for these agents.
Acknowledgment
This work was supported by RO1AR049010 (to L.J.C.) from the National Institutes of Health. Partial support for analytical chemistry was provided by NIEHS RO1 ES ES002710 and NIEHS Superfund Research Program grant P42 ES004699, or NIH U24DK097154 (to B.D.H.). B.D.H. is a George and Judy Marcus senior fellow of the American Asthma Foundation.
ABBREVIATIONS
- ARA
arachidonic acid
- RA
rheumatoid arthritis
- MPO
myeloperoxidase
- IL-6
interleukin 6
- LC/MS/MS
liquid chromatography/tandem mass spectrometry
- PGE2
prostaglandin E2
- PGD2
prostaglandin D2
- PGF2α
prostaglandin F2α
- 5-HETE
5-Hydroxyeicosatetraenoic acid
- 9-HETE
9-Hydroxyeicosatetraenoic acid
- 11-HETE
11-Hydroxyeicosatetraenoic acid
- 15-HETE
15-Hydroxyeicosatetraenoic acid
- 9,10-DiHOME
(±)9,10-dihydroxy-12Z-octadecenoic acid
- 14,15-DiHETrE
(±)14,15-dihydroxy-5Z,8Z,11Z-eicosatrienoic acid
- 9,10-EpOME
(±)9(10)-epoxy-12Z-octadecenoic acid
- 14,15-EpETrE
(±)14(15)-epoxy-5Z,8Z,11Z-eicosatrienoic acid.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Harris SG, Padilla J, Koumas L, Ray D, Phipps RP. Prostaglandins as modulators of immunity. Trends Immunol. 2002;123:44–150. doi: 10.1016/s1471-4906(01)02154-8. [DOI] [PubMed] [Google Scholar]
- 2.Ricciotti E, Fitzgerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31:986–1000. doi: 10.1161/ATVBAHA.110.207449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burke JE, Dennis EA. Phospholipase A2 structure/function, mechanism, and signaling. J Lipid Res. 2009;50(Suppl):S237–S242. doi: 10.1194/jlr.R800033-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fritsche KL. Too much linoleic acid promotes inflammation-doesn't it? Prostaglandins Leukot Essent Fatty Acids. 2008;79:173–175. doi: 10.1016/j.plefa.2008.09.019. [DOI] [PubMed] [Google Scholar]
- 5.Rouzer CA, Marnett LJ. Cyclooxygenases: structural and functional insights. J Lipid Res. 2009;50(Suppl):S29–S34. doi: 10.1194/jlr.R800042-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smyth EM, Grosser T, Wang M, Yu Y, Fitzgerald GA. Prostanoids in health and disease. J Lipid Res. 2009;50(Suppl):S423–S428. doi: 10.1194/jlr.R800094-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Myers LK, Kang AH, Postlethwaite AE, Rosloniec EF, Morham SG, Shlopov BV, Goorha S, Ballou LR. The genetic ablation of cyclooxygenase 2 prevents the development of autoimmune arthritis. Arthritis Rheum. 2000;43:2687–2693. doi: 10.1002/1529-0131(200012)43:12<2687::AID-ANR8>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 8.Robinson DR, Tashjian AH, Jr., Levine L. Prostaglandin-induced bone resorption by rheumatoid synovia. Trans Assoc Am Physicians. 1975;88:146–160. [PubMed] [Google Scholar]
- 9.Crofford LJ. Is there a place for non-selective NSAIDs in the treatment of arthritis? Joint Bone Spine. 2002;69:4–7. doi: 10.1016/s1297-319x(01)00334-7. [DOI] [PubMed] [Google Scholar]
- 10.Shibata-Nozaki T, Ito H, Mitomi H, Akaogi J, Komagata T, Kanaji T, Maruyama T, Mori T, Nomoto S, Ozaki S, Yamada H. Endogenous prostaglandin E2 inhibits aberrant overgrowth of rheumatoid synovial tissue and the development of osteoclast activity through EP4 receptor. Arthritis Rheum. 2011;63:2595–2605. doi: 10.1002/art.30428. [DOI] [PubMed] [Google Scholar]
- 11.Melnikova I. Future of COX2 inhibitors. Nat Rev Drug Discov. 2005;4:453–454. doi: 10.1038/nrd1755. [DOI] [PubMed] [Google Scholar]
- 12.van Vollenhoven RF. Treatment of rheumatoid arthritis: state of the art 2009. Nat Rev Rheumatol. 2009;5:531–541. doi: 10.1038/nrrheum.2009.182. [DOI] [PubMed] [Google Scholar]
- 13.Blaho VA, Buczynski MW, Brown CR, Dennis EA. Lipidomic analysis of dynamic eicosanoid responses during the induction and resolution of Lyme arthritis. J Biol Chem. 2009;284:21599–21612. doi: 10.1074/jbc.M109.003822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chan MM, Moore AR. Resolution of inflammation in murine autoimmune arthritis is disrupted by cyclooxygenase-2 inhibition and restored by prostaglandin E2-mediated lipoxin A4 production. J. Immunol. 2010;184:6418–6426. doi: 10.4049/jimmunol.0903816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith RJ. Modulation of phagocytosis by and lysosomal enzyme secretion from guinea-pig neutrophils: effect of nonsteroid anti-inflammatory agents and prostaglindins. J Pharmacol Exp Ther. 1977;200:647–657. [PubMed] [Google Scholar]
- 16.Nemeth K, Leelahavanichkul A, Yuen PS, Mayer B, Parmelee A, Doi K, Robey PG, Leelahavanichkul K, Koller BH, Brown JM, Hu X, Jelinek I, Star RA, Mezey E. Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat Med. 2009;15:42–49. doi: 10.1038/nm.1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heusinkveld M, de Vos van Steenwijk PJ, Goedemans R, Ramwadhdoebe TH, Gorter A, Welters MJ, van HT, van der Burg SH. M2 macrophages induced by prostaglandin E2 and IL-6 from cervical carcinoma are switched to activated M1 macrophages by CD4+ Th1 cells. J Immunol. 2011;187:1157–1165. doi: 10.4049/jimmunol.1100889. [DOI] [PubMed] [Google Scholar]
- 18.Qian X, Zhang J, Liu J. Tumor-secreted PGE2 inhibits CCL5 production in activated macrophages through cAMP/PKA signaling pathway. J Biol Chem. 2011;286:2111–2120. doi: 10.1074/jbc.M110.154971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Faour WH, Alaaeddine N, Mancini A, He QW, Jovanovic D, Di Battista JA. Early growth response factor-1 mediates prostaglandin E2-dependent transcriptional suppression of cytokine-induced tumor necrosis factor-alpha gene expression in human macrophages and rheumatoid arthritis-affected synovial fibroblasts. J Biol Chem. 2005;280(9):536–9546. doi: 10.1074/jbc.M414067200. [DOI] [PubMed] [Google Scholar]
- 20.D'Acquisto F, Sautebin L, Iuvone T, Di RM, Carnuccio R. Prostaglandins prevent inducible nitric oxide synthase protein expression by inhibiting nuclear factor-kappaB activation in J774 macrophages. FEBS Lett. 1998;440(7):6–80. doi: 10.1016/s0014-5793(98)01407-0. [DOI] [PubMed] [Google Scholar]
- 21.Gomez PF, Pillinger MH, Attur M, Marjanovic N, Dave M, Park J, Bingham CO, III, Al-Mussawir H, Abramson SB. Resolution of inflammation: prostaglandin E2 dissociates nuclear trafficking of individual NF-kappaB subunits (p65, p50) in stimulated rheumatoid synovial fibroblasts. J Immunol. 2005;175:6924–6930. doi: 10.4049/jimmunol.175.10.6924. [DOI] [PubMed] [Google Scholar]
- 22.Ziegler-Heitbrock HW, Wedel A, Schraut W, Strobel M, Wendelgass P, Sternsdorf T, Bauerle PA, Haas JG, Riethmuller G. Tolerance to lipopolysaccharide involves mobilization of nuclear factor kappa B with predominance of p50 homodimers. J Biol Chem. 1994;269:17001–17004. [PubMed] [Google Scholar]
- 23.Porta C, Rimoldi M, Raes G, Brys L, Ghezzi P, Di LD, Dieli F, Ghisletti S, Natoli G, De BP, Mantovani A, Sica A. Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor kappaB. Proc Natl Acad Sci U. S. A. 2009;106:14978–14983. doi: 10.1073/pnas.0809784106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Conner JR, Smirnova II, Moseman AP, Poltorak A. IRAK1BP1 inhibits inflammation by promoting nuclear translocation of NF-kappaB p50. Proc Natl Acad Sci U. S. A. 2010;107:11477–11482. doi: 10.1073/pnas.1006894107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. 2008;8:349–361. doi: 10.1038/nri2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sampey AV, Monrad S, Crofford LJ. Microsomal prostaglandin E synthase-1: the inducible synthase for prostaglandin E2. Arthritis Res Ther. 2005;7:114–117. doi: 10.1186/ar1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jakobsson PJ, Thoren S, Morgenstern R, Samuelsson B. Identification of human prostaglandin E synthase: a microsomal, glutathione-dependent, inducible enzyme, constituting a potential novel drug target. Proc Natl Acad Sci U. S. A. 1999;96:7220–7225. doi: 10.1073/pnas.96.13.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Samuelsson B, Morgenstern R, Jakobsson PJ. Membrane prostaglandin E synthase-1: a novel therapeutic target. Pharmacol. Rev. 2007;59:207–224. doi: 10.1124/pr.59.3.1. [DOI] [PubMed] [Google Scholar]
- 29.Kojima F, Kapoor M, Yang L, Fleishaker EL, Ward MR, Monrad SU, Kottangada PC, Pace CQ, Clark JA, Woodward JG, Crofford LJ. Defective generation of a humoral immune response is associated with a reduced incidence and severity of collagen-induced arthritis in microsomal prostaglandin E synthase-1 null mice. J Immunol. 2008;180:8361–8368. doi: 10.4049/jimmunol.180.12.8361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trebino CE, Stock JL, Gibbons CP, Naiman BM, Wachtmann TS, Umland JP, Pandher K, Lapointe JM, Saha S, Roach ML, Carter D, Thomas NA, Durtschi BA, McNeish JD, Hambor JE, Jakobsson PJ, Carty TJ, Perez JR, Audoly LP. Impaired inflammatory and pain responses in mice lacking an inducible prostaglandin E synthase. Proc Natl Acad Sci U. S. A. 2003;100:9044–9049. doi: 10.1073/pnas.1332766100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Asquith DL, Miller AM, McInnes IB, Liew FY. Animal models of rheumatoid arthritis. Eur J Immunol. 2009;39:2040–2044. doi: 10.1002/eji.200939578. [DOI] [PubMed] [Google Scholar]
- 32.Kannan K, Ortmann RA, Kimpel D. Animal models of rheumatoid arthritis and their relevance to human disease. Pathophysiology. 2005;12:167–181. doi: 10.1016/j.pathophys.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 33.Ozaki N, Suzuki S, Ishida M, Harada Y, Tanaka K, Sato Y, Kono T, Kubo M, Kitamura D, Encinas J, Hara H, Yoshida H. Syk-dependent signaling pathways in neutrophils and macrophages are indispensable in the pathogenesis of anti-collagen antibody-induced arthritis. Int Immunol. 2012;24:539–550. doi: 10.1093/intimm/dxs078. [DOI] [PubMed] [Google Scholar]
- 34.Charles RL, Burgoyne JR, Mayr M, Weldon SM, Hubner N, Dong H, Morisseau C, Hammock BD, Landar A, Eaton P. Redox regulation of soluble epoxide hydrolase by 15-deoxy-delta-prostaglandin J2 controls coronary hypoxic vasodilation. Circ Res. 2011;108:324–334. doi: 10.1161/CIRCRESAHA.110.235879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bradley PP, Priebat DA, Christensen RD, Rothstein G. Measurement of cutaneous inflammation: estimation of neutrophil content with an enzyme marker. J Invest Dermatol. 1982;78:206–209. doi: 10.1111/1523-1747.ep12506462. [DOI] [PubMed] [Google Scholar]
- 36.van LM, Gijbels MJ, Duijvestijn A, Smook M, van de Gaar MJ, Heeringa P, de Winther MP, Tervaert JW. Accumulation of myeloperoxidase-positive neutrophils in atherosclerotic lesions in LDLR−/− mice. ArteriosclerThromb Vasc Biol. 2008;28:84–89. doi: 10.1161/ATVBAHA.107.154807. [DOI] [PubMed] [Google Scholar]
- 37.Melani C, Mattia GF, Silvani A, Care A, Rivoltini L, Parmiani G, Colombo MP. Interleukin-6 expression in human neutrophil and eosinophil peripheral blood granulocytes. Blood. 1993;81:2744–2749. [PubMed] [Google Scholar]
- 38.Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol. 2006;177:7303–7311. doi: 10.4049/jimmunol.177.10.7303. [DOI] [PubMed] [Google Scholar]
- 39.Maloney CG, Kutchera WA, Albertine KH, McIntyre TM, Prescott SM, Zimmerman GA. Inflammatory agonists induce cyclooxygenase type 2 expression by human neutrophils. J Immunol. 1998;160:1402–1410. [PubMed] [Google Scholar]
- 40.Tomlinson A, Appleton I, Moore AR, Gilroy DW, Willis D, Mitchell JA, Willoughby DA. Cyclo-oxygenase and nitric oxide synthase isoforms in rat carrageenin-induced pleurisy. Br J Pharmacol. 1994;113:693–698. doi: 10.1111/j.1476-5381.1994.tb17048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gleissner CA, Shaked I, Little KM, Ley K. CXC chemokine ligand 4 induces a unique transcriptome in monocyte-derived macrophages. J Immunol. 2010;184:4810–4818. doi: 10.4049/jimmunol.0901368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Imig JD, Hammock BD. Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat Rev Drug Discov. 2009;8:794–805. doi: 10.1038/nrd2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zurier RB, Sayadoff DM, Torrey AB, Rothfield NF. Prostaglandin E treatment of NZB/NZW mice. Arthritis Rheum. 1977;20:723–728. doi: 10.1002/art.1780200213. [DOI] [PubMed] [Google Scholar]
- 44.Zurier RB, Quagliata F. Effect of prostaglandin E 1 on adjuvant arthritis. Nature. 1971;234:304–305. doi: 10.1038/234304a0. [DOI] [PubMed] [Google Scholar]
- 45.Kamei D, Yamakawa K, Takegoshi Y, Mikami-Nakanishi M, Nakatani Y, Oh-Ishi S, Yasui H, Azuma Y, Hirasawa N, Ohuchi K, Kawaguchi H, Ishikawa Y, Ishii T, Uematsu S, Akira S, Murakami M, Kudo I. Reduced pain hypersensitivity and inflammation in mice lacking microsomal prostaglandin e synthase-1. J Biol Chem. 2004;279:33684–33695. doi: 10.1074/jbc.M400199200. [DOI] [PubMed] [Google Scholar]
- 46.Mosca M, Polentarutti N, Mangano G, Apicella C, Doni A, Mancini F, De BM, Coletta I, Polenzani L, Santoni G, Sironi M, Vecchi A, Mantovani A. Regulation of the microsomal prostaglandin E synthase-1 in polarized mononuclear phagocytes and its constitutive expression in neutrophils. J Leukoc Biol. 2007;82:320–326. doi: 10.1189/jlb.0906576. [DOI] [PubMed] [Google Scholar]
- 47.Chan MM-Y, Moore AR. Resolution of inflammation in murine autoimmune arthritis is disrupted by cyclooxygenase-2 inhibition and restored by prostaglandin E2-mediated lipoxin A4 production. J Immunol. 2010;184:6418–6426. doi: 10.4049/jimmunol.0903816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gilroy DW, Colville-Nash PR, Willis D, Chivers J, Paul-Clark MJ, Willoughby DA. Inducible cyclooxygenase may have anti-inflammatory properties. Nat Med. 1999;5:698–701. doi: 10.1038/9550. [DOI] [PubMed] [Google Scholar]