

Overview
The importance of inflammation in promoting carcinogenesis and tumor progression is well recognized. Chronic inflammation caused by a variety of infectious agents can lead to the development of several common malignancies. Similarly, inflammatory bowel disease is a well known risk factor for colorectal cancer. Much less is known about the link between inflammation and the development of breast cancer. Recent data suggest that obesity causes both in-breast and systemic inflammation that contribute to the development and progression of breast cancer. This observation has potentially important implications in terms of prevention and treatment of breast cancer, especially given the rising worldwide overweight and obesity rates.
Inflamed white adipose tissue (WAT) within the breast is associated with elevated levels of proinflammatory mediators, enhanced expression of aromatase (the rate-limiting enzyme for estrogen biosynthesis), and increased estrogen receptor-α (ER-α)-dependent gene expression. Systemic consequences of obesity including altered adipokine levels, elevated circulating estrogen levels, and insulin resistance are also believed to play a role in the pathogenesis of breast cancer. Collectively, these findings suggest a significant role for inflammation in the pathogenesis of breast cancer in obese and overweight patients.
Introduction
Amongst its wide range of clinical consequences, obesity is a well-known risk factor for the development of several common epithelial malignancies.1 In postmenopausal women, the risk of developing breast cancers that express the estrogen and progesterone receptors (ER and PR), is significantly elevated for those who are obese or overweight.2 Furthermore, obese and overweight patients, once diagnosed, suffer from worse disease-related outcomes than their leaner counterparts, regardless of breast cancer subtype.3 After menopause, estrogen is mostly derived peripherally from the non-cyclical conversion of androgen precursors within adipose tissue. The rate-limiting step in this conversion is catalyzed by the cytochrome P450 enzyme, aromatase, which is encoded by the CYP19 gene. Circulating estrogens, such as estradiol, are known to stimulate the proliferation of breast epithelial cells and potentially exert a mutagenic effect. Higher levels of circulating estradiol as a result of increased adiposity and aromatase expression are thought to contribute, in part, to the greater risk of ER/PR-positive breast cancer in obese postmenopausal women.2 However, compared to the premenopausal state, circulating estradiol levels are significantly lower as the ovaries no longer produce substantial amounts of estrogen. Nevertheless, the incidence of ER-positive disease rises with age, approaching nearly 85% of breast cancers diagnosed in women during their ninth decade of life.4 This seemingly paradoxical association between the increasing incidence of hormone sensitive tumors in aging women whose circulating estradiol levels have declined after menopause highlights an important scientific question: Why are hormone sensitive breast cancers more prevalent after circulating levels of tumor-stimulating estrogens have significantly and naturally declined? One possibility is that the microenvironment in which the neoplasm arises and grows plays an important role and that it compensates for the decrease in circulating estrogens. Indeed, locally produced estrogens and, possibly, ligand-independent activation of ER-α have been associated with carcinogenesis.5 Local effects may also be important for understanding the link between obesity and increased risk of hormone receptor-positive breast cancer in obese postmenopausal women.
Additional evidence suggests that there are likely to be several estrogen-independent mechanisms involved in the link between obesity and breast carcinogenesis (Figure 1). In addition to the elevated risk of hormonally sensitive breast tumors, obesity has also been associated with an increased risk of ER-negative breast cancers in some studies.6 This observation, in particular, points to the involvement of estrogen-independent pathways (although these may also be operative in hormone receptor-positive cancers). Obesity is associated with chronic, systemic inflammation characterized by elevated levels of circulating proinflammatory mediators known to promote tumorigenesis and growth.1,7,8 Both the systemic and local consequences of chronic adipose inflammation thus provide key potential mechanistic links between obesity and breast cancer. In addition to inflammation, other obesity-related effects promote cell proliferation and survival. These include hyperinsulinemia and increased insulin-like growth factor-1 (IGF-1) signaling, as well as altered levels of the adipokines, adiponectin and leptin. This review addresses the complex biological interactions between obesity and breast cancer, with a particular focus on inflammation as a key mediator.
Fig. 1. Pathways linking obesity and breast cancer.30.

Abbreviations: IGF-1, insulin-like growth factor-1; IL-6, interleukin-6; IL-1β, interleukin-1β; TNFα, tumor necrosis factor α; SHBG, sex hormone binding globulin; VEGF, vascular endothelial growth factor. Reprinted with permission. © 2010 American Society of Clinical Oncology. All rights reserved.
Connecting obesity and breast cancer via inflammation
Tumor microenvironment
The links between inflammation and cancer have long been described. In the mid nineteenth century, Virchow first hypothesized that tissue injury and the ensuing inflammation promote enhanced cellular proliferation thereby predisposing cells to neoplastic transformation.9 Subsequent observations that many cancers arise at sites of chronic inflammation, coupled with epidemiologic data demonstrating increased cancer rates in patients with underlying inflammatory conditions, have led to significant efforts to better understand the complex role of inflammation. Adding urgency to this ongoing investigation, are reports linking infection and the inflammatory response to 15–20% of all cancer-related deaths worldwide.9 A critical observation is that the tumor microenvironment closely resembles that of a healing wound, including the influx of immune cells with resultant production of proinflammatory mediators as well as tissue remodeling and angiogenesis.10 Furthermore, malignant cells may co-opt the inflammatory mechanism leading to increased growth, invasion, and metastasis.11 Together, the epidemiologic and histologic observations strongly suggest that inflammation plays a key role in tumor biology, and is not simply a result of the development of cancer.
While many cancers including colon, lung, bladder, esophagus, liver, and others, are known to have infectious and/or inflammation-related etiologies, this link has been far less clear for breast cancer. Recently, inflammation, characterized by the hallmarks of wound healing, has been demonstrated to occur in breast tumors.12 Emerging evidence points to a causal relationship between obesity, chronic in-breast inflammation, and tumorigenesis. Increased body mass index has been associated with adipocyte hypertrophy in the breast.13 Moreover, a positive correlation has been observed between increased adipocyte size and cell death in the human breast. Adipocytes can release a variety of proinflammatory cytokines and adipokines which are likely to contribute to the recruitment and activation of immune cells including macrophages.14 Dying adipocytes also release free fatty acids (FFA) which can stimulate the production of proinflammatory cytokines via toll-like receptor-4 (TLR-4) and NFκB signaling.
Adipocyte interactions
A growing understanding of the complex interactions between adipocytes and immune cells within the WAT stromal vascular fraction is shedding new light on the role of adipose inflammation in tumor development and progression. The activated macrophage is a key mediator of adipose inflammation, and the adipocyte-macrophage interaction is emerging as a central theme. Adipocyte death leads to myeloid cell recruitment in a characteristic pattern whereby macrophages form a crown surrounding the dead adipocyte. This formation is histologically apparent as crown-like structures (CLS), which have been observed in subcutaneous and visceral fat in association with the metabolic syndrome.15 More recently, these inflammatory lesions were discovered to occur in the mammary gland of obese mice and the WAT of the human breast, termed CLS-B.13,16 Notably, CLS-B are detected more efficiently using immunohistochemical staining for CD68, a macrophage marker, rather than with standard hematoxylin and eosin (H&E) staining (Figure 2).13 For this reason as well as the usual focus on tumor, rather than benign tissue, CLS-B and their associated biology may not have been previously appreciated. As in the preclinical mouse models, the presence of CLS-B in women was associated with activation of NFκB and increased levels of TNFα, IL-1β, IL-6, and COX-2-derived PGE2. Relating to the epidemiological association of obesity and postmenopausal hormone receptor-positive breast cancer, a critical consequence of CLS-B and the associated elevation in tissue levels of proinflammatory cytokines is increased transcription of the CYP19 gene encoding aromatase.19 Indeed, several of the proinflammatory mediators associated with CLS-B, including TNFα, IL-1β, IL-6, and PGE2, are known to up regulate aromatase expression via specific promoters that give rise to unique mRNA species found in breast tissue.19–21 Increased levels and activity of aromatase lead to enhanced estrogen biosynthesis and up regulation of PR, an ER target gene. Additionally, increased circulating levels of proinflammatory mediators in obese breast cancer patients correlate with poor prognosis.17,18 For example, elevated levels of IL-6 in serum have been associated with worse survival in patients with metastatic hormone-refractory breast cancer.17
Fig. 2. Crown-like structures of the breast (CLS-B, arrow).
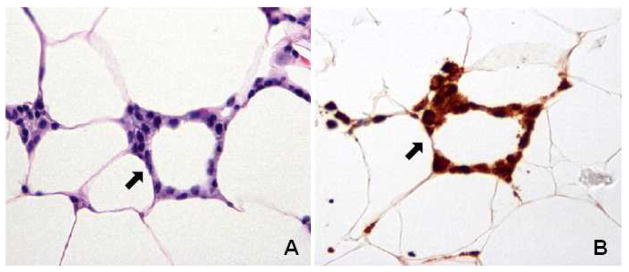
A. Hematoxylin and eosin stain. B. Anti-CD68 immunostain identifies macrophages. Reprinted from Morris P et al.: Cancer Prev Res Vol. 4(7), 2011:1021–9.
Given the significant biological consequences of these inflammatory pathways and the need to further explore the histologic structures associated with their activation, a CLS-B index was developed to quantify the severity of breast WAT inflammation. This index is defined as the number of slides with histologic evidence of CLS-B compared to the number of slides examined, reported on a scale ranging from 0 to 1.0. Consistent with the hypothesis that the obesity→inflammation→aromatase axis is operative, the severity of breast inflammation as defined by the CLS-B index correlated with aromatase activity.13 Furthermore, the presence of CLS-B was not merely a surrogate for increasing BMI. In support of this point, CLS-B were found in approximately 75% of obese women. Inflammatory foci can also be found in a minority of lean women. These findings are consistent with the observation that not all obese individuals suffer from the metabolic syndrome and that other factors may also induce localized inflammation in WAT. Overall, the discovery of CLS-B implicates inflammation specifically, rather than obesity alone, as a key driver of aromatase activity in the breast. Taken together, these findings demonstrate an obesity→inflammation→aromatase signaling axis, characterized by a complex system of paracrine interactions between macrophages and other cells, e.g. preadipocytes, in human breast WAT (Figure 3). This axis places inflammation at the center of ER-positive breast cancer pathogenesis for many patients, and provides an important mechanistic and targetable link between obesity and breast cancer.
Fig. 3. Paracrine interactions involving activated macrophages establish an obesity→inflammation→aromatase axis in the breast tissue of obese/overweight women.
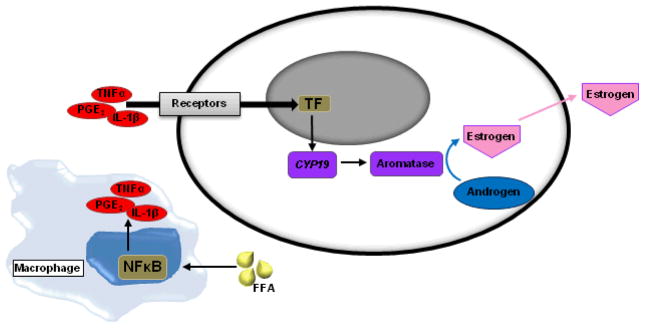
Abbreviations: FFA, free fatty acids; TNFα, tumor necrosis factor α; IL-1β, interleukin-1β; PGE2, prostaglandin E2; TF, transcription factors.
Metabolic dysfunction and inflammation
In addition to activation of estrogen signaling pathways via inflammation-mediated up regulation of aromatase, obesity promotes breast cancer through the systemic effects of dysregulated metabolism. A primary function of adipose tissue is to store energy in the form of triglycerides within the WAT adipocytes. Hyperadiposity is characterized by a state of energy imbalance, which plays an important role in immune activation. In addition to cell wall stretch and subsequent release of proinflammatory cytokines, adipocyte hypertrophy is associated with increased lipolysis resulting in the release of FFA. FFA are believed to stimulate inflammatory pathways in adipose tissue, but can also be deposited in various organs contributing to atherosclerosis, hypertension, insulin resistance and glucose intolerance. Indeed, excess adiposity is associated with higher circulating triglyceride levels, reflecting the state of energy imbalance that occurs in many obese/overweight patients. As noted, FFA stimulate multiple inflammatory signaling pathways, ultimately leading to activation of the transcription factor, NFκB, which plays a key role in the immune response and adipose inflammation.22 Activation of NFκB can be mediated by TLR-4, a member of the TLR family of pattern recognition molecules that are centrally involved in the immune response.23 Lipopolysaccharide (LPS), a component of the cell wall of gram negative bacteria, is a prototypic TLR-4 agonist and stimulates the innate immune response. In a similar manner, TLR-4 is thought to orchestrate inflammation in response to excess endogenous lipids.23,24 FFA signaling through TLR-4 can lead to macrophage activation, resulting in elevated levels of TNFα, IL-6, and other proinflammatory mediators as detailed above.24 These cytokines stimulate further lipolysis and release of FFA, thus establishing a reinforced feedback cycle of sustained inflammation.
Connecting obesity and breast cancer via endocrine dysfunction
Endocrine-mediated consequences of obesity also contribute to breast carcinogenesis and tumor progression. These include elevated levels of insulin and bioavailable insulin-like growth factor-1 (IGF-1), partly related to alterations in levels of the adipokines, adiponectin and leptin (Figure 1). Both of these adipokines have angiogenic properties. Elevated leptin levels, as occur in obesity, stimulate breast tumor cell proliferation through several signal transduction pathways and by altering cell-cycle checkpoints via up regulation of cyclin D1 and cdk2.25,26 In contrast to leptin, adiponectin levels are diminished in obesity. Although the underlying mechanisms are not completely understood, decreased adiponectin levels are associated with hyperinsulinemia and increased breast cancer risk. These observations may be partly explained by the effects of adiponectin on the PTEN/PI3K/mTOR, MAPK, and AMPK pathways, leading to enhanced cell proliferation and inhibition of apoptosis.1,25
Insulin resistance, characterized by elevated plasma insulin levels, occurs in association with obesity-related inflammation and is likely to play an important role in breast cancer progression.27 Both insulin and IGF-1 promote cell proliferation and inhibition of apoptosis by stimulating the PI3K/Akt and Ras/Raf/MAPK systems.28 Additionally, IGF-1 and insulin, to a lesser degree, interact with estrogen signaling pathways to promote hormone sensitive breast cancers. IGF-1 has been shown to stimulate aromatase activity.29 Insulin, on the other hand, acts systemically by inhibiting hepatic synthesis of sex-hormone-binding globulin (SHBG) which binds and transports the hormones testosterone, dihydrotestosterone, and estradiol, in a biologically inactive state.1 Increased free estradiol, as a result of diminished SHBG levels, is then available to stimulate growth of ER expressing breast tumors. Together, these findings suggest that obesity-associated insulin resistance has multiple local and systemic consequences that help to explain the link between obesity and breast cancer.
Conclusion
Rates of obesity and overweight are predicted to climb in the coming years and as such, we are likely to see a proportional increase in neoplastic disease and obesity-related mortality. Understanding the biological underpinnings of this relationship is critical to the development of preventative and adjunctive therapies that could disrupt the obesity-cancer link even if obesity itself remains. To this end, it is becoming increasingly apparent that activation of multiple inflammatory signaling pathways provides a critical link between obesity and cancer. Recent observations reporting the interactions between adipocytes and immune cells leading to a proinflammatory local milieu are providing new insights into the wound-like tumor microenvironment described by Virchow more than a century ago. Specifically, the discovery of an obesity→inflammation→aromatase axis highlights inflammation as a central mediator of breast carcinogenesis for many patients. Identifying patients in whom this axis is active could uncover a specific population that may benefit from anti-inflammatory interventions. The potential role of behavioral interventions, nutritional changes, and pharmacological strategies to attenuate obesity-related inflammation need to be explored. Because WAT inflammation is found in most but not all overweight and obese women, the development of non-invasive methods to detect chronic in-breast inflammation to identify the at-risk population is a key research objective.
Key Points.
In obese women, low grade or “smoldering” inflammation occurs in multiple white adipose tissue (WAT) depots including the breast.
Overweight and obesity are characterized by adipocyte hypertrophy and adipocyte death in association with recruitment of immune cells into WAT.
Infiltrating macrophages surround dead adipocytes in breast WAT to form inflammatory crown-like structures (CLS-B) which are associated with NFκB activation and increased levels of several proinflammatory mediators that are known inducers of aromatase and thereby estrogen biosynthesis.
Breast inflammation, quantified by the CLS-B index, correlates with increased aromatase activity thus establishing an obesity→inflammation→aromatase signaling axis.
Excess adiposity is associated with a state of metabolic dysregulation and systemic inflammation, which have been linked to breast carcinogenesis.
Acknowledgments
Grant Support:
This work was supported by NIH R01CA154481, the Breast Cancer Research Foundation, and grant UL1TR000457 of the Clinical and Translational Science Center at Weill Cornell Medical College.
Contributor Information
Neil M. Iyengar, Email: iyengarn@mskcc.org.
Clifford A. Hudis, Email: hudisc@mskcc.org.
Andrew J. Dannenberg, Email: ajdannen@med.cornell.edu.
References
- 1.van Kruijsdijk RC, van der Wall E, Visseren FL. Obesity and cancer: the role of dysfunctional adipose tissue. Cancer Epidemiol Biomarkers Prev. 2009;18:2569–2578. doi: 10.1158/1055-9965.EPI-09-0372. [DOI] [PubMed] [Google Scholar]
- 2.Cleary MP, Grossmann ME. Minireview: Obesity and breast cancer: the estrogen connection. Endocrinology. 2009;150:2537–2542. doi: 10.1210/en.2009-0070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Calle EE, Rodriguez C, Walker-Thurmond K, et al. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. 2003;348:1625–1638. doi: 10.1056/NEJMoa021423. [DOI] [PubMed] [Google Scholar]
- 4.Li CI, Daling JR, Malone KE. Incidence of invasive breast cancer by hormone receptor status from 1992 to 1998. J Clin Oncol. 2003;21:28–34. doi: 10.1200/JCO.2003.03.088. [DOI] [PubMed] [Google Scholar]
- 5.Lorincz AM, Sukumar S. Molecular links between obesity and breast cancer. Endocr Relat Cancer. 2006;13:279–292. doi: 10.1677/erc.1.00729. [DOI] [PubMed] [Google Scholar]
- 6.Daling JR, Malone KE, Doody DR, et al. Relation of body mass index to tumor markers and survival among young women with invasive ductal breast carcinoma. Cancer. 2001;92:720–729. doi: 10.1002/1097-0142(20010815)92:4<720::aid-cncr1375>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 7.Pierce BL, Ballard-Barbash R, Bernstein L, et al. Elevated biomarkers of inflammation are associated with reduced survival among breast cancer patients. J Clin Oncol. 2009;27:3437–3444. doi: 10.1200/JCO.2008.18.9068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol. 2010;72:219–246. doi: 10.1146/annurev-physiol-021909-135846. [DOI] [PubMed] [Google Scholar]
- 9.Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–545. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 10.Mantovani A, Allavena P, Sica A, et al. Cancer-related inflammation. Nature. 2008;454:436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 11.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell. 2005;7:211–217. doi: 10.1016/j.ccr.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 13.Morris PG, Hudis CA, Giri D, et al. Inflammation and increased aromatase expression occur in the breast tissue of obese women with breast cancer. Cancer Prev Res (Phila) 2011;4:1021–1029. doi: 10.1158/1940-6207.CAPR-11-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Monteiro R, Azevedo I. Chronic inflammation in obesity and the metabolic syndrome. Mediators Inflamm. 2010;2010 doi: 10.1155/2010/289645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cinti S, Mitchell G, Barbatelli G, et al. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J Lipid Res. 2005;46:2347–2355. doi: 10.1194/jlr.M500294-JLR200. [DOI] [PubMed] [Google Scholar]
- 16.Subbaramaiah K, Howe LR, Bhardwaj P, et al. Obesity is associated with inflammation and elevated aromatase expression in the mouse mammary gland. Cancer Prev Res (Phila) 2011;4:329–346. doi: 10.1158/1940-6207.CAPR-10-0381. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 17.Bachelot T, Ray-Coquard I, Menetrier-Caux C, et al. Prognostic value of serum levels of interleukin 6 and of serum and plasma levels of vascular endothelial growth factor in hormone-refractory metastatic breast cancer patients. Br J Cancer. 2003;88:1721–1726. doi: 10.1038/sj.bjc.6600956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dandona P, Weinstock R, Thusu K, et al. Tumor necrosis factor-alpha in sera of obese patients: fall with weight loss. J Clin Endocrinol Metab. 1998;83:2907–2910. doi: 10.1210/jcem.83.8.5026. [DOI] [PubMed] [Google Scholar]
- 19.Subbaramaiah K, Morris PG, Zhou XK, et al. Increased levels of COX-2 and prostaglandin E2 contribute to elevated aromatase expression in inflamed breast tissue of obese women. Cancer Discov. 2012;2:356–365. doi: 10.1158/2159-8290.CD-11-0241. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 20.Zhao Y, Agarwal VR, Mendelson CR, et al. Estrogen biosynthesis proximal to a breast tumor is stimulated by PGE2 via cyclic AMP, leading to activation of promoter II of the CYP19 (aromatase) gene. Endocrinology. 1996;137:5739–5742. doi: 10.1210/endo.137.12.8940410. [DOI] [PubMed] [Google Scholar]
- 21.Irahara N, Miyoshi Y, Taguchi T, et al. Quantitative analysis of aromatase mRNA expression derived from various promoters (I.4, I.3, PII and I.7) and its association with expression of TNF-alpha, IL-6 and COX-2 mRNAs in human breast cancer. Int J Cancer. 2006;118:1915–1921. doi: 10.1002/ijc.21562. [DOI] [PubMed] [Google Scholar]
- 22.Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–917. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi H, Kokoeva MV, Inouye K, et al. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. 2006;116:3015–3025. doi: 10.1172/JCI28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nguyen MT, Favelyukis S, Nguyen AK, et al. A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. J Biol Chem. 2007;282:35279–35292. doi: 10.1074/jbc.M706762200. [DOI] [PubMed] [Google Scholar]
- 25.Jarde T, Perrier S, Vasson MP, et al. Molecular mechanisms of leptin and adiponectin in breast cancer. Eur J Cancer. 2011;47:33–43. doi: 10.1016/j.ejca.2010.09.005. [DOI] [PubMed] [Google Scholar]
- 26.Okumura M, Yamamoto M, Sakuma H, et al. Leptin and high glucose stimulate cell proliferation in MCF-7 human breast cancer cells: reciprocal involvement of PKC-alpha and PPAR expression. Biochim Biophys Acta. 2002;1592:107–116. doi: 10.1016/s0167-4889(02)00276-8. [DOI] [PubMed] [Google Scholar]
- 27.Goodwin PJ, Ennis M, Pritchard KI, et al. Fasting insulin and outcome in early-stage breast cancer: results of a prospective cohort study. J Clin Oncol. 2002;20:42–51. doi: 10.1200/JCO.2002.20.1.42. [DOI] [PubMed] [Google Scholar]
- 28.Pollak MN, Schernhammer ES, Hankinson SE. Insulin-like growth factors and neoplasia. Nat Rev Cancer. 2004;4:505–518. doi: 10.1038/nrc1387. [DOI] [PubMed] [Google Scholar]
- 29.Su B, Wong C, Hong Y, et al. Growth factor signaling enhances aromatase activity of breast cancer cells via post-transcriptional mechanisms. J Steroid Biochem Mol Biol. 2011;123:101–108. doi: 10.1016/j.jsbmb.2010.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sinicrope FA, Dannenberg AJ. Obesity and breast cancer prognosis: weight of the evidence. J Clin Oncol. 2011;29:4–7. doi: 10.1200/JCO.2010.32.1752. [DOI] [PubMed] [Google Scholar]