

Abstract
Fatty acid-derived biofuels and biochemicals can be produced in microbes using β-oxidation pathway engineering. In this study, the β-oxidation pathway of Saccharomyces cerevisiae was engineered to accumulate a higher ratio of medium chain fatty acids (MCFAs) when cells were grown on fatty acid-rich feedstock. For this purpose, the haploid deletion strain Δpox1 was obtained, in which the sole acyl-CoA oxidase encoded by POX1 was deleted. Next, the POX2 gene from Yarrowia lipolytica, which encodes an acyl-CoA oxidase with a preference for long chain acyl-CoAs, was expressed in the Δpox1 strain. The resulting Δpox1 [pox2+] strain exhibited a growth defect because the β-oxidation pathway was blocked in peroxisomes. To unblock the β-oxidation pathway, the gene CROT, which encodes carnitine O-octanoyltransferase, was expressed in the Δpox1 [pox2+] strain to transport the accumulated medium chain acyl-coAs out of the peroxisomes. The obtained Δpox1 [pox2+, crot+] strain grew at a normal rate. The effect of these genetic modifications on fatty acid accumulation and profile was investigated when the strains were grown on oleic acids-containing medium. It was determined that the engineered strains Δpox1 [pox2+] and Δpox1 [pox2+, crot+] had increased fatty acid accumulation and an increased ratio of MCFAs. Compared to the wild-type (WT) strain, the total fatty acid production of the strains Δpox1 [pox2+] and Δpox1 [pox2+, crot+] were increased 29.5% and 15.6%, respectively. The intracellular level of MCFAs in Δpox1 [pox2+] and Δpox1 [pox2+, crot+] increased 2.26- and 1.87-fold compared to the WT strain, respectively. In addition, MCFAs in the culture medium increased 3.29-fold and 3.34-fold compared to the WT strain. These results suggested that fatty acids with an increased MCFAs ratio accumulate in the engineered strains with a modified β-oxidation pathway. Our approach exhibits great potential for transforming low value fatty acid-rich feedstock into high value fatty acid-derived products.
Introduction
The renewable synthesis of fatty acid-derived biofuels and chemicals has gained considerable attention in recent years. In several studies, fatty acids are overproduced in microbes and later converted to fuels or chemicals through chemical conversion [1] or bioconversion [2], [3]. Other studies directly utilize fatty acid-rich feedstock as a carbon source to produce biofuels and chemicals through the complete oxidation of fatty acids [4] or using the intermediates of β-oxidation [5], [6]. The availability of fatty acids from nonedible fatty acid-rich crops, oleaginous fungi/algae and industrial by-products encourage the use of fatty acid feedstock as a cheap carbon source. In addition, fatty acids have a product yield advantage over sugars when used as a carbon source due to the efficient β-oxidation metabolism process [4], which has 100% carbon recovery. In short, fatty acids can be exploited as an alternative feedstock to lignocellulosic sugars for microbial bioconversion platforms.
Saccharomyces cerevisiae has been commonly used in industrial fermentation. It has better tolerance to organic solvent than Escherichia coli and also owns a well-studied β-oxidation pathway. Aiming to utilize fatty acid-rich feedstock to produce biofuels or biochemicals, we investigated fatty acid metabolism in S. cerevisiae. We intended to generate more medium chain fatty acids (MCFAs), which are of higher value compared to other fatty acids [7]. Because fatty acid-rich feedstock usually contains sugars and fatty acids in carbon length at C16 and C18 [8], we used oleic acid as a glucose co-substrate to test the bioconversion ability of engineered strains during culture.
Fatty acid β-oxidation is the main pathway for fatty acid degradation in yeast. It enables yeast cells to grow on medium containing fatty acids as the sole carbon and energy source. This process is carried out in the peroxisomes of S. cerevisiae [9], [10]. The first and rate-limiting step is catalyzed by acyl-CoA oxidase using acyl-CoA as the initial substrate. Before the degradation process begins, long chain fatty acids (LCFAs) are activated into long chain acyl-CoAs by acyl-CoA synthetases in the cytosol and then are transported into peroxisomes by Pxa1p/Pxa2p. In contrast to LCFAs, MCFAs can freely enter peroxisomes and become medium chain fatty acyl-CoAs with the help of acyl-CoA synthetases. In S. cerevisiae, the POX1 gene encodes the only acyl-CoA oxidase, which can oxidize short to long chain acyl-CoAs. Disruption of POX1 prevents yeast survival on medium using oleic acid as the sole carbon source [11]. In contrast to S. cerevisiae, the yeast Yarrowia lipolytica (Y. lipolytica) has several acyl-CoA oxidases with different chain length specificities [12], [13]. It has been demonstrated that modification of POX genotype can affect β-oxidation and further influence lipid accumulation in Y. lipolytica [14], but there is no data about changes specifically to MCFAs.
To use fatty acid-rich feedstock to generate tailored fatty acids for biofuel production, we modified the β-oxidation pathway of S. cerevisiae, both to maintain LCFA oxidation and to prevent MCFA oxidation. The strain was engineered with the intention to accumulate more fatty acids with a higher ratio of MCFAs when cells were grown on fatty acid-rich feedstock. To achieve this aim, the S. cerevisiae deletion strain Δpox1 was obtained in which the POX1 gene, the sole acyl-CoA oxidase, was deleted. Then, acyl-CoA oxidase Aox2p (encoded by POX2) from Y. lipolytica [15], which has a preference for long-chain acyl-CoA substrates, was expressed. The replacement of POX1 with POX2 led to the blocking of β-oxidation pathway. To unblock the β-oxidation pathway in the Aox2p-expressing strain, peroxisomal carnitine octanoyltransferase (CROT) [16] from Mus musculus was also expressed. This transferase moves medium chain fatty acyl-CoAs out of peroxisomes by producing medium chain fatty acyl carnitine esters. The modified pathway is depicted in Figure 1. The effect of β-oxidation cycle modification on fatty acid profile and accumulation was investigated and discussed.
Figure 1. Genetic modification of the β-oxidation pathway in Saccharomyces cerevisiae.

The dashed line represents the original pathway; the solid line represents the modified pathway. The only acyl-CoA oxidase (encoded by the gene POX1) in the S. cerevisiae genome was deleted, and the POX2 gene from Yarrowia lipolytica, which encodes acyl-CoA oxidase with a preference for long chain acyl-CoAs, was expressed. To unblock the β-oxidation pathway, peroxisomal carnitine octanoyltransferase (CROT) from Mus musculus was also expressed to transport medium chain fatty acyl-CoAs out of peroxisomes.
Materials and Methods
Yeast strains and growth conditions
The yeast strains used in this study were derived from S. cerevisiae BY4741 (Mat a; his3Δ1; leu2Δ0; met15Δ0; ura3Δ0, Acc. noY00000). The WT strain BY4741, the Δpox1 deletion strain (BY4741; Mat a; his3Δ1; leu2Δ0; met15Δ0; ura3Δ0; YGL205w::kanMX4, acc. no Y04571) and the plasmid pVTU260 were obtained from EUROSCARF (Germany). The strains were cultivated at 30°C in YPD complete medium [1% Bacto yeast extract, 2% Bacto peptone, 2% dextrose], supplemented with 200 µg/ml G418 (Sigma, Singapore) when necessary. SC Minimal Medium (YNBD) was comprised of 0.67% yeast nitrogen base (without amino acids but with ammonium sulfate, Invitrogen), 2% dextrose and amino acid drop out (without URA, Clontech). YNBD0.5O3 (YNB supplemented with 0.5% dextrose and 3% oleic acid (from Fluka 75096)) was used for the optimum accumulation of fatty acids. Minimal oleic acid medium (YNBO) was used to test the fatty acid degradation ability of the engineered strains. YNBO was YNB supplemented with 1% oleic acid as the sole carbon source.
Construction of plasmids pVTU260-POX2 and pVTU260-POX2-CROT
Molecular cloning was performed according to standard procedures. The QIAprep Spin Miniprep Kit was used for plasmid preparation. The QIAquick Gel Extraction Kit and QIAquick PCR Purification Kit (QIAGEN) were used for purification of gene fragments.
The gene POX2 (GenBank: accession no. CR382132.1) encoding acyl-CoA oxidase 2 (Aox2p) was synthesized by Geneart (American) with optimized codons. The GenBank accession number for the codon-optimized sequence of POX2 is KC912711. The primers used for PCR were POX2F-pvtu260 (5′ CGGCTAGCCGCAAAATGAATCCAAACAATAC) and POX2R-pvtu260 (5′ CGGGATCCGGTCAATGGTGATGGTGATGATGTTCTTCATCCAATTC). The forward primer introduced a unique NheI restriction site and a yeast consensus sequence (CAAA) before the 5′ initiation codon. The reverse primer introduced a His-tag sequence before the 3′ termination codon and a unique BamHI restriction site after the termination codon. The PCR products were digested with NheI and BamHI and then ligated into the pVTU260 vector, which had also been digested with NheI and BamHI. The ligation product was transformed into E. coli Top10. The resulting plasmid pVTU260-POX2 was verified by restriction analysis and sequencing.
The gene CROT (NM_023733.3) was also codon optimized and synthesized by Geneart. The GenBank accession number for the codon-optimized sequence of CROT is KC912712. Codon-optimized CROT was amplified using the primers TEFp-CROTF (5′ GTAGGAGGGCTTTTGTAGATGCTAGCCAAAGGTAAGCCTATCCCTAACCCTCTCCTCGGTCTCGATTCTACGATGGA) and CROTR-TEFt (5′ ACAACACTCCCTTCGTGCTTCCCCCCGGGGGGTCACAAGTGGGCGGTATTCATC). An NheI restriction site, yeast consensus sequence and V5-tag was introduced before the 5′ initiation codon. Because Crot contains a peroxisome-targeting signal peptide (AHL) at the C-terminal end, the V5-tag was placed at the N-terminal to prevent interference with the peroxisome-targeting signal peptide. The Ashbya gossypii TEF2 promoter and terminator were PCR-amplified individually from pUG27 [17] using the primer pair TEFp-pUG27-F 5′ GAAGATCGTCGTTTTGCCAGGTGACCGACAACCCTTAATATA and TEFp-R 5′ GGCTTACCTTTGGCTAGCATCTACAAAAGCCCTCCTAC and the primer pair TEFt-F 5′ CCGCCCACTTGTGACCCCCCGGGGGGAAGCACGAAGGGAGTGTTGT and TEFt-R 5′ TTAATTATATCAGTTATTACCCGGGTGAGCGAGGAAGCGGAAGA. The CROT gene expression cassette containing the three PCR fragments was assembled into pVTU260-POX2 digested with BstEII and XmaI using the Cold Fusion Cloning Kit (SBI). The resulting plasmid pVTU260-POX2-CROT was verified through restriction analysis and sequencing.
Cell transformation and control cell development
The plasmids pVTU260-POX2 and pVTU260-POX2-CROT were transformed separately into Δpox1 yeast competent cells using the lithium acetate method [18]. Control cells were created by transforming the empty vector pVTU260 into the WT and Δpox1 strain. To examine if expression of Aox2p caused growth defects, pVTU260-POX2 was also transformed into the WT strain as a control. Colonies on the screening plates were verified by colony PCR.
Western blot analysis
After 24 h, 5 ml of cell culture was collected by centrifugation at 8000 rpm. After washing once with lysis buffer (50 mM HEPES, 5% glycerol, 1 mM DTT, 1 mM PMSF, 1 mM EDTA), the pellet was resuspended in lysis buffer to an OD600 of 100. The peroxisome proteins were isolated using a peroxisome isolation kit (Sigma). Total proteins were mixed with loading buffer and boiled at 95°C for 5 min and separated on a 10% SDS%-PAGE (Bio-Rad). The separated proteins were transferred to a nitrocellulose (NC) membrane (GE Hybond, USA) at 21 V for 1 h 20 min using a semi-dry apparatus. After incubating the NC membrane in blocking buffer (PBST+5% nonfat, dry milk, w/v) for 1 hour at room temperature, the membrane was probed with an anti-V5 antibody or an anti-His antibody (Sigma) at 4°C overnight. Then, the membrane was incubated with an anti-mouse IgG HRP-conjugated secondary antibody (#31430, Pierce) at the dilution suggested by the manufacturer. After washing with PBST buffer, the signal was detected with the ECL Western blot reagent.
Growth tests of control and engineered strains
Yeast cells were collected after overnight culture in liquid YNBD medium at 30°C. The cells were resuspended at a cell density of 5×103 cells/µl after washing twice in sterile distilled H2O. Cells (5 µl) were spotted onto YNBD or YNBO-agar plates. The first drop contained 2.5×104 cells, and each subsequent drop was diluted six fold more than the previous one.
Sample preparation for metabolite and fatty acid analysis
Five milliliters of liquid YNBD medium was inoculated with a single colony from agar plate and cultured overnight at 30°C with shaking. Then, the overnight culture was used to inoculate 50 ml liquid YNBD or YNBD0.5O3 medium. The initial OD600 was adjusted to approximately 0.3. The cells were cultured by shaking at 250 rpm and 30°C. Same amounts of yeast cells (8×107 cells) were separately collected at different growth time points. The intracellular metabolites were extracted from control and engineered cells based on previously published work [19], [20]. In general, the culture sample was centrifuged for 5 min at 4°C. The supernatant was collected for extracellular metabolite analysis. The cell pellet was washed twice with cold methanol (<−40°C) and then collected.
Fatty acid analysis of cells and culture supernatants
The lipids were extracted from yeast cells using an adjusted chloroform-methanol 2∶1 method [21]. The cell pellet was resuspended in 1000 µl of 0.9% NaCl solution and then acidified with 200 µl of acetic acid. As an internal standard (IS) to correct for metabolite loss during sample preparation, 10 µl of 10 mg/ml heptadecanoic acid and heptanoic acid dissolved in ethanol was added to the extraction solvent. After adding an equal volume of glass beads, the cells were disrupted in a FastPrep®-24 Instrument (6004-500, MP Biomedicals) for 30 s and cooled in ice for 30 s; this procedure was repeated 4 times. Then, 3 ml of a chloroform-methanol 2∶1 mixture was added, and the samples were inverted several times, vortexed vigorously, and centrifuged at 10000 g for 10 min at 4°C. The aqueous layer and cell debris were transferred to a new tube using aspiration. The chloroform layer (lower) was collected. An additional 3 ml of the chloroform-methanol 2∶1 mixture was added to the aqueous layer to further extract lipids. Then, the chloroform layer was rotary evaporated to near dryness overnight using a TurboVapH LV Concentration Workstation.
For the fatty acids in the culture medium, the Solid-Phase Extraction (SPE) method [22] was adopted. First, the SPE column (Strata C18-E 200 mg/3 ml) was activated with 2.5 ml methanol and washed with 5 ml water. Then, 10 ml culture supernatant to which 0.5 ml 1 M HCl, 2 µl 10 mg/ml heptadecanoic acid and 2 µl 10 mg/ml heptanoic acid were added was passed through the SPE column. The SPE column was washed with 5 ml water and dried at room temperature. Next, 1.5 ml chloroform was used for fatty acid elution. The chloroform in elution products was also evaporated to near dryness.
Fatty acid analysis was performed according to previous work [22]. Fatty acid methyl esters (FAMEs) mix C8–C24 (SUPELCO, 18918) was used as a standard. Three independent experiments were conducted. The dried lipid residue was redissolved in 500 µl BF3-methanol 10% (FLUKA, 15716) and incubated in a sealed vial in a 95°C heater for 20 min. FAMEs were extracted with 300 µl n-hexane after the addition of 300 µl saturated NaCl in water. All of the samples were analyzed using an Agilent Technologies 7890A-5975C GC-MS system equipped with a HP-5MS capillary column (30 m×0.250 mm i.d.; film thickness: 0.25 µm; Agilent J&W Scientific, Folsom, CA, USA). Helium was used as a carrier gas at 1.1 mL/min. The inlets and MS source temperatures were maintained at 250 and 230°C, respectively. For fatty acid analysis, the oven temperature was maintained at 80°C for 1 min and ramped to 250°C at a rate of 7°C.min−1, then held at 250°C for 10 min. Data were acquired in a full scan from 35 to 600 m/z. The detected FAME peaks were integrated. Amounts were calculated with reference to the IS heptadecanoic acid, and the relative response factors were calculated.
Metabolic profiling
The sample for metabolic profiling was derivatized according to previous work [23]. For metabolic profiling, the same GC-MS system was used. The oven temperature was maintained at 75°C for 4 min and raised at 4°C.min−1 to a final temperature of 280°C and held for 2 min. Mass spectra were recorded from 35 to 600 m/z with a scan time of 0.2 s. Chromatogram acquisition and mass spectra identification were processed using the Agilent MSD Chemstation Data Analysis software. Chemical identification of the detected metabolite peaks was performed by searching the NIST08 mass spectral library. The compounds were quantified from the peak area relative to the IS ribitol. No response factors were made.
Results
Gene modification of the β-oxidation pathway in S. cerevisiae
The modification of the β-oxidation pathway is based on the deletion of one gene and the expression of two proteins. The deletion strain Δpox1 was obtained from EUROSCARF and tested for growth on YNBO. The Δpox1 strain was unable to grow on medium in which oleic acid was the only carbon source. This is consistent with a previous report [11]. To express acyl-CoA oxidase 2 (Aox2p), which has a preference for long chain substrates, the multi-copy plasmid pVTU260 was used. The plasmid pVTU260-POX2 encoding the POX2 gene under the control of the constitutive promoter ADH1 was transformed into Δpox1, resulting in the strain Δpox1 [pox2+]. To express carnitine O-octanoyltransferase, the gene CROT was cloned into pVTU260-POX2 under the control of the constitutive promoter TEF. The resulting plasmid pVTU260-POX2-CROT was transformed into strain Δpox1, creating strain Δpox1 [pox2+, crot+]. These two enzymes were successfully expressed in the Δpox1 strain. Western results are shown in Figure 2. Aox2p is approximately 78 kDa; Crot is approximately 72 kDa.
Figure 2. Western for Aoxp2 and Crot.
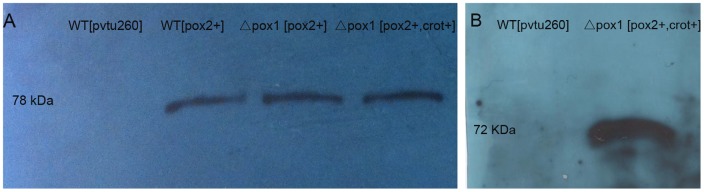
A. Expression of Aoxp2 in WT [pox2+], Δpox1 [pox2+] and Δpox1 [pox2+, crot+]. B. Expression of Crot in Δpox1 [pox2+, crot+].
Growth phenotype analysis
Growth tests were performed to evaluate the potential negative effects of the genetic modification on the growth of yeast cells. The Δpox1 strain did not grow on the oleic acid-based (YNBO) agar plate. The Δpox1 [pox2+] and WT [pox2+] strains showed a severe growth defect (Figure 3), whereas Δpox1 [pox2+, crot+] grew similar to the WT. When all the strains were cultured in glucose-based (YNBD) medium (as shown in Figure 4A), there was no severe growth defect observed in the engineered strains. It was observed that Δpox1 [pox2+] grew more slowly than the WT strain. Δpox1 [pox2+, crot+] showed a growth rate similar to the WT strain. However, in oleic acid-glucose-based (YNBD0.5O3) medium (Figure 4B), Δpox1 showed reduced growth compared to the WT strain. The growth of Δpox1 [pox2+], expressing Aox2p, was more severely affected than the Δpox1 strain. The Δpox1 [pox2+, crot+] strain did not show a growth defect. The Δpox1 growth defect in oleic acid-containing medium was due to defective fatty acid degradation. Oleic acid could not be utilized by Δpox1 as a carbon source. The growth defect in strain Δpox1 [pox2+] indicated that β-oxidation was not fully rescued by Aox2p. Because Aox2p has very low activity towards acyl-CoAs with fewer than ten carbons [14], the over-accumulation of medium chain acyl-CoAs from LCFA degradation in the peroxisome prevented further fatty acid oxidation, which restricted the carbon flux toward β-oxidation. The defective growth of the WT [pox2+] strain further supported this conclusion. In strain Δpox1 [pox2+, crot+], the medium chain acyl-CoAs accumulated were transferred to carnitine by Crot [24] and subsequently removed from peroxisomes, allowing unobstructed carbon flux.
Figure 3. Growth tests on the engineered strains and the WT strain in YNBO medium.

WT and Δpox1 transformed with the empty vector pvtu260, WT transformed with pvtu260-pox2, Δpox1 transformed with pvtu260-pox2 and pvtu260-pox2-crot were all cultured overnight in liquid YNBD medium at 30°C. Yeast pellets were collected and resuspended at a cell density of 5×103 cells/µl after being washed twice in sterile distilled H2O. To test growth, 5 µl were spotted on YNBO-agar plates. The first drop contained 2.5×104 cells, and each subsequent drop was diluted six fold more than the previous one.
Figure 4. Growth curves of the engineered strains and the WT strain.

A. Growth curves of the engineered strains and the WT strain in YNBD, B. Growth curves of the engineered strains and the WT strain in YNBD0.5O3. The results are the mean values of three independent experiments.
Analysis of fatty acid composition in cell extracts and culture supernatants
To examine the effect of Aox2p and Crot on fatty acid accumulation and composition, the intracellular and extracellular fatty acids of cultured cells at the stationary phase were analyzed. When the engineered strains were cultured in YNBD medium, the total fatty acids in the engineered strains increased less than 10% compared with the WT strain. There was not an obvious change in the fatty acid profile and C8:0 and C10:0 were not detected in the culture supernatant of any of the engineered strains (data not shown).
When cells were cultured in YNBD0.5O3, the fatty acid accumulation and composition of the engineered strains were different from WT strain. For intracellular fatty acids from the cell extract at stationary phase (Table 1 and Table 2), the total fatty acids in strains Δpox1 [pox2+] and Δpox1 [pox2+, crot+] increased 29.5% and 15.6% compared to the WT strain, respectively. There is no increase of total fatty acids in the Δpox1 strain. The unsaturated fatty acids ratio (unsaturated fatty acids compared to saturated fatty acids) increased to 0.083 (Δpox1), 0.073 (Δpox1 [pox2+]) and 0.078 (Δpox1 [pox2+, crot+]) compared to 0.041 (WT). In terms of MCFAs, there was an increase in MCFA content (present only as lauric acid in the cell extract) in the strains Δpox1, Δpox1 [pox2+] and Δpox1 [pox2+, crot+] compared with the WT strain. Lauric acid (C12:0) content in Δpox1 (0.486 µg/OD), Δpox1 (pox2+) (0.77 µg/OD) and Δpox1 [pox2+, crot+] (0.64 µg/OD) were increased 1.42-, 2.26- and 1.87-fold compared to the WT strain (0.34 µg/OD) (Figure 5A). In the strains Δpox1, Δpox1 [pox2+] and Δpox1 [pox2+, crot+], lauric acid constituted 0.85%, 1.0% and 0.92% of the total fatty acids (Figure 5B) compared to 0.57% in the WT, which corresponds to a 1.47-, 1.75- and 1.61-fold increase. The data demonstrated that modification of the β-oxidation pathway changed the total fatty acids, unsaturated fatty acids/saturated fatty acids ratio and the MCFAs ratio in yeast cells.
Table 1. Fatty acid production in the cell extract of the WT and the engineered strainsa.
| Fatty acid type | WT | Δpox1 | Δpox1 [pox2+] | Δpox1[pox2+ crot+] |
| C12:0 | 0.341 | 0.486 | 0.772 | 0.637 |
| C14:0 | 1.183 | 1.590 | 2.143 | 1.855 |
| C16:1 | 0.881 | 1.384 | 1.878 | 1.788 |
| C16:0 | 30.623 | 27.675 | 35.794 | 32.535 |
| C18:1 | 1.449 | 2.977 | 3.334 | 3.170 |
| C18:0 | 24.889 | 22.959 | 32.890 | 28.548 |
| C20:0 | 0.120 | 0.143 | 0.202 | 0.227 |
| Total fatty acids | 59.486 | 57.214 | 77.014 | 68.760 |
a Data represent fatty acid composition in µg/OD cell when WT (wild-type) and engineered strains were cultured in YNBD0.5O3 medium. The values are the means from three experiments examining the cell extracts at 24 h. The standard deviations were <5% of the values.
Table 2. Fatty acid composition changes in cell extract comparing the WT and the engineered strains in YNBD0.5O3 mediumb.
| Fold changes of fatty acids | Δpox1 | Δpox1 [pox2+] | Δpox1[pox2+ crot+] |
| C12:0 | 1.422 | 2.262 | 1.866 |
| C14:0 | 1.344 | 1.812 | 1.568 |
| C16:1 | 1.570 | 2.132 | 2.029 |
| C16:0 | 0.904 | 1.169 | 1.062 |
| C18:1 | 2.054 | 2.300 | 2.187 |
| C18:0 | 0.923 | 1.322 | 1.147 |
| C20:0 | 1.194 | 1.681 | 1.889 |
| Total fatty acids | 0.962 | 1.29 | 1.156 |
b The fold change data for the engineered strains were analyzed compared to the WT (wild-type) strain. The amount of fatty acids in the WT strain was set as 1. The values are the means from three experiments examining cell extracts at 24 h. The standard deviations were <5% of the values.
Figure 5. MCFAs analysis of the cell extracts.

A. MCFAs content in the cell extracts from the engineered strains and the WT strain after 240.5O3 medium. B. Composition of MCFAs from the total fatty acids in the cell extracts from the engineered strains and the WT strain at 24 h when cultured in YNBD0.5O3 medium. Cells were collected after 24 h of growth in YNBD0.5O3 medium, and the cell pellet was separated by centrifugation. The total fatty acids were extracted and detected. There was C12:0 but not C8:0 and C10:0 in the cell extract, so the MCFAs were only C12:0 in this case.
Another observed change was an increase in the MCFAs content of the culture supernatants from engineered strains compared to the WT strain. MCFAs (chain length 8 to 12 carbons) are usually a minor constituent of total fatty acids present at a low density in the cell, and C8:0 and C10:0 are not generally detectable in culture supernatant. Because MCFAs can freely traverse the membrane of a cell, when MCFAs were produced, they were secreted into the culture medium. As shown in Figure 6, there was no detectable C8:0 or C10:0 in the WT strain, only C12:0. In contrast, C8:0, C10:0 and C12:0 were all detected in the engineered strains. MCFAs content in the three engineered strains increased 2.84-fold (Δpox1, 0.419 µg/ml), 3.29-fold (Δpox1 [pox2+], 0.484 µg/ml) and 3.34-fold (Δpox1 [pox2+, crot+], 0.493 µg/ml) compared to the WT strain (0.147 µg/ml).
Figure 6. MCFAs content in the culture supernatant of the engineered strains and the WT strain grown for 240.5O3 medium.

Cells were collected after growth for 240.5O3 medium, and the culture supernatant was separated by centrifugation. MCFAs were detected with the SPE method (detailed information referred to in the fatty acids analysis section). Data are the mean values of three independent experiments. The error bar indicates the S.D. C8:0: open bar, C10:0: striped bars, C12:0: gridded bar, MCFAs: solid bar.
Metabolic profiling of control and engineered strains
S. cerevisiae was modified to maintain the ability to degrade LCFAs but limited ability to degrade MCFAs. To analyze the difference between the engineered strains in the oxidation of MCFAs, GC-MS-based metabolic profiling (Figure 7) was performed. The metabolic responses of the engineered strains were useful in understanding the changes to the fatty acid degradation pathway after gene modification. It was found that there were obvious differences in the cellular metabolite levels between engineered strains and the WT strain, as shown in Figure 8. Fewer dicarboxylic acids were present in the engineered strains compared with the WT strain when cultured in YNBD0.5O3 medium. This observation is consistent with a previous study [25] reporting that dicarboxylic acids (hexanedioic acid, heptanedioic acid, octanedioic acid, azelaic acid and sebacic acid) are formed when fatty acid oxidation is increased or inhibited. Because the formation of dicarboxylic acids could facilitate the degradation process, there are higher levels of dicarboxylic acids when fatty acid oxidation is increased than when it is inhibited. In the WT strain, dicarboxylic acids were at the highest level, which was due to increased β-oxidation compared to the engineered strains. There were much lower levels of dicarboxylic acids in the Δpox1, Δpox1 [pox2+] and Δpox1 [pox2+, crot+] strains, which had negligible β-oxidation of MCFAs. This difference was consistent with the fatty acid metabolism of several strains. With regard to L-proline, glutamine, and L-ornithine, these changes might be related to alterations in the metabolism of the tricarboxylic acid cycle (TCA) cycle. These amino acids are synthesized from intermediates of the TCA. When a strain has a growth defect, the carbon flow toward the TCA cycle becomes restricted, further decreasing the concentration of amino acids related to the TCA cycle. In the strain Δpox1 [pox2+, crot+], there was no observed growth defect, and the amino acid levels were upregulated relative to normal levels. As for the increase of ornithine in Δpox1 [pox2+], this may be due to the very low growth rate. Thus, there was a relative excess of nitrogen driving ornithine synthesis.
Figure 7. The representative GC-MS spectra for azelaic acid derived from total ion chromatograms.

Intracellular metabolites were extracted from the engineered strains and the WT strain, and metabolic profiling was conducted. WT (black), Δpox1 (blue), Δpox1 [pox2+] (red), and Δpox1[pox2+, crot+] (green).
Figure 8. Differential expression levels of intracellular metabolites in the engineered strains compared to the WT strain.

Discussion
Impact of Aox2p and Crot on fatty acids composition in the engineered strains
Our study showed that when oleic acid was used as a co-substrate of glucose, the total fatty acids were increased in engineered strains compared to the WT strain. As shown in Table 1, the total fatty acids from strains Δpox1 [pox2+] and Δpox1 [pox2+, crot+] were increased compared to the WT strain. In contrast, the total fatty acids of strains Δpox1 were not increased. This indicated that the β-oxidation defect had a side effect on fatty acid accumulation other than fatty acid uptake. Modification of the β-oxidation pathway not only affected the total fatty acids content but also changed the composition. There was MCFAs (C8:0, C10:0 and C12:0) accumulation, and C8:0 and C10:0 were secreted into the medium. For the initial gene modification step, we obtained the deletion strain Δpox1. It was observed that there was an increase in C8:0 and C10:0 in Δpox1. From a previous report [26], it had been shown that medium chain acyl-CoAs can be released from the fatty acid synthetase (FAS) complex due to the accumulation of saturated LCFAs during the fermentation conditions. Medium chain acyl-CoAs are hydrolyzed to recycle CoA-SH. MCFAs can diffuse passively through the cytoplasmic membrane. Our findings therefore suggested that medium chain acyl-CoAs might be released from the FAS complex in the Δpox1 strain when cultured in oleic acid-containing medium, even under respiratory conditions. In contrast, C8:0 and C10:0 did not accumulate in the WT strain. In Δpox1 [pox2+], the increase of MCFAs was partly due to the effect of POX1 deletion and partly due to the expression of POX2 from Y. lipolytica. In the peroxisomes of Δpox1 [pox2+], only long-chain acyl-CoAs were degraded to medium-chain acyl-CoAs and acetyl-CoA. The degradation product acetyl-CoA was activated to acetyl carnitine and transported out of the peroxisome by carnitine acetyltransferase (Cat2p) [9]. However, there is no transferase in S. cerevisiae that is involved in transporting medium chain acyl-CoAs out of the peroxisome and recycling CoA-SH. Although there is a peroxisomal acyl-CoA thioesterase (PTE1) [27] in S. cerevisiae to hydrolyze acyl-CoAs of all chain lengths to generate free CoA-SH, this enzyme does not appear to possess sufficient activity to hydrolyze all of the medium chain acyl-CoAs accumulated in peroxisomes in the strain Δpox1 [pox2+]. Owing to the expression of Aox2p and thioesterase in the peroxisome, there was an increase of MCFAs in Δpox1 [pox2+]. However, the accumulated medium chain acyl-CoAs led to depletion of free CoA-SH and eventually the termination of the β-oxidation pathway, causing the growth defect. When Crot was expressed in the strain Δpox1 [pox2+, crot+], the carbon flux through β-oxidation became unobstructed because the accumulated medium chain acyl-CoAs could be transported out of the peroxisome, balancing the CoA concentration in peroxisomes. Therefore, the growth rate was recovered to the levels of the WT strain. However, it should be noted that there was not a continuous increase of MCFAs in the strain Δpox1 [pox2+, crot+]. It was assumed that there was an increase of cytosolic medium chain acyl-CoAs in the engineered strains. In future studies, we will detect medium chain acyl-CoAs, as well [28].
Metabolic engineering of S. cerevisiae for MCFAs production
It has been shown that MCFAs accumulation is related to fermentative metabolism in S. cerevisiae [26], [29]. During fermentation, fatty acid synthesis is inhibited by the lack of oxygen. As a result, medium chain acyl-CoAs are released from the FAS and hydrolyzed to recycle CoA-SH. Cell growth ceases due to the inhibition of fatty acid synthesis. During our investigation of the role of Aox2p and Crot in fatty acid profile and MCFAs accumulation, we expressed Aox2p and Crot in the Δpox1 strain. As expected, the total fatty acids and MCFAs were increased in the engineered strains compared to the WT strain. We showed that in aerobic conditions, MCFAs could be produced in S. cerevisiae through modification of the β-oxidation pathway. The metabolic responses in different strains helped us to further investigate the impact of Aox2p and Crot. In the WT strain, there was a high concentration of medium chain dicarboxylic acids detected, which facilitated β-oxidation in the cells [25]. In contrast, the MCFAs oxidation ability decreased dramatically in the engineered strains. A much lower concentration of medium chain dicarboxylic acids were detected. On the contrary, MCFAs were secreted into the culture medium.
In summary, we observed that the production of both intracellular and extracellular MCFAs increased in the engineered stains compared to the WT strain when grown in YNBD0.5O3. It was demonstrated that modification of β-oxidation was capable of turning low value fatty acid feedstock into higher value fatty acids containing more MCFAs. However, MCFAs remained a minor component of the total fatty acids. The observed increase in total fatty acids indicated that most of the oleic acid in the medium went into the storage pathway instead of degradation pathway. Thus, further investigation is needed to improve the oxidation of LCFAs and the formation of MCFAs. Future studies will aim at the expression of other β-oxidation-related genes and medium chain acyl-CoA hydrolases, such as Acot5 (medium chain acyl-CoA thioesterase) [30]. Meanwhile, endogenously produced lipase can be used [31] to increase the uptake of fatty acid-rich feedstock, such as triacylglycerols (TAG), in the engineered strains. The current work terminated the β-oxidation of yeast in peroxisomes at medium chain acyl-CoAs. It was demonstrated that heterologous expression of β-oxidation enzymes in S. cerevisiae system is a promising tool to exploit bioconversion platforms.
Acknowledgments
We thank Prof. J. H. Hegemann for kindly providing the plasmid pUG27.
Funding Statement
National Research Foundation, Singapore. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Lennen RM, Braden DJ, West RM, Dumesic JA, Pfleger BF (2010) A Process for Microbial Hydrocarbon Synthesis: Overproduction of Fatty Acids in Escherichia coli and Catalytic Conversion to Alkanes. Biotechnol Bioeng 106: 193–202 10.1002/bit.22660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Steen EJ, Kang YS, Bokinsky G, Hu ZH, Schirmer A, et al. (2010) Microbial production of fatty-acid-derived fuels and chemicals from plant biomass. Nature 463: 559–U182 10.1038/nature08721 [DOI] [PubMed] [Google Scholar]
- 3. Schirmer A, Rude MA, Li XZ, Popova E, del Cardayre SB (2010) Microbial Biosynthesis of Alkanes. Science 329: 559–562 10.1126/science.1187936 [DOI] [PubMed] [Google Scholar]
- 4. Dellomonaco C, Rivera C, Campbell P, Gonzalez R (2010) Engineered Respiro-Fermentative Metabolism for the Production of Biofuels and Biochemicals from Fatty Acid-Rich Feedstocks. Appl Environ Microbiol 76: 5067–5078 10.1128/aem.00046-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Haddouche R, Delessert S, Sabirova J, Neuveglise C, Poirier Y, et al. (2010) Roles of multiple acyl-CoA oxidases in the routing of carbon flow towards beta-oxidation and polyhydroxyalkanoate biosynthesis in Yarrowia lipolytica . FEMS Yeast Res 10: 917–927 10.1111/j.1567-1364.2010.00670.x [DOI] [PubMed] [Google Scholar]
- 6. Poirier Y, Erard N, Petétot JM-C (2001) Synthesis of Polyhydroxyalkanoate in the Peroxisome of Saccharomyces cerevisiae by Using Intermediates of Fatty Acid β-Oxidation. Appl Environ Microbiol 67: 5254–5260 10.1128/aem.67.11.5254-5260.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lee SK, Chou H, Ham TS, Lee TS, Keasling JD (2008) Metabolic engineering of microorganisms for biofuels production: from bugs to synthetic biology to fuels. Curr Opin Biotechnol 19: 556–563. [DOI] [PubMed] [Google Scholar]
- 8. Meng X, Yang J, Xu X, Zhang L, Nie Q, et al. (2009) Biodiesel production from oleaginous microorganisms. Renew Energ 34: 1–5 doi:http://dx.doi.org/10.1016/j.renene.2008.04.014 [Google Scholar]
- 9. Hiltunen JK, Mursula AM, Rottensteiner H, Wierenga RK, Kastaniotis AJ, et al. (2003) The biochemistry of peroxisomal beta-oxidation in the yeast Saccharomyces cerevisiae . FEMS Microbiol Rev 27: 35–64 10.1016/s0168-6445(03)00017-2 [DOI] [PubMed] [Google Scholar]
- 10. van Roermund CWT, Waterham HR, Ijlst L, Wanders RJA (2003) Fatty acid metabolism in Saccharomyces cerevisiae . Cell Mol Life Sci 60: 1838–1851 10.1007/s00018-003-3076-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dmochowska A, Dignard D, Maleszka R, Thomas DY (1990) Structure and transcriptional control of the Saccharomyces cerevisiae POX1 gene encoding acylcoenzyme A oxidase. Gene 88: 247–252 10.1016/0378-1119(90)90038-s [DOI] [PubMed] [Google Scholar]
- 12. Wang HJ, Le Dall MT, Waché Y, Laroche C, Belin JM, et al. (1999) Evaluation of acyl coenzyme A oxidase (Aox) isozyme function in the n- alkane-assimilating yeast Yarrowia lipolytica . J Bacteriol 181: 5140–5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mlickova K, Roux E, Athenstaedt K, d'Andrea S, Daum G, et al. (2004) Lipid accumulation, lipid body formation, and acyl coenzyme A oxidases of the yeast Yarrowia lipolytica . Appl Environ Microbiol 70: 3918–3924 10.1128/aem.70.7.3918-3924.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mlickova K, Luo Y, d'Andrea S, Pec P, Chardot T, et al. (2004) Acyl-CoA oxidase, a key step for lipid accumulation in the yeast Yarrowia lipolytica . J Mol Catal B: Enzym 28: 81–85 10.1016/j.molcatb.2004.01.007 [DOI] [Google Scholar]
- 15. Luo YS, Nicaud JM, Van Veldhoven PP, Chardot T (2002) The acyl-CoA oxidases from the yeast Yarrowia lipolytica: characterization of Aox2p. Arch Biochem Biophys 407: 32–38. [DOI] [PubMed] [Google Scholar]
- 16. Westin MAK, Hunt MC, Alexson SEH (2008) Short- and medium-chain carnitine acyltransferases and acyl-CoA thioesterases in mouse provide complementary systems for transport of beta-oxidation products out of peroxisomes. Cell Mol Life Sci 65: 982–990 10.1007/s00018-008-7576-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gueldener U, Heinisch J, Koehler GJ, Voss D, Hegemann JH (2002) A second set of loxP marker cassettes for Cre-mediated multiple gene knockouts in budding yeast. Nucleic Acids Res 30 doi:e23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gietz RD, Schiestl RH (2007) Frozen competent yeast cells that can be transformed with high efficiency using the LiAc/SS carrier DNA/PEG method. Nat Protoc 2: 1–4 10.1038/nprot.2007.17 [DOI] [PubMed] [Google Scholar]
- 19. Mal M, Koh PK, Cheah PY, Chan ECY (2009) Development and validation of a gas chromatography/mass spectrometry method for the metabolic profiling of human colon tissue. Rapid Commun Mass Spectrom 23: 487–494 10.1002/rcm.3898 [DOI] [PubMed] [Google Scholar]
- 20. Villas-Boas SG, Hojer-Pedersen J, Akesson M, Smedsgaard J, Nielsen J (2005) Global metabolite analysis of yeast: evaluation of sample preparation methods. Yeast 22: 1155–1169 10.1002/yea.1308 [DOI] [PubMed] [Google Scholar]
- 21. Browse J, McCourt PJ, Somerville CR (1986) Fatty acid composition of leaf lipids determined after combined digestion and fatty acid methyl ester formation from fresh tissue. Anal Biochem 152: 141–145 10.1016/0003-2697(86)90132-6 [DOI] [PubMed] [Google Scholar]
- 22. Horak T, Culik J, Cejka P, Jurkova M, Kellner V, et al. (2009) Analysis of Free Fatty Acids in Beer: Comparison of Solid-Phase Extraction, Solid-Phase Microextraction, and Stir Bar Sorptive Extraction. J Agric Food Chem 57: 11081–11085 10.1021/jf9028305 [DOI] [PubMed] [Google Scholar]
- 23. Wang MX, Bai J, Chen WN, Ching CB (2010) Metabolomic Profiling of Cellular Responses to Carvedilol Enantiomers in Vascular Smooth Muscle Cells. Plos One 5 10.1371/journal.pone.0015441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Farrell SO, Fiol CJ, Reddy JK, Bieber LL (1984) Properties of purified carnitine acyltransferases of mouse liver peroxisomes. J Biol Chem 259: 13089–13095. [PubMed] [Google Scholar]
- 25. Mortensen PB (1992) Formation and degradation of dicarboxylic acids in relation to alterations in fatty acid oxidation in rats. Biochim Biophys Acta 1124: 71–79 10.1016/0005-2760(92)90128-i [DOI] [PubMed] [Google Scholar]
- 26. Bardi L, Cocito C, Marzona M (1999) Saccharomyces cerevisiae cell fatty acid composition and release during fermentation without aeration and in absence of exogenous lipids. Int J Food Microbiol 47: 133–140. [DOI] [PubMed] [Google Scholar]
- 27. Jones JM, Nau K, Geraghty MT, Erdmann R, Gould SJ (1999) Identification of Peroxisomal Acyl-CoA Thioesterases in Yeast and Humans. J Biol Chem 274: 9216–9223 10.1074/jbc.274.14.9216 [DOI] [PubMed] [Google Scholar]
- 28. Kopka J, Ohlrogge JB, Jaworski JG (1995) Analysis of in Vivo Levels of Acyl-Thioesters with Gas Chromatography/Mass Spectrometry of the Butylamide Derivative. Anal Biochem 224: 51–60 10.1006/abio.1995.1007 [DOI] [PubMed] [Google Scholar]
- 29. Torija MJ, Beltran G, Novo M, Poblet M, Guillamon JM, et al. (2003) Effects of fermentation temperature and Saccharomyces species on the cell fatty acid composition and presence of volatile compounds in wine. Int J Food Microbiol 85: 127–136 10.1016/s0168-1605(02)00506-8 [DOI] [PubMed] [Google Scholar]
- 30. Hunt MC, Alexson SEH (2008) Novel functions of acyl-CoA thioesterases and acyltransferases as auxiliary enzymes in peroxisomal lipid metabolism. Prog Lipid Res 47: 405–421 doi:http://dx.doi.org/10.1016/j.plipres.2008.05.001 [DOI] [PubMed] [Google Scholar]
- 31. Shockey J, Chapital D, Gidda S, Mason C, Davis G, et al. (2011) Expression of a lipid-inducible, self-regulating form of Yarrowia lipolytica lipase LIP2 in Saccharomyces cerevisiae . Appl Microbiol Biotechnol 92: 1207–1217 10.1007/s00253-011-3505-y [DOI] [PubMed] [Google Scholar]