

Abstract
The complement system is tightly regulated to safeguard against tissue damage that results from unwanted activation. The key step of C3 cleavage to C3b is regulated by multiple mechanisms that control the initiation and extent of activation. This study demonstrated that C3b:plasma protein complexes form in the fluid-phase during complement activation. Several different plasma proteins displayed a discrete high molecular SDS-resistant band when any of the three complement activating pathways were triggered in normal human serum or plasma. Serum depleted of individual complement proteins revealed that C3, factors B and D were essential for complex formation. Inactivation of the thioester bond in C3 also prevented complex formation. In vitro, complexes could be generated using four purified proteins: C3, factor B, factor D, target protein and Mg2+ to allow C3 convertase formation. These studies showed that the complexes consisted of a plasma protein covalently bound to C3b in a 1:1 molar ratio; the C3b portion was rapidly degraded by factors H and I. Analysis of plasma samples from patients with dense deposit disease (DDD) and C3 glomerulonephritis (C3GN) demonstrated that C3b:protein complexes form spontaneously in the blood of patients with DDD and to a lesser extent in C3GN patients, but not in healthy controls. This finding supports the underlying hypothesis that these C3 glomerulopathies are diseases of fluid-phase complement dysregulation. These complexes could normally function as a passive mechanism to intercept C3b from depositing on host cells. However, excessive generation and/or defective clearance of fluid-phase C3b:protein complexes may have pathological consequences.
INTRODUCTION
The complement system is the primary humoral component of innate immunity and activation of the cascade by recognition pathways (classical, lectin and alternative) all converge at the crucial step of C3 cleavage (1). The alternative pathway is unique in that it is constitutively active due to the slow spontaneous hydrolysis of the labile thioester bond in the thioester domain (TED) of C3 (2). This “tick-over” mechanism forms an active “C3b-like” C3 (C3-H2O) that binds factor B, assembles a C3 convertase and generates additional C3b to amplify the pathway. Proteolytic cleavage of C3 to C3b exposes the unstable thioester bond, which then reacts very quickly with available amine or hydroxyl groups to covalently attach C3b on a target surface (3). This initial ‘tagging’ step is amplified on pathogens but regulated on host cells, and controlled in both the fluid-phase and on host cell surfaces by regulators of complement activation (RCA), a class of proteins that regulate C3 cleavage (4, 5). Because covalent attachment of C3b to targets is indiscriminate and somewhat inefficient, only about 10% of activated C3b attaches to intended targets (6, 7). The majority of C3b thioesters react with water molecules in the fluid-phase, neutralizing the reactive thioester and limiting host cell (and target cell) deposition (7).
Dysregulation of C3 and the ensuing spontaneous activation of the alternative pathway have been associated with several ultra-rare complement-mediated renal diseases. One such example is the C3 glomerulopathies (C3Gs), the two prototypical examples of which are C3 glomerulonephritis (C3GN) and dense deposit disease (DDD) (8). Both C3GN and DDD are defined by their shared C3-dominant glomerular immunofluorescence and differentiated by the location and appearance of glomerular deposits that can be resolved on electron microscopy. Genetic studies have identified loss-of-function mutations in RCA proteins or gain-of-function mutations in C3 in these diseases (9). In addition, DDD (and less often C3GN) is often associated with the development of autoantibodies to the alternative pathway C3 convertase (C3bBb) that are known as C3 nephritic factors (C3Nefs) (10–13). C3Nefs stabilize the C3bBb enzyme complex dramatically increasing its half-life (14). Persistence of the C3 convertase consumes C3 leading to the very low plasma C3 levels characteristic of this disease (15).
The current study initially investigated the effect of complement activation on the vitamin D binding protein (DBP) and made the unexpected observation that DBP forms covalent complexes with C3b. We hypothesized that this interaction would not be unique to DBP and explored the interaction of C3b with several other plasma proteins of diverse size, charge and abundance. All proteins formed complexes with C3b that could be degraded by the fluid-phase RCA proteins factors H and I. Although it is intuitive that nascently generated C3b should be able to attach to proteins in close proximity, there have been very few descriptions of these complexes and the observations that have been reported focus on C3b binding to other complement proteins or activators (16–18). Thus, the existence of fluid-phase C3b:plasma protein complexes generally is not recognized and their physiological significance has not been described. In this study we observed these complexes in plasma samples from DDD patients and to a lesser extent in plasma samples from C3GN patients. Their presence further supports the pathophysiological basis of these two C3Gs as fluid-phase dysregulation of the C3 convertase. Our findings also suggest that in the normal state, covalent attachment of C3b to plasma proteins may be a passive mechanism to minimize host cell deposition at sites of complement activation. Excessive generation or defective clearance of these circulating C3b:plasma protein complexes in the C3Gs may contribute to disease pathology but also offer therapeutic targets to limit the observed renal damage.
MATERIALS AND METHODS
Reagents
Purified cobra venom factor (CVF), pre-activated Zymosan A and sheep erythrocytes were obtained from Complement Technology, Inc. (Tyler, TX). Human serum depleted of individual complement components (C3, C4, C5, factor B, factor D, properdin, factor H and factor I), and purified human complement proteins (C3, C3b, factor B, factor D, factor H and factor I) were all purchased from Complement Technology, Inc. The following purified human proteins were all obtained from Athens Research and Technology (Athens, GA): alpha-1 proteinase inhibitor (α1PI), alpha-1 acid glycoprotein (α1AG), immunoglobulin G (IgG) and vitamin D binding protein (DBP).
The IgG fraction of goat polyclonal anti-human DBP was purchased from DiaSorin (Stillwater, MN) and then affinity-purified in our lab using immobilized DBP. Chicken polyclonal anti-α1PI antibody was obtained from ProSci Inc. (Poway, CA). Biotinylated rabbit polyclonal anti-α1AG, goat polyclonal anti-human albumin, and rabbit polyclonal anti-kininogen antibodies were all obtained from Abcam (Cambridge, MA). Mouse monoclonal anti-human factor B (clone D33/3) was obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Polyclonal anti-human C1 inhibitor antibody was developed in rabbits. Chicken polyclonal anti-human C3 was obtained from Gallus Immunotech (Cary, NC). Rabbit IgG antibody developed against epitopes on human C3d (#ab15981) was purchased from Abcam. Mouse monoclonal anti-human factor I neutralization antibody (#A247) was obtained from Quidel (San Diego, CA). William’s plasma (deficient in high and low molecular weight kininogen) was a generous gift from Dr. Alvin Schmaier, Case Western Reserve University.
Collection of human blood and in vitro activation of complement
Blood was collected from healthy, medication-free human subjects who gave informed consent using a protocol approved by the Stony Brook University IRB. Vacutainer tubes (BD, Franklin Lakes, NJ) containing either 3.2% sodium citrate (for plasma) or a silica clot activator (for serum) were utilized. Individual serum and plasma samples from at least 5 subjects were pooled, aliquoted and frozen at −80°C. Pooled serum or citrated plasma (0.3 ml) was activated either with CVF (416 U/ml), Zymosan A (10 mg/ml) or 50 μl heat-aggregated (heated at 63°C for 20 minutes) human IgG (10 mg/ml) and incubated at 37°C for the time indicated in each experiment. For activation of citrated plasma samples, 2 mM Mg2+ was added to overcome the chelation effect of sodium citrate. Serum depleted of the complement regulators factors H and I spontaneously activates the alternative pathway and hence was stored in 0.1 mM EDTA. These samples were activated by adding just 0.5 mM Mg2+ and incubating at 37°C. In certain experiments, depleted serum samples were reconstituted by adding back the purified protein to achieve its mean plasma concentration (factor B: 210 μg/ml; factor D: 1 μg/ml; C3: 1.3 mg/ml) along with 0.4 mM Mg2+ and then incubating at 37°C for 15 minutes.
Human Subjects with C3G
12 patients with biopsy-proven C3G (6 DDD and 6 C3GN) were selected from our C3G registry for inclusion in this study based on histopathological data (light microscopy, IF, EM) and the availability of sufficient plasma samples to complete all assays multiple times. The human research institutional review board at the University of Iowa approved all procedures, and all patients gave informed consent.
SDS-PAGE and Immunoblotting
All samples were separated using 8% polyacrylamide gels with SDS at 80 V for the stacking gel and 100 V for the resolving gel. Resolved gels then were transferred onto an Immobilon PVDF membrane (Millipore, Bedford, MA) at 100 V for 75 minutes. The PVDF membrane was blocked with 5% non-fat dry milk (NFDM) in Tris-buffered saline with 0.1% Tween 20 (TBS-T) for 30 minutes, followed by primary and HRP-labeled secondary antibody incubations in 5% NFDM. Finally, blots were developed using HyGLO Quick Spray chemiluminescent detection reagent (Denville Scientific, Denville, NJ) and X-ray film.
In-vitro complex formation and breakdown
To evaluate the role of activated C3 in complex formation, the alternative pathway was assembled in vitro using the purified proteins C3 (1.3 mg/ml), factor B (200 μg/ml) factor D (1 μg/ml) and DBP (400 μg/ml) or α1AG (1 mg/ml) with 0.5 mM Mg2+ and incubated at 37°C for the specified amount of time. In control experiments, factor B was eliminated from the mixture to prevent activation. The molar ratios of the various components were maintained at physiological levels even when the exact concentrations could not be maintained due to dilution effects. The roles of regulatory proteins factors H and I in the breakdown of C3 complexes was determined by adding purified factor H alone (340 μg/ml), factor I alone (54 μg/ml) or both together along with C3 (1 mg/ml), factor B (177 μg/ml), factor D (0.76 μg/ml), DBP (307 μg/ml) and 0.5 mM Mg2+ at 37°C along with CVF (416 U/ml) to activate the protein mixture. Complexes were evaluated by SDS-PAGE and immunoblotting.
C5a Sandwich ELISA
A sandwich ELISA to detect human C5a and C5a des Arg was developed as previously described in detail (19). Briefly, Maxi-sorb 96 well plates were coated with 500 ng of mouse monoclonal anti-human C5a (clone 295003; R&D Systems, Minneapolis, MN) capture antibody at 4°C overnight. The coating solution was removed and the wells were blocked with 300 μl of blocking solution (3% NFDM in PBS-T) for 1 hour at room temperature. After 3 washes, 100 μl of standards (78 pg/ml to 5 ng/ml of purified natural human C5a, Complement Technology, Inc.) or experimental samples were added and incubated at room temperature with shaking for 90 minutes. This was followed by 4 washes and incubation with 100 ng of the detection antibody, biotinylated mouse monoclonal anti-human C5a (clone 295009, R&D Systems) for 60 minutes at room temperature with shaking. Finally, 40 ng of HRP-conjugated streptavidin (KPL, Gaithersburg, MD) was added to each well and incubated at room temperature for 30 minutes. After 5 washes, 100 μl of HRP substrate solution (KPL) was added until color development followed by addition of 100 μl stop solution (KPL) and the absorbance was measured at 450 nm using a SpectraMax M2 plate reader (Molecular Devices, Sunnyvale, CA).
Hydrolysis of the C3 thioester
The thioester bond in C3 was inactivated by treating 250 μg C3 with 0.5 M hydroxylamine (NH2OH) pH 7.5 for 2 hours at 21°C followed by exhaustive dialysis against PBS (1.8 L, 3x) at 4°C for a total of 18 hours (20). Inactivation of thioester was confirmed by hemolytic assays using both rabbit erythrocytes for the alternative pathway, and antibody-coated sheep erythrocytes for classical pathway, as well as a C5a ELISA of the supernatant from the hemolysis assay with antibody-coated sheep erythrocytes. C3- depleted serum alone or C3- depleted serum reconstituted with either native C3 or C3-NH2OH (at 0.5 mg/ml) was taken in a final dilution of 1:100 in GVB++ buffer along with 50 μl EAs (5 × 107/ml) and incubated at 37°C for 1 hour, after which the cells were centrifuged, supernatant collected and C5a levels quantified.
Antibody neutralization of factor I
Mouse monoclonal anti-human factor I, that inhibits the serine protease domain, was used to determine if enzyme activity of factor I is required to cleave the C3b:plasma protein complexes. Anti-factor I (100 μg/ml) was added to factor H depleted serum and incubated on ice for 15 minutes to allow antibody binding, followed by incubation at 37°C for specified amount of time.
RESULTS
Complement activation correlates with generation of high molecular weight SDS-resistant forms of several plasma proteins
Because DBP has been shown to function as a chemotactic cofactor for C5a in vitro, the initial goal of this study was to determine if complement activation converts DBP into an active chemotactic cofactor (21). Possible structural changes in DBP were investigated in normal human serum (NHS) by treating with three distinct pathway activators and then analyzing the samples by SDS-PAGE and immunoblotting. There was no apparent diminution in the 56 kDa native DBP band and neither were lower molecular weight DBP bands detected (Fig. 1 and not shown). However, figure 1 shows that activation of any complement pathway did generate an SDS-resistant DBP band at approximately 200 kDa that was not observed in untreated serum. Moreover, simultaneous addition of EDTA with CVF to serum inhibited complement activation and prevented formation of this high molecular weight band. To determine if formation of this high molecular weight band is specific to DBP, two other plasma proteins, α1-proteinase inhibitor (α1PI) and α1-acid glycoprotein (α1AG), both of similar size but greater abundance than DBP, were examined in the C-activated serum samples. Figure 1 shows that activation of any complement pathway also induced formation of high molecular weight SDS-resistant bands of α1PI and α1AG; these bands were not observed in untreated NHS or CVF-activated serum treated with EDTA. Factor B cleavage is shown in the bottom panel of figure 1 to verify complement activation. Thus, activation of serum complement in vitro correlates with the appearance of high molecular weight SDS-resistant bands of DBP, α1PI and α1AG.
Figure 1. Complement activation induces formation of high molecular weight SDS-resistant complexes with plasma proteins.
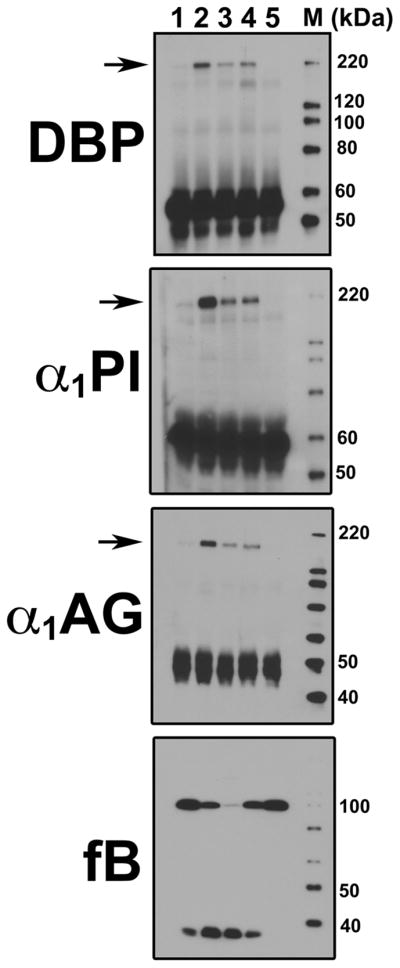
Pooled normal human serum was sham-treated with PBS (lane 1) or complement was activated by incubating serum at 37°C using either 416 U/ml CVF (lane 2), 10 mg/ml zymosan A (lane 3), 0.5 mg/300 μl heat-aggregated human IgG (lane 4). As a control to inhibit complement activation, 10 mM EDTA was added to serum prior to addition of CVF (lane 5). Serum aliquots were separated on an 8% SDS-PAGE and then immunoblotted for DBP, α1PI or α1AG. Arrows indicate the position of high molecular weight SDS-resistant bands formed during complement activation. The same samples also were blotted for factor B (fB) to verify cleavage as an indicator of complement activation (bottom panel).
The temporal correlation between complement activation and generation of these high molecular weight bands was investigated in serum and citrated plasma to determine if the clotting process alters band formation. Samples were blotted for several proteins with different abundance, molecular weights and isoelectric points. All proteins examined formed similar SDS-resistant bands upon complement activation in both serum and plasma (Fig 2A), suggesting that this process is not affected by blood clotting and is not restricted to a certain class of proteins with a common structural motif. The temporal appearance of these SDS-resistant bands correlated with complement activation in both CVF-activated serum and plasma (Fig. 2A). In serum, high molecular weight bands begin appearing at the 5-minute time point (Fig. 2A), which is the precise time that complement activation products begin to appear in the CVF-treated serum: Bb (Fig. 2A) and C5a generation (Fig. 2B). Peak band formation occurs at approximately 15 minutes and then gradually decreases in intensity (Fig. 2A). In CVF-treated citrated plasma (Fig. 2A), complement activation is delayed due to chelation of divalent cations by sodium citrate, but bands do consistently appear at the 30-minute time point (Fig. 2A), their appearance correlating with factor B cleavage (Fig. 2A) and C5a generation (Fig. 2C). Interestingly, complement activation generates a single kininogen band in serum but a doublet in plasma, reflecting the presence of two forms of kininogen in blood, high molecular weight kininogen (HK) and low molecular weight kininogen (LK). In serum, in contrast, HK is cleaved during the clotting process and only the LK band appears (Fig. 2A). Thus the formation of high molecular weight SDS-resistant bands of several plasma proteins is not unique to specific blood proteins and correlates temporally with in vitro activation of complement.
Figure 2. Formation of SDS-resistant bands correlates temporally with complement activation in vitro.

Pooled normal human serum or citrated plasma were activated at 37°C with 416 U/ml CVF for the indicated times. In addition to CVF, 2 mM Mg2+ was added to plasma to overcome the chelation effects of sodium citrate. At each time-point, activation was stopped by placing the sample on ice. Panel A: Aliquots were separated on an 8% SDS-PAGE and then immunoblotted for the indicated proteins. Gels were cropped to focus only on the high molecular weight SDS-resistant bands. Samples also were blotted for factor B to verify cleavage as an indicator of complement activation. The 75 kDa β-chain of C3 was utilized as a loading control. Panel B: C5a ELISA of complement activation in serum. Panel C: C5a ELISA of complement activation in citrated plasma.
The role of erythrocytes in the generation of these high molecular weights bands was investigated using citrated whole blood. Figure 3 demonstrates that SDS-resistant bands of α1PI and α1AG form in whole blood during complement activation, indicating that surface expression of RCA proteins on erythrocytes does not prevent high molecular weight band formation. Using William’s plasma (deficient in both HK and LK) we also found that these bands form independent of each other. As seen in Figure 4, although no kininogen bands formed in C-activated William’s plasma, the generation of SDS-resistant bands of DBP, α1PI and α1AG was not altered.
Figure 3. High molecular weight SDS-resistant bands form in whole blood with complement activation.

Citrated whole blood or citrated plasma from the same blood donor was treated with 2 mM Mg2+ and CVF (416 U/ml) for 30 minutes at 37°C. PBS was added to plasma to compensate for blood cell volume and equalize the protein concentrations between the plasma and whole blood samples. After CVF-activation, whole blood was centrifuged to pellet the cells and aliquots of both plasma samples were separated using an 8% SDS-PAGE and immunoblotted for α1PI, α1AG and factor B.
Figure 4. High molecular weight SDS-resistant bands of the different plasma proteins can form independent of each other.
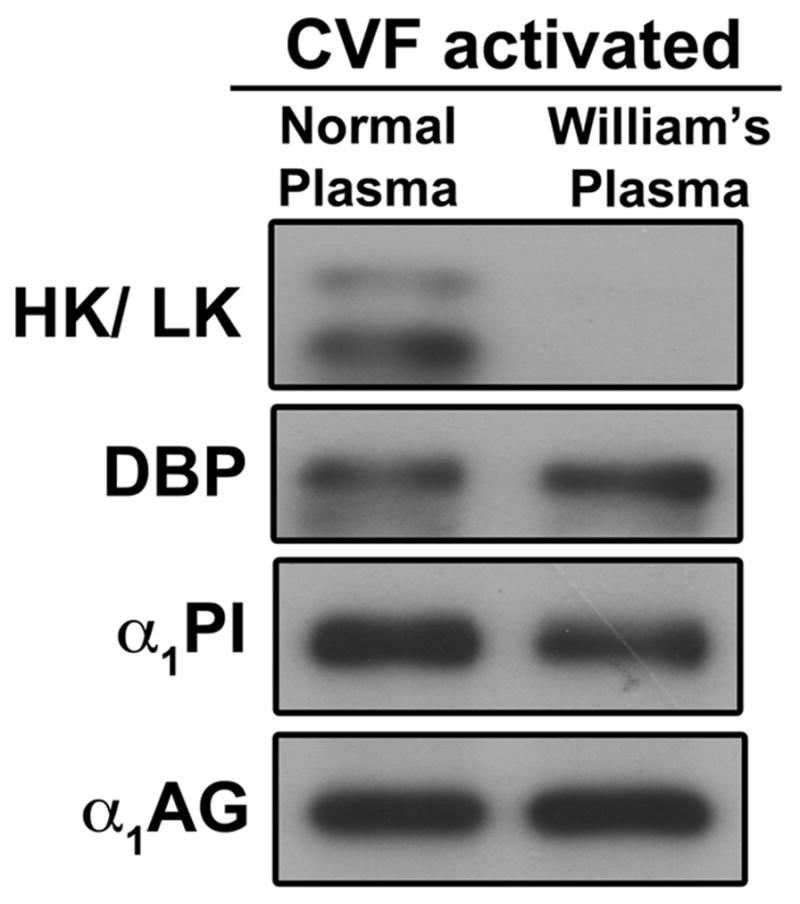
Normal human citrated plasma or citrated William’s plasma (deficient in both HK and LK) were activated with CVF (416 U/ml) and 2 mM Mg2+ for 60 minutes at 37°C. Aliquots were separated using 8% SDS-PAGE and immunoblotted for Kininogen, DBP, α1PI, α1AG and factor B.
Activation of the C3 thioester during C-activation causes formation of covalent complexes of C3b with plasma proteins
The evidence presented above indicates a strong correlation between complement activation and formation of high molecular weight SDS-resistant bands of several plasma proteins. To identify the steps involved in the generation of these complexes, sera depleted of different complement components were utilized. Figure 5A shows the formation of high molecular weight SDS-resistant bands of DBP, α1PI and α1AG in various depleted serum samples activated with CVF; reconstitution of depleted serum with the purified missing component was performed in samples where depletion abolished band formation. C3, factor B and factor D depleted sera were unable to generate high molecular weight bands upon addition of CVF (Fig. 5A), but formation was restored with addition of the deficient proteins. In contrast, generation was not altered in serum depleted of properdin, C4 or C5, clearly demonstrating that C3 cleavage is essential to this process.
Figure 5. Generation of C3 convertases and C3 are both essential for formation of high molecular weight SDS-resistant bands.

Panel A: Serum samples depleted of individual complement components (fB, fD, fP, C3, C5 and C4) were activated with CVF (416 U/ml) for 15 minutes at 37°C. Samples where complex formation was abolished were reconstituted with physiological levels of missing purified protein (fB, fD or C3) followed by activation with CVF. Aliquots were separated using 8% SDS-PAGE and immunoblotted for DBP, α1PI and α1AG. Samples also were immunoblotted for C3 β-chain band as a loading control and factor B to show complement activation. Panel B: Factor B-depleted and C3-depleted sera were reconstituted with purified C3b (1.3 mg/ml) and incubated at 37°C for 1, 3.5 or 5 minutes, separated using 8% SDS-PAGE and immunoblotted for DBP, α1PI and α1AG. Samples also were blotted for factor B to show complement activation in C3-depleted serum reconstituted with C3b. CVF activated normal human serum was included as a positive control.
The role of the C3 cleavage product C3b was examined next (Fig. 5B). Purified C3b added to factor B-depleted serum showed no complex formation. Addition of C3b to C3-depleted serum generated a C3 convertase but was not able to induce formation of high molecular weight forms of DBP, α1PI and α1AG, even though robust factor B cleavage could be demonstrated, indicating the requirement for both native C3 and the components needed to form C3 convertase (factors B and D). Since native C3 is essential for this process, the role of the thioester bond was investigated by treating C3 with hydroxylamine (NH2OH) to inactivate this bond and then using this chemically modified C3 to reconstitute C3 depleted serum (Fig. 6). Hydroxylamine-treated C3, like C3b, could not attach to an activating cell surface and generate C5a indicating effective thioester inactivation (Fig. 6A). This inactivated C3 also failed to induce formation of high molecular weight complexes of DBP, α1PI or α1AG upon addition of CVF (Fig. 6B) indicating that an intact thioester bond is essential for this process. Thus the SDS-resistant high molecular weight bands of plasma proteins formed during complement activation are covalent complexes with nascently generated C3b (note high molecular weight bands in the C3 blot of Fig. 6B). Consistent with this mechanism, multiple high molecular weight C3 bands were observed when the samples described in figures 1 and 2 were blotted for C3. Figure 6C shows immunoreactive C3 bands above the 110 kDa C3 α-chain in the samples from Figure 1. Furthermore, the temporal appearance and intensity of these multiple C3 bands correlate precisely with the individual proteins detected in CVF-activated serum and plasma in Figure 2 (shown in Fig. 6D). Moreover, both figures 6C and 6D show that the appearance of high molecular weight C3 bands correlate with a reduction in the intensity of the native C3 α-chain (110 kDa) that contains the thioester bond.
Figure 6. Reaction with the thioester bond in C3 during complement activation causes formation of covalent complexes with plasma proteins.

Panel A: The amount of C5a generated in reconstituted C3-depleted serum samples measured by ELISA. Antibody coated sheep erythrocytes were treated with either C3-depleted serum alone or C3-depleted serum reconstituted with 1.3 mg/ml native C3, hydroxylamine treated C3 (C3-NH2OH) or C3b and incubated at 37°C for 1 hour. The supernatant was removed and C5a levels quantified by C5a ELISA. Panel B: C3-depleted serum alone or reconstituted with native C3 or hydroxylamine treated C3 (C3-NH2OH), both at 1.3 mg/ml, were activated with CVF (416 U/ml) plus 0.4 mM Mg2+ for 15 minutes at 37°C. CVF activated normal human serum was included as a positive control. Aliquots were separated using 8% SDS-PAGE and immunoblotted for DBP, α1PI, α1AG, factor B and C3. Panel C: Pooled normal human serum was sham-treated with PBS (lane 1) or complement was activated by incubating serum at 37°C using either 416 U/ml CVF (lane 2), 10 mg/ml zymosan A (lane 3), 0.5 mg/300 μl heat-aggregated human IgG (lane 4). As a control to inhibit complement activation, 10 mM EDTA was added to serum prior to addition of CVF (lane 5). Serum aliquots were separated on an 8% SDS-PAGE and then immunoblotted for C3. Panel D: Pooled normal human serum or citrated plasma were activated at 37°C with 416 U/ml CVF for the indicated times. In addition to CVF, 2 mM Mg2+ was added to plasma to overcome the chelation effects of sodium citrate. At each time-point, activation was stopped by placing the sample on ice. Aliquots were separated on an 8% SDS-PAGE and then immunoblotted C3.
Factors H and I degrade the C3b:plasma protein complexes
The time course of complex formation (Fig. 2A) shows that band intensity decreases over time, consistent with the breakdown of these complexes. Since C3b is cleaved by factors H and I, their possible role in this process was examined. Factor H-depleted and factor I-depleted sera were incubated at 37°C for 15 minutes to allow for C3b:protein complex formation following which breakdown was monitored for 16 hours with or without the addition of purified factor H or factor I (Fig. 7A). No breakdown was observed in the factor I-depleted serum but addition of purified factor I induced complete degradation of each complex consistent with the known role of this protease (Fig 7A). In factor H-depleted serum, there was significant breakdown of C3b:protein complexes that was only marginally increased by the addition of purified factor H (Fig 7A). Next, factor H depleted serum was examined at very early time points to assess its role in complex breakdown. DBP, α1PI and α1AG complexes spontaneously formed in factor H depleted serum (without a complement activator) and the complexes appeared as doublets within one minute but were all cleaved to the lower molecular weight form by 5 minutes (Fig. 7B). No doublets were observed in normal serum even one minute after CVF was added (data not shown), indicating that complex cleavage is delayed in the absence of factor H, further confirming that, although factor H facilitates complex breakdown, other serum cofactors (factor H-like 1, factor H related proteins) can substitute in the absence of factor H (22–24). To confirm the role of factor I in the initial rapid cleavage reaction, purified factor I was added to factor I depleted serum. As expected, reconstitution of factor I to the depleted serum restored the initial cleavage (Fig. 7C). Furthermore, an antibody that specifically blocks the serine protease domain in factor I also inhibits complex cleavage (Fig. 7D). These results indicate that factor I is essential for the initial cleavage and subsequent breakdown of these complexes, a process that can be facilitated by the cofactor activity of factor H. C3b:protein complexes are initially generated as higher molecular weight forms and factor I along with cofactors mediates the cleavage of C3b to iC3b and smaller C3b cleavage products. This degradation pattern is readily observed on a C3 blot of CVF-activated normal serum (CVFAS) versus factor I-depleted serum (Fig. 7E), where in CVFAS there is a distinct 67 kDa iC3b product of the alpha chain, whereas in factor I depleted serum the prominent C3 cleavage product is the 100 kDa alpha prime chain of C3b.
Figure 7. C3b:protein complexes are cleaved in serum by factor I with the help of a cofactor.

Panel A: Factor I-depleted serum and factor H-depleted serum were reconstituted with 0.5 mM Mg2+ and incubated at 37°C for 15 minutes, to allow for complex formation, followed by addition of 10 mM EDTA to stop the reaction. The samples were then reconstituted with either PBS or the purified depleted component (factor I or factor H) and incubated for 16 hours at 37°C. Samples were separated using 8% SDS-PAGE and immunoblotted for DBP, α1PI and α1AG. Panel B: Factor H-depleted serum was reconstituted with 0.5 mM Mg2+ and incubated at 37°C for the indicated times. Serum aliquots were separated using 8% SDS-PAGE and immunoblotted for DBP, α1PI, α1AG and factor B. Panel C: Factor I-depleted serum alone or reconstituted with 25% (0.25X) or 100% (1X) of the plasma concentration of factor I (70 μg/ml) was activated with CVF (416 U/ml) for 15 minutes at 37°C. CVF activated normal human serum was included as a positive control for complex formation. Serum aliquots were separated using 8% SDS-PAGE and immunoblotted for DBP, α1PI and α1AG. Panel D: Factor H-depleted serum was treated with 100 μg/ml of either an irrelevant mouse IgG or an antibody that neutralizes the serine protease domain of factor I for 15 minutes on ice. Serum samples then were incubated with 0.5 mM Mg2+ at 37°C for the indicated times. Immunoblots for DBP and α1PI complex formation are shown. Panel E: Pooled normal human serum and C3 depleted serum were activated with CVF for 15 minutes, while factor I depleted serum was incubated at 37°C for 15 minutes. The samples were separated using 8% SDS-PAGE and immunoblotted for C3.
In vitro generation of C3b:DBP complexes using purified proteins
The preceding data demonstrate that formation of C3b:protein complexes requires native C3, a C3 convertase and a target plasma protein. To examine whether this system can be assembled in vitro, purified C3, factor B, factor D and a target protein of interest (DBP) were mixed at physiological ratios in the presence of Mg2+. Figure 8A shows that a C3b:DBP complex was formed upon incubation at 37°C for either 15 or 30 minutes. This complex was the same size as the uncleaved complex in factor I depleted serum (Fig. 8A). Moreover, hydroxylamine-treated C3 was capable of activating the alternative pathway as evidenced by factor B cleavage but, in contrast to native C3, it could not form a complex with DBP (Fig 8B). These results confirm that only four components are necessary and sufficient for C3b:protein complex formation.
Figure 8. C3b:protein complexes can be formed in-vitro using four purified proteins.
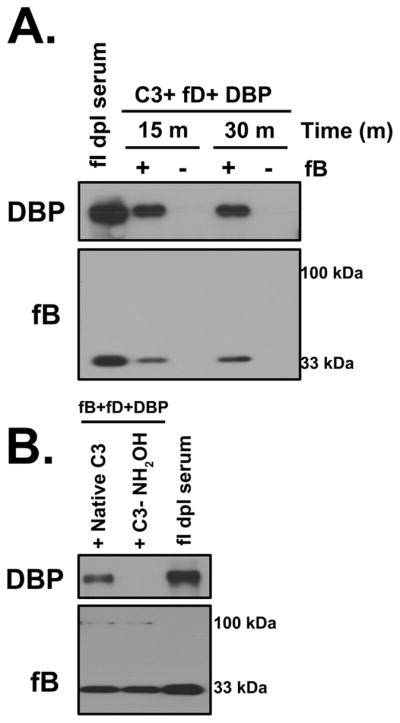
Panel A: Purified C3, fD, DBP and 0.5 mM Mg2+ were mixed in the presence or absence of fB at physiological concentrations and incubated at 37°C for either 15 or 30 minutes. Factor I-depleted serum sample incubated at 37°C for 15 minutes in the presence of Mg2+ was included as a positive control. Mixtures were immunoblotted for DBP (for complex formation) and factor B (for complement activation). Panel B: Purified fB, fD, DBP and 0.5 mM Mg2+ were mixed with either native C3 or hydroxylamine treated C3 (C3-NH2OH), at physiological ratios were incubated at 37°C for 15 minutes. Factor I depleted serum sample incubated at 37°C for 15 minutes in the presence of Mg2+ was used as a positive control. Samples were immunoblotted for DBP to assess complex formation and factor B to verify complement activation.
Since the use of purified proteins provides a well-defined in vitro model, the breakdown of C3b:protein complexes was investigated using purified factors H and I. Figure 9A shows C3b:DBP complexes after 15 minutes, 2 hours or 16 hours with no regulators, factor H alone, factor I alone, or both factors H and I. Only the combination of both factor H and factor I, could initiate cleavage and breakdown of the C3b:DBP complex. These results using purified proteins confirm that C3b:DBP (or other plasma proteins) are initially generated as higher molecular weight complexes that are rapidly cleaved by factor I with the help of a cofactor to a lower molecular weight form, and that this complex represents the relatively stable SDS-resistant complex observed in serum and plasma following complement activation (Figures 1–5). Although the iC3b:DBP band completely disappeared after a 16 hour incubation, a intermediate cleavage product, which could potentially be C3dg:DBP appeared at 15 minutes and was not further degraded even after 16 hours (Fig. 9B). These results were confirmed using purified α1AG as the target protein, where a prominent band at an approximate molecular weight corresponding to C3dg:α1AG also appeared at 15 minutes and was present at 16 hours (Fig. 9C, left panel). Similar results were also observed using purified α1PI (data not shown). This putative C3dg:protein band does not appear in factor I depleted CVFAS reconstituted with purified factor I (Fig. 9C, left panel), indicating that in serum there is a factor I independent mechanism that further cleaves the C3dg portion (25, 26). This factor I-independent process is consistent with the known cleavage of C3dg by trypsin and trypsin-like proteases (25, 26). In order to verify that the C3d fragment (containing the TED covalent linkage site) is indeed attached to α1AG, the same samples were blotted using an antibody against C3d specific epitopes (Fig. 9C, right panel). Results show that the size and temporal appearance of C3d correlated with iC3b:α1AG and C3dg:α1AG complexes. Because no other intermediate cleavage products could be detected in serum samples, even when utilizing a broad range gradient gel (data not shown), it is reasonable to surmise that following the initial rapid factor I-mediated cleavage of C3b (to iC3b), a second slower factor I-dependent cleavage removes most of the remaining C3 (C3c fragment) leaving C3dg attached to the plasma protein. In serum or plasma this is then followed by a rapid factor I independent cleavage removing the remaining C3d and perhaps leaving a small C3 peptide tag attached to the plasma protein, which would be indistinguishable from the native protein by immunoblotting.
Figure 9. The breakdown of C3b:plasma protein complexes requires cleavage by factor I in the presence of factor H.

Panel A: Purified C3, fB, fD and DBP were mixed with either factor H alone, factor I alone or a combination of factors H and factor I at physiological ratios and then activated with CVF in the presence of 0.5 mM Mg2+ for the indicated times. Samples were immunoblotted for DBP to assess complex formation and factor B to verify complement activation. C3 β-chain was used as a loading control. Panel B: The uncropped DBP immunoblot of the last six lanes of panel A to show intermediate cleavage products. Panel C: Purified C3, fB, fD and α1AG were mixed with either factor I alone or a combination of factors H and factor I at physiological ratios and then activated with CVF in the presence of 0.5 mM Mg2+ for the indicated times. Samples were immunoblotted for α1AG (center panel) or C3d (right panel) to assess complex formation and breakdown. The positions of C3b, iC3b and C3dg complexes with α1AG are indicated. To compare complex degradation in serum to the purified protein system, factor I depleted serum alone, or factor I depleted serum reconstituted with factor I and activated with CVF for 15 minutes, were separated and immunoblotted for α1AG (left panel).
C3b:plasma protein complexes form spontaneously in patients with dense deposit disease
The data presented above show that C3b:protein complexes are generated by complement activation and then degraded by the actions of factors H and I. Defective regulation may allow excessive generation of C3b:plasma protein complexes in the circulation. The C3 glomerulopathies are a type of ultra-rare renal diseases characterized by fluid-phase dysregulation of the alternative pathway of complement. Aliquots of EDTA plasma from six C3GN and six DDD patients were analyzed by SDS-PAGE and immunoblotting for C3b:protein complexes but no sample showed high molecular weight SDS-resistant complexes when blotted for C3 and α1AG (Fig. 10A). To determine whether complexes could form in these patient plasmas, samples were incubated at 37°C for 60 minutes in the presence of 7.5 mM Mg2+ to overcome the EDTA chelation, and in three samples (DDD patient samples 1, 4 and 12) complexes were then observed (Fig. 10B). No complex formation was observed in normal controls and C3GN samples although multiple high molecular weight bands that did not correspond to complexes on the α1PI and α1AG blots were present on the C3 blot in C3GN sample 5 (Fig. 10B).
Figure 10. C3b:plasma protein complexes form spontaneously in DDD patient samples.

Panel A: EDTA-plasma from DDD patients (n=6) and C3GN patients (n=6) were separated using 8% SDS-PAGE and immunoblotted for C3, factor B, and α1AG. As a reference for complex size, factor H and factor I depleted serum samples were included. Panel B: EDTA-plasma from DDD patients (n=6) and C3GN patients (n=6) or healthy controls (n=2) were reconstituted with 7.5 mM Mg2+ and incubated at 37°C for 60 minutes. The samples were then separated using 8% SDS-PAGE and immunoblotted for C3, factor B, α1PI and α1AG. Panel C: 1.3 mg/ml of C3 and 7.5 mM Mg2+ were added to the EDTA-plasma from DDD patients (n=6) and C3GN patients (n=6) and incubated at 37°C for either 30 minutes or 60 minutes. As a reference for complex size, factor H and factor I depleted serum samples were included. The samples were separated using 8% SDS-PAGE and immunoblotted for C3, factor B and α1AG.
Since many of the patient samples were significantly depleted of C3, purified C3 (1.3 mg/ml) and 7.5 mM Mg2+ were added to all patient samples followed by 37°C incubation for 30 minutes for 60 minutes. Five of six DDD samples (1, 4, 6, 11 and 12) formed complexes (Fig. 10C). Sample 8, the only DDD sample that showed no complex formation, was also the only DDD sample negative for C3Nefs. The C3GN sample 7 formed a clear complex with α1AG whereas sample 2, 9 formed weak complexes and samples 3, 5 and 10 were essentially negative. Interestingly, the bands formed in the patient plasma samples corresponded to the approximate size of bands in factor H-depleted serum (Fig. 10C). The presence of C3b:protein complexes in the blood of patients with DDD and to a lesser extent in C3GN patients, but not in plasma samples from healthy controls, indicates that there is dysregulation of the C3 convertase.
DISCUSSION
Excessive activation and/or poor regulation of the complement system can trigger significant host cell damage, the possibility of which is minimized by multiple levels of regulation that control both the initiation and extent of complement activation. Amongst the key steps in complement activation is the cleavage of C3 to C3b, which exposes the labile thioester bond in the TED of C3b (27). The extremely rapid ability of the thioester to react with available nucleophile acceptor groups (amino or hydroxyl) covalently attaches C3b to the surface of cells or tissue debris. This essential step of C3b-tagging marks foreign targets for destruction and/or removal by phagocytes (28). However, the C3b thioester also reacts with H2O in plasma resulting in fluid-phase hydrolysis. In this paper, we demonstrate that upon complement activation in serum (or plasma), C3b also forms transient covalent complexes with at least 10 different plasma proteins of various sizes, isoelectric points and abundance, and that these C3b:protein complexes are subsequently degraded by factors H and I. Covalent binding of C3b to IgG (18), C4 (16) and properdin (17) has been reported previously but these studies were examining formation of C3 convertases or immune complex clearance, and not the role of plasma proteins in binding and neutralizing C3b. Given our current knowledge of the reactivity of the thioester in C3, this process of generating C3b:plasma protein complexes is predictable, but surprisingly, is neither well-recognized nor documented.
The amount of C3b:plasma protein complexes generated in complement activated normal serum, and particularly in factor I depleted serum, is substantial, accounting for a large percentage of the total immunoreactive C3 bands (Figs. 6C, D and 7E). Complexes form despite the fact that 91.5% of plasma is water (55 Molar), and only 7.5% is protein. Given that water reacts with the thioester bond and is in great molar excess as compared to plasma proteins, it is noteworthy that C3b:protein complexes form and can be detected in serum or plasma after complement activation. Furthermore, the C3b:protein complex attachment must have some level of selectivity because if the reaction was entirely nonspecific then C3b:albumin would be prevalent, since albumin is the most abundant plasma protein. However, formation of a high molecular weight albumin band was considerably weaker than other much less abundant plasma proteins (Fig. 2A). Thus, the binding reaction is not entirely indiscriminate even though the C3b thioester has a wide range of protein targets in plasma. The results suggest that it may prefer available nucleophile acceptor groups in certain proteins.
The covalent attachment of C3b to multiple plasma proteins reflects the highly reactive nature of the thioester bond, but very few descriptions of these complexes have been reported (16–18) and their breakdown has not been described. This collateral by-product of complement activation could serve a passive regulatory role by restraining unintended deposition of C3b on host cells and to limit complement-mediated tagging to the intended target at sites of activation. Moreover, we have noted that C3b binds with a much greater propensity to chemically or thermally-denatured plasma proteins, suggesting that C3 may be involved in clearing non-native proteins from the circulation (Ramadass et. al., manuscript in preparation). The C3b:protein complexes we have described are first produced as higher molecular weight forms at a 1:1 molar ratio of C3b-to-plasma-protein. Consistent with what is known about C3b sequential cleavage following complement activation, the C3b:protein complexes are then cleaved very rapidly by factor I and its cofactors to iC3b and then more slowly to C3dg, which is followed by a rapid factor I independent cleavage possibly leaving a very small peptide tag attached to the plasma protein (25, 26). Factor I is absolutely essential to the process of complex degradation. That we could detect no further cleavage products, even utilizing a broad range gradient gel, suggests that a second slower factor I-dependent cleavage reaction resulting in C3dg bound to the plasma protein is followed by a rapid factor I independent cleavage reaction that leaves behind a small C3 peptide tag (a portion of C3d) attached to the protein (25, 26), which would be indistinguishable from the native protein band by immunoblotting.
Uncontrolled alternative pathway activation caused by deficiency of factor H has been associated with several diseases including atypical hemolytic uremic syndrome (aHUS), C3 glomerulopathies (C3GN and DDD) and age-related macular degeneration (AMD) (29, 30). aHUS and AMD are surface-phase diseases, with loss of tight complement regulation at the level of either the endothelial cell or Bruch’s membrane, respectively. The C3Gs, in contrast, are fluid-phase diseases in which complement dysregulation occurs in the plasma. The consequence of fluid phase dysregulation is massive conversion of C3 to C3b, which would lead to the formation of multiple different C3b:protein complexes. However, complexes were only detected when plasma was either incubated at 37°C or spiked with native C3 in vitro (in patient samples where C3 was depleted), indicating that although dysregulation allows complexes to spontaneously form, they must be rapidly degraded and/or cleared from the circulation in vivo. Furthermore, these results are consistent with the clinical finding of extremely low plasma C3 levels in these patients. It is intriguing to speculate whether these C3b:protein complexes have a direct role in the pathogenesis of the C3Gs. Clearly, factor H deficiency has been associated with aHUS, AMD and C3Gs but it is also relevant to consider the clinical phenotype associated with factor I deficiency (30–34). This very rare primary immunodeficiency is inherited as an autosomal recessive trait and has been reported in only about 30 families. In the absence of factor I, C3 is depleted, and any C3b:protein complexes that form cannot be broken down (Fig. 7). Because iC3b and its smaller cleavage product C3d are not generated, consequently there is a lack of recognition by the C3 receptors CR2 (binds C3d on B lymphocytes) and CR3 (binds iC3b on phagocytic cells), which are needed to stimulate phagocytosis and link the innate and adaptive immune responses. Therefore, defective opsonization makes factor I-deficient patients susceptible to recurrent pyogenic infections and aseptic meningitis (31–34). But what is notable is that although renal disease has been described in these patients, it is very uncommon, and associated with deposition of immune complexes suggesting activation of the classical pathway (35, 36). Thus the clinical picture of factor I deficiency suggests that C3b:protein complexes alone do not promote C3G and that functional factor I is required for the disease to develop, perhaps by generating iC3b and C3dg fragments of the C3b:plasma protein complexes.
Interestingly, factor I is also required for the development of C3G-like disease in mice. Factor H deficient mice develop kidney disease that is similar to human DDD, whereas factor I deficient mice, as well as factors H and I double knock-out animals, do not develop kidney disease (37). The authors conclude that in order to develop DDD, factor I is required to generate C3 fragments in the circulation of factor H deficient mice (37). This study also showed that plasma samples from factor I deficient as well as H and I double knockout mice, but not factor H deficient mice, showed multiple high molecular weight SDS-resistant forms of C3 that correspond to the uncleaved C3b:plasma protein complexes reported herein. However, these bands were noted but were thought to be C3b oligomers (37). These in vivo findings are consistent with our results and support the premise that C3b:protein complexes can be detected in the circulation. Moreover, a follow up study analyzed the glomerular C3 deposits (obtained by laser capture microdissection) in factor I deficient mouse kidneys (38). Analysis by SDS-PAGE showed no C3 α-chain band (that contains the thioester bond), but a prominent C3 β-chain, in factor I deficient mouse kidneys. However, a higher SDS-resistant band was observed, indicating that C3 α-chain was covalently linked to other molecules (38). Another study analyzed glomerular dense deposits by mass spectroscopy isolated by laser capture microdissection from patients with DDD and showed that deposits contain various complement components and other plasma proteins such as ApoE, fibronectin and fibrinogen (39). These in vivo studies are consistent with the results reported herein and suggest that under normal physiological conditions, covalent attachment of C3b to plasma proteins in the fluid-phase may be a passive mechanism to minimize host cell deposition at sites of complement activation. However, excessive generation and/or defective clearance of these circulating C3b:plasma protein complexes may have pathological consequences in conditions such as the C3Gs.
Currently, there are minimal disease-specific treatment options for the C3Gs (40). Based on our current understanding of pathogenesis, therapies that warrant consideration include drugs that can prevent ongoing dysregulation of the C3 convertase or drugs that can potentially ‘mop up’ the C3 breakdown products. Interestingly, intravenous immunoglobulin (IVIG) therapy has been used to treat autoimmune and inflammatory disorders and IVIG is known to scavenge activated complement fragments including C3b (41–43). This raises the intriguing prospect that IVIG could interfere with the formation of C3b:plasma protein complexes, but this possibility remains to be investigated. In addition, a major question for future studies will be to determine if different C3b:protein complexes have different clearance rates and associations with specific diseases. Furthermore, the fate and function of plasma proteins with an attached C3 peptide tag remains to be investigated. We believe that further work on characterizing C3b:plasma protein complexes is warranted, and these complexes possibly could be useful as a marker of dysregulated or excessive fluid-phase complement activation.
Acknowledgments
This work was supported by National Institutes of Health Grants GM063769 (to R.R.K.), AI060866 and AI084178 (to B.G.).
The authors wish to thank Dr. Alvin Schmaier, Case Western Reserve University for generously providing the William’s plasma sample, and Dr. Viviana Ferreira of University of Toledo for helpful discussions and suggestions.
Abbreviations used in this article
- α1AG
α1-acid glycoprotein
- α1PI
α1-proteinase inhibitor
- C3G
C3 glomerulopathy
- C3GN
C3 glomerulonephritis
- C3Nef
C3 nephritic factor
- CVF
cobra venom factor
- CVFAS
CVF-activated serum
- DBP
vitamin D binding protein
- DDD
dense deposit disease
- HK
high molecular weight kininogen
- LK
low molecular weight kininogen
- TED
thioester domain of C3
References
- 1.Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11:785–797. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pangburn MK, Schreiber RD, Muller-Eberhard HJ. Formation of the initial C3 convertase of the alternative complement pathway. Acquisition of C3b-like activities by spontaneous hydrolysis of the putative thioester in native C3. J Exp Med. 1981;154:856–867. doi: 10.1084/jem.154.3.856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pangburn MK, Muller-Eberhard HJ. Relation of putative thioester bond in C3 to activation of the alternative pathway and the binding of C3b to biological targets of complement. J Exp Med. 1980;152:1102–1114. doi: 10.1084/jem.152.4.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pangburn MK, V, Ferreira P, Cortes C. Discrimination between host and pathogens by the complement system. Vaccine. 2008;26(Suppl 8):I15–21. doi: 10.1016/j.vaccine.2008.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zipfel PF, Skerka C. Complement regulators and inhibitory proteins. Nat Rev Immunol. 2009;9:729–740. doi: 10.1038/nri2620. [DOI] [PubMed] [Google Scholar]
- 6.Mullereberhard HJ, Dalmasso AP, Calcott MA. The reaction mechanism of beta-1C-globulin (C′3) in immune hemolysis. J Exp Med. 1966;123:33–54. doi: 10.1084/jem.123.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Law SK, Dodds AW. The internal thioester and the covalent binding properties of the complement proteins C3 and C4. Protein science : a publication of the Protein Society. 1997;6:263–274. doi: 10.1002/pro.5560060201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sethi S, Nester CM, Smith RJ. Membranoproliferative glomerulonephritis and C3 glomerulopathy: resolving the confusion. Kidney international. 2012;81:434–441. doi: 10.1038/ki.2011.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Y, Meyer NC, Wang K, Nishimura C, Frees K, Jones M, Katz LM, Sethi S, Smith RJ. Causes of alternative pathway dysregulation in dense deposit disease. Clin J Am Soc Nephrol. 2012;7:265–274. doi: 10.2215/CJN.07900811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cameron JS, Turner DR, Heaton J, Williams DG, Ogg CS, Chantler C, Haycock GB, Hicks J. Idiopathic mesangiocapillary glomerulonephritis. Comparison of types I and II in children and adults and long-term prognosis. The American journal of medicine. 1983;74:175–192. doi: 10.1016/0002-9343(83)90606-x. [DOI] [PubMed] [Google Scholar]
- 11.Schwertz R, Rother U, Anders D, Gretz N, Scharer K, Kirschfink M. Complement analysis in children with idiopathic membranoproliferative glomerulonephritis: a long-term follow-up. Pediatr Allergy Immunol. 2001;12:166–172. doi: 10.1034/j.1399-3038.2001.012003166.x. [DOI] [PubMed] [Google Scholar]
- 12.Sethi S, Fervenza FC, Zhang Y, Zand L, Vrana JA, Nasr SH, Theis JD, Dogan A, Smith RJ. C3 glomerulonephritis: clinicopathological findings, complement abnormalities, glomerular proteomic profile, treatment, and follow-up. Kidney international. 2012;82:465–473. doi: 10.1038/ki.2012.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Servais A, Noel LH, Roumenina LT, Le Quintrec M, Ngo S, Dragon-Durey MA, Macher MA, Zuber J, Karras A, Provot F, Moulin B, Grunfeld JP, Niaudet P, Lesavre P, Fremeaux-Bacchi V. Acquired and genetic complement abnormalities play a critical role in dense deposit disease and other C3 glomerulopathies. Kidney international. 2012;82:454–464. doi: 10.1038/ki.2012.63. [DOI] [PubMed] [Google Scholar]
- 14.Mold C, Medof ME. C3 nephritic factor protects bound C3bBb from cleavage by factor I and human erythrocytes. Mol Immunol. 1985;22:507–512. doi: 10.1016/0161-5890(85)90173-7. [DOI] [PubMed] [Google Scholar]
- 15.Martinez-Barricarte R, Heurich M, Valdes-Canedo F, Vazquez-Martul E, Torreira E, Montes T, Tortajada A, Pinto S, Lopez-Trascasa M, Morgan BP, Llorca O, Harris CL, Rodriguez de Cordoba S. Human C3 mutation reveals a mechanism of dense deposit disease pathogenesis and provides insights into complement activation and regulation. J Clin Invest. 2010;120:3702–3712. doi: 10.1172/JCI43343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim YU, Carroll MC, Isenman DE, Nonaka M, Pramoonjago P, Takeda J, Inoue K, Kinoshita T. Covalent binding of C3b to C4b within the classical complement pathway C5 convertase. Determination of amino acid residues involved in ester linkage formation. J Biol Chem. 1992;267:4171–4176. [PubMed] [Google Scholar]
- 17.Whiteman LY, Purkall DB, Ruddy S. Covalent linkage of C3 to properdin during complement activation. Eur J Immunol. 1995;25:1481–1484. doi: 10.1002/eji.1830250555. [DOI] [PubMed] [Google Scholar]
- 18.van Dam AP, Hack CE. Formation of C3-IgG complexes in serum by aggregated IgG and by non-immunoglobulin activators of complement. Immunology. 1987;61:105–110. [PMC free article] [PubMed] [Google Scholar]
- 19.Trujillo G, Habiel DM, Ge L, Ramadass M, Cooke NE, Kew RR. Neutrophil Recruitment to the Lung in Both C5a- and CXCL1-Induced Alveolitis Is Impaired in Vitamin D-Binding Protein-Deficient Mice. J Immunol. 2013 doi: 10.4049/jimmunol.1202941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Law SK, Lichtenberg NA, Levine RP. Covalent binding and hemolytic activity of complement proteins. Proc Natl Acad Sci U S A. 1980;77:7194–7198. doi: 10.1073/pnas.77.12.7194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kew RR, Webster RO. Gc-globulin (vitamin D-binding protein) enhances the neutrophil chemotactic activity of C5a and C5a des Arg. J Clin Invest. 1988;82:364–369. doi: 10.1172/JCI113596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pangburn MK, Schreiber RD, Muller-Eberhard HJ. Human complement C3b inactivator: isolation, characterization, and demonstration of an absolute requirement for the serum protein beta1H for cleavage of C3b and C4b in solution. J Exp Med. 1977;146:257–270. doi: 10.1084/jem.146.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Misasi R, Huemer HP, Schwaeble W, Solder E, Larcher C, Dierich MP. Human complement factor H: an additional gene product of 43 kDa isolated from human plasma shows cofactor activity for the cleavage of the third component of complement. Eur J Immunol. 1989;19:1765–1768. doi: 10.1002/eji.1830190936. [DOI] [PubMed] [Google Scholar]
- 24.McRae JL, Duthy TG, Griggs KM, Ormsby RJ, Cowan PJ, Cromer BA, McKinstry WJ, Parker MW, Murphy BF, Gordon DL. Human factor H-related protein 5 has cofactor activity, inhibits C3 convertase activity, binds heparin and C-reactive protein, and associates with lipoprotein. J Immunol. 2005;174:6250–6256. doi: 10.4049/jimmunol.174.10.6250. [DOI] [PubMed] [Google Scholar]
- 25.Seya T, Nagasawa S. Limited proteolysis of complement protein C3b by regulatory enzyme C3b inactivator: isolation and characterization of a biologically active fragment, C3d,g. J Biochem. 1985;97:373–382. doi: 10.1093/oxfordjournals.jbchem.a135064. [DOI] [PubMed] [Google Scholar]
- 26.Ross GD, Lambris JD, Cain JA, Newman SL. Generation of three different fragments of bound C3 with purified factor I or serum. I. Requirements for factor H vs CR1 cofactor activity. J Immunol. 1982;129:2051–2060. [PubMed] [Google Scholar]
- 27.Janssen BJ, Christodoulidou A, McCarthy A, Lambris JD, Gros P. Structure of C3b reveals conformational changes that underlie complement activity. Nature. 2006;444:213–216. doi: 10.1038/nature05172. [DOI] [PubMed] [Google Scholar]
- 28.Sim RB, Twose TM, Paterson DS, Sim E. The covalent-binding reaction of complement component C3. Biochem J. 1981;193:115–127. doi: 10.1042/bj1930115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zipfel PF, Heinen S, Jozsi M, Skerka C. Complement and diseases: defective alternative pathway control results in kidney and eye diseases. Mol Immunol. 2006;43:97–106. doi: 10.1016/j.molimm.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 30.Zipfel PF, Smith RJ, Skerka C. Factor I and factor H deficiency in renal diseases: similar defects in the fluid phase have a different outcome at the surface of the glomerular basement membrane. Nephrol Dial Transplant. 2009;24:385–387. doi: 10.1093/ndt/gfn652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vyse TJ, Spath PJ, Davies KA, Morley BJ, Philippe P, Athanassiou P, Giles CM, Walport MJ. Hereditary complement factor I deficiency. QJM. 1994;87:385–401. [PubMed] [Google Scholar]
- 32.Alba-Dominguez M, Lopez-Lera A, Garrido S, Nozal P, Gonzalez-Granado I, Melero J, Soler-Palacin P, Camara C, Lopez-Trascasa M. Complement factor I deficiency: a not so rare immune defect: characterization of new mutations and the first large gene deletion. Orphanet J Rare Dis. 2012;7:42. doi: 10.1186/1750-1172-7-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moller Rasmussen J, Teisner B, Jepsen HH, Svehag SE, Knudsen F, Kirstein H, Buhl M. Three cases of factor I deficiency: the effect of treatment with plasma. Clin Exp Immunol. 1988;74:131–136. [PMC free article] [PubMed] [Google Scholar]
- 34.Nilsson SC, Sim RB, Lea SM, Fremeaux-Bacchi V, Blom AM. Complement factor I in health and disease. Mol Immunol. 2011;48:1611–1620. doi: 10.1016/j.molimm.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 35.Solal-Celigny P, Laviolette M, Hebert J, Atkins PC, Sirois M, Brun G, Lehner-Netsch G, Delage JM. C3b inactivator deficiency with immune complex manifestations. Clin Exp Immunol. 1982;47:197–205. [PMC free article] [PubMed] [Google Scholar]
- 36.Sadallah S, Gudat F, Laissue JA, Spath PJ, Schifferli JA. Glomerulonephritis in a patient with complement factor I deficiency. Am J Kidney Dis. 1999;33:1153–1157. doi: 10.1016/S0272-6386(99)70155-1. [DOI] [PubMed] [Google Scholar]
- 37.Rose KL, Paixao-Cavalcante D, Fish J, Manderson AP, Malik TH, Bygrave AE, Lin T, Sacks SH, Walport MJ, Cook HT, Botto M, Pickering MC. Factor I is required for the development of membranoproliferative glomerulonephritis in factor H-deficient mice. J Clin Invest. 2008;118:608–618. doi: 10.1172/JCI32525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Paixao-Cavalcante D, Hanson S, Botto M, Cook HT, Pickering MC. Factor H facilitates the clearance of GBM bound iC3b by controlling C3 activation in fluid phase. Mol Immunol. 2009;46:1942–1950. doi: 10.1016/j.molimm.2009.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sethi S, Gamez JD, Vrana JA, Theis JD, Bergen HR, 3rd, Zipfel PF, Dogan A, Smith RJ. Glomeruli of Dense Deposit Disease contain components of the alternative and terminal complement pathway. Kidney international. 2009;75:952–960. doi: 10.1038/ki.2008.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nester CM, Smith RJ. Treatment options for C3 glomerulopathy. Curr Opin Nephrol Hypertens. 2013;22:231–237. doi: 10.1097/MNH.0b013e32835da24c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spycher M, Matozan K, Minnig K, Zehnder R, Miescher S, Hoefferer L, Rieben R. In vitro comparison of the complement-scavenging capacity of different intravenous immunoglobulin preparations. Vox Sang. 2009;97:348–354. doi: 10.1111/j.1423-0410.2009.01217.x. [DOI] [PubMed] [Google Scholar]
- 42.Basta M. Ambivalent effect of immunoglobulins on the complement system: activation versus inhibition. Mol Immunol. 2008;45:4073–4079. doi: 10.1016/j.molimm.2008.07.012. [DOI] [PubMed] [Google Scholar]
- 43.Frank MM, Basta M, Fries LF. The effects of intravenous immune globulin on complement-dependent immune damage of cells and tissues. Clin Immunol Immunopathol. 1992;62:S82–86. doi: 10.1016/0090-1229(92)90045-p. [DOI] [PubMed] [Google Scholar]