

Abstract
Introduction
IFNα has been largely implicated in the ethiopathogenesis of autoimmune diseases but only recently it has been linked to endothelial damage and accelerated atherosclerosis in autoimmunity. In addition, proinflammatory conditions are supposed to be implicated in the cardiovascular status of these patients. Since a role for IFNα in endothelial damage and impaired Endothelial Progenitor Cell (EPC) number and function has been reported in other diseases, we aimed to evaluate the potential associations of IFNα serum levels on EPC populations and cytokine profiles in Rheumatoid Arthritis (RA) patients.
Methods
pre-EPC, EPC and mature EPC (mEPC) populations were quantified by flow cytometry analyzing their differential CD34, CD133 and VEGFR2 expression in blood samples from 120 RA patients, 52 healthy controls (HC), and 83 systemic lupus erythematosus (SLE) patients as disease control. Cytokine serum levels were measured by immunoassays and clinical and immunological data, including cardiovascular (CV) events and CV risk factors, were retrospectively obtained by reviewing clinical records.
Results
Long-standing, but not recent onset RA patients displayed a significant depletion of all endothelial progenitor populations, unless high IFNα levels were present. In fact, the IFNhigh RA patient group (n = 40, 33%), showed increased EPC levels, comparable to SLE patients. In addition, high IFNα serum levels were associated with higher disease activity (DAS28), presence of autoantibodies, higher levels of IL-1β, IL-6, IL-10 and MIP-1α, lower amounts of TGF-β, and increased mEPC/EPC ratio, thus suggesting higher rates of endothelial damage and an endothelial repair failure. Finally, the relationship between high IFNα levels and occurrence of CV events observed in RA patients seems to support this hypothesis.
Conclusions
IFNα serum marker could be used to identify a group of RA patients with increased disease activity, EPC imbalance, enhanced proinflammatory profile and higher cardiovascular risk, probably due, at least in part, to an impaired endothelial repair.
Introduction
Rheumatoid Arthritis (RA) is associated with increased cardiovascular (CV) disease morbidity and mortality rates that cannot be explained by traditional risk factors [1], [2]. Moreover, endothelial dysfunction, the main cause of premature atherosclerosis, has been found even in young RA patients without traditional CV risk factors [3], thus suggesting the involvement of disease-related pathways.
Endothelial damage leads to denuded sites at the endothelial wall that must be repaired. In this sense, bone marrow-derived Endothelial Progenitor Cells (EPCs) carry out vasculogenesis and endothelial repair functions, contributing to vascular homeostasis [4]. Although there is no consensus on their precise phenotypic definition, functional EPC are characterized by the expression of Vascular Endothelial Growth Factor Receptor-2 (VEGFR-2 or CD309), CD34 and CD133 [5], [6]; whereas those lacking CD34 expression are considered a pre-EPC subpopulation [7]. During EPC differentiation, CD133 expression is lost and they begin to express mature endothelial-specific markers, becoming mature EPC (mEPC) with lower vasculogenic functionality [6]. As endothelial status depends on injury and repair, the balance between EPC populations could be a surrogate marker which may be used as a potential CV risk factor. In fact, some studies have shown that circulating EPC could serve as a predictor of CV events in several conditions [8], [9]. EPC studies in RA patients, however, are contradictory.
On the other hand, disease-related risk factors have been identified [10], [11], suggesting that immune dysregulation could play a role in RA endothelial damage. Although the specific pathways remains unclear, a number of inflammatory and immune mediators seem to have a role, including C-reactive protein (CRP), cytokines, chemokines and growth factors [12], [13], most of them dysregulated in RA patients and implicated in the pathogenesis of autoimmune diseases. Among these mediators, it is worth noting the case of IFNα, since type-I interferons play a role in the pathogenesis of SLE and probably other autoimmune diseases [14], and recent evidence suggests their involvement in endothelial damage and EPC dysfunction. It has been reported that IFNα impair EPC function in vitro as well as in vivo and, as a consequence, endothelial repair [15]–[18]. Moreover, type I IFNs have been linked to atherothrombosis by acting on platelets and foam cells [19]. In addition, IFNα-signature has been linked to vasculopathy in systemic sclerosis patients [20].
Since previous studies suggest that circulating EPC populations and type I IFNs could be involved in increasing cardiovascular risk in autoimmune diseases, the main aim of this study is to determine EPCs frequency in RA patients’ peripheral blood and evaluate the potential associations with IFNα serum levels and clinical and immunological features.
Patients and Methods
Patients and Controls
Our study involved 120 RA patients fulfilling the 1987 revised criteria of the American College of Rheumatology, recruited from the Rheumatology outpatient clinic of the Hospital Universitario Central de Asturias, and 52 sex- and age-matched unrelated healthy controls (47 women, age (mean±SD): 44.74±11.04 years). Eighty-three SLE patients (79 women, age: 48.28±16.30 years, disease duration: 12.3±8.9 years, SLEDAI: 4.02±4.11) were included as disease controls. Routine clinical examination, information on clinical and immunological manifestations, therapies received in the previous three months and 28-joint disease activity score (DAS28) were obtained at the time of sampling. Clinical response to anti-TNFα therapy, in a six-month period, was analyzed using EULAR response criteria [21]. Patients were classified on having a “good” “moderate” or “no response” according to DAS28 change from baseline (6-month previous clinical visit). Patients’ clinical records were exhaustively revised in order to register the history of CV events and traditional CV risk factors (diabetes mellitus, hypercholesterolemia, hypertension and smoking habits). A CV event was considered if the patient suffered from heart failure, ischemic heart disease, cerebrovascular accident or peripheral arteriopathy from their RA diagnosis. Clinical definition of CV events and risk factors was performed as previously stated [22], [23].
Ethics Statement
Approval for the study was obtained from the Regional Ethics Committee for Clinical Investigation (Servicio de Salud del Principado de Asturias, Hospital Universitario Central de Asturias), according to the Declaration of Helsinki. All procedures were performed with an informed written consent from all individuals.
Flow Cytometry EPCs Quantification
Blood samples were immediately transported to the laboratory and processed. EPC were analyzed by FACS as described previously [24], following EUSTAR recommendations [25] with few modifications. Briefly, 100 µl of peripheral blood were preincubated with 10 µl of FcR Blocking Reagent (Miltenyi Biotech) for 20 minutes, followed by 30-minutes triple-labelling with anti-VEGFR2-phycoerythrin (PE, R&D Systems), anti-CD34-fluorescein isothiocyanate (FITC, BD Pharmigen) and anti-CD133-allophycocyanin (APC, Miltenyi Biotech) or with identical isotype antibodies (BD Pharmigen). Labelled cells were lysed with 2 ml BD Lysing Solution (BD Biosciences) for 5 minutes and washed twice with PBS. Finally, samples were analyzed in a BD FACSCanto II flow cytometer. After gating the lymphocyte population, CD34-positive events were selected and analyzed in a CD133 vs. VEGFR2 dot plot, thus considering CD34/VEGFR2/CD133 triple-positive cells as EPCs while CD34+VEGFR2+CD133− cells were identified as mature EPCs (mEPCs) [24] (Figure 1A). On the other hand, VEGFR2-positive events within the lymphocyte gate were analyzed for CD34/CD133 expression and CD34−VEGFR2+CD133+ cells were considered as pre-EPCs. At least 100,000 events in the lymphocyte gate and more than 100 CD34+ cells were acquired per sample. Cell counts were expressed as the number of positive cells per 100,000 events in the lymphocyte gate.
Figure 1. EPC analyses in RA patients by flow cytometry.

(A) Gating strategy of EPC, mEPC and pre-EPC analysis by flow cytometry in peripheral blood samples. Representative dot plots of a HC are shown. (B) The size of endothelial precursor populations is influenced by disease duration. Pre-EPC, EPC and mEPC counts in early (ERA, disease duration <1 year, n = 36) and long-standing (LRA, n = 84) Rheumatoid Arthritis patients and healthy controls (HC, n = 52). Differences were measured by Mann-Withney U-test.
Cytokine Serum Levels Quantifications
Serum aliquots were stored at −80°C until cytokine immunoassay measurement. Levels of IL-1β, IL-6, IL-8, IL-10, IFNα, MIP-1α (CCL3) and VEGF-A165were quantified using a Cytometric Bead Array Flex Set (BD) and analyzed in a BD FACS Canto II flow cytometer using FCAP Array v.1.0.1. For IL-1β, IL-6 and IL-10, an Enhanced Sensitivity Flex Set was needed. Technical detection limits were 48.4 fg/ml for IL-1β, 68.4 fg/ml for IL-6, 1.2 pg/ml for IL-8, 13.7 fg/ml for IL-10, 1.5 pg/ml for IFNα, 0.2 pg/ml for MIP-1α and 4.0 pg/ml for VEGF-A165. TGF-β1 and TNFα serum levels were quantified using ELISA kits (OptEIA, BD Bioscience), in accordance with the manufacturer’s instructions. Detection limits for these cytokines were 5 ng/ml and 0.48 pg/ml, respectively.
Statistical Analysis
All data are presented as median (Interquartile Range) unless otherwise stated. Comparisons were performed by non-parametric tests (Mann-Whitney U, Kruskal-Wallis tests and Spearman’s rank) as data were not normally distributed. Categorical variables were compared with a chi-squared test. The association between categorical variables and the CV events was assessed and adjusted for other factors (sex, age, traditional CV risk factors and disease activity) using multiple logistic regression analysis. Adjusted odds ratios (OR) and 95% confidence intervals (95% CI) were calculated so as to evaluate the strength of the associations. A p-value <0.05 was considered statistically significant. All data were analyzed with SPSS v.15.0 software.
Results
Circulating EPC Populations and IFNα Serum Levels in RA Patients
We aimed to investigate the possible relationship between EPC populations and IFNα levels in RA patients. To this end, IFNα serum levels and circulating pre-EPC, EPC and mEPC populations were quantified in 52 healthy controls (HC) and 120 RA patients with different disease duration (range 0–219 months) (Table 1). No significant differences in any endothelial progenitor population were found between patients and HC. However, disease duration was negatively correlated with EPC (r = −0.316, p<0.001) and mEPC (r = −0.342, p<0.001), suggesting an EPC depletion associated with disease progression. In fact, patients at recent onset (less than one year, n = 36, early RA, ERA) showed similar levels of these populations than HC, whereas those with longer disease duration (n = 84, long-standing RA, LRA) exhibited a significant depletion of all EPC populations (Figure 1B). Neither associations with age at sampling, age at diagnosis, nor autoantibodies status were found.
Table 1. Demographic, immunological and clinical parameters of the RA patients.
| RA patients | IFNα | ||
| (n = 120) | IFNlow (n = 80) | IFNhigh (n = 40) | |
| Sex (female/male) | 101/19 | 70/10 | 31/9 |
| Age at sampling, years | 55.33±15.23 | 55.24±15.04 | 55.56±16.00 |
| Age at diagnosis, years | 53.09±18.00 | 52.78±14.97 | 53.77±16.23 |
| Disease duration, months | 21.02±20.50 | 23.33±20.83 | 16.00±19.26 |
| Clinical features | |||
| Number of tender joints | 5.24±5.25 | 4.29±5.59 | 6.86±7.00 |
| Number of swollen joints | 2.50±3.31 | 1.83±3.15 | 3.64±3.38 |
| Patient global assessment (0–100) | 32.05±23.63 | 28.13±25.20 | 38.79±19.69 |
| Pain of patient’s assessment (0–10) | 3.21±2.42 | 2.71±2.40 | 4.07±2.30 |
| Duration of morning stiffness, min | 48.42±70.93 | 46.25±78.30 | 52.14±58.72 |
| DAS28 | 3.81±1.61 | 3.35±1.52 | 4.59±1.49* |
| HAQ | 0.77±0.70 | 0.68±0.70 | 0.95±0.67 |
| CRP, mg/dl | 0.39±0.63 | 0.28±0.45 | 0.59±0.83 |
| ESR, mm/h | 23.47±19.94 | 16.87±10.64 | 34.78±26.71* |
| RF positivity, n (%) | 68 (56.7) | 36 (45.0) | 32 (80.0)*** |
| Anti-CCP positivity, n (%) | 69 (57.5) | 36 (45.0) | 33 (82.5)*** |
| ANA positivity, n (%) | 46 (38.3) | 25 (31.2) | 21 (52.5)* |
| Smoking habit, n (%) | 49 (40.8) | 32 (40.0) | 17 (42.5) |
| Hypertension, n (%) | 30 (25.0) | 19 (23.7) | 11 (27.5) |
| Hypercholesterolemia, n (%) | 11 (9.1) | 8 (6.6) | 3 (2.5) |
| Diabetes mellitus, n (%) | 15 (12.5) | 8 (10.0) | 7 (17.5) |
| Treatments n (%) | |||
| None or NSAIDs | 19 (15.8) | 10 (10.1) | 9 (22.5) |
| Glucocorticoids | 58 (48.3) | 40 (50.0) | 18 (45.0) |
| Methotrexate | 79 (65.8) | 57 (71.3) | 22 (55.0) |
| Leflunomide | 16 (13.3) | 11 (13.7) | 5 (12.5) |
| TNF-α blockers | 26 (21.6) | 14 (17.5) | 12 (30.0) |
| Cardiovascular events, n(%) | |||
| Cardiovascular events | 27 (22.5) | 12 (15.0) | 15 (37.5)** |
| Ischemic heart disease | 10 (8.33) | 4 (5.0) | 6 (15.0) |
| Cerebrovascular accidents | 4 (3.33) | 2 (2.5) | 2 (5.0) |
| Heart failure | 12 (10.0) | 6 (7.5) | 6 (15.0) |
| Peripheral arteriopathy | 1 (0.83) | 0 (0.0) | 1 (2.5) |
Data of the whole RA patients group and classified according to IFNα serum levels. Data are expressed as (mean ± SD) unless otherwise was stated. Differences between categorical variables were evaluated by chi-square test, whereas Mann-Withney U test was used for continuous ones. *p<0.05, **p<0.01, ***p<0.001. IFNlow: serum levels <90th percentile in HC (4.092 pg/ml); IFNhigh: serum levels ≥90th percentile in HC.
On the other hand, IFNα serum levels were increased in RA patients compared with HC (20.25±47.61 vs. 1.76±3.08 pg/ml, p = 0.001), and positively correlated with all endothelial progenitor populations in patients (EPC: r = 0.294, p<0.001; mEPC r = 0.265, p<0.001, pre-EPC r = 0.367, p<0.001) but not in controls. Of note, this cytokine was associated with DAS-28 score (r = 0.253, p = 0.023) but unrelated to disease duration (r = 0.055, p = 0.547). In fact, ERA and LRA patients showed similar IFNα levels (14.43±31.16 vs. 22.74±53.10, p = 0.718).
In spite of the IFNα increase in RA, Figure 2 evidences that only a fraction of patients showed high levels of this molecule, whereas the other group presented low levels, similar to HC. Thus, we classified RA patients in IFNlow and IFNhigh using the HC 90th percentile (P90th = 4.092 pg/ml) as cut off. As shown in Table 1, these RA groups did not differ in age, disease duration or treatment followed, but IFNhigh patients (n = 40, 33%) exhibited higher disease activity (DAS28) and ESR as well as increased positivity for autoantibodies. Other clinical markers such as Tender Joint Count, Patient Global Assessment or CRP were slightly augmented.
Figure 2. IFNα serum levels are increased in a subgroup of RA patients.
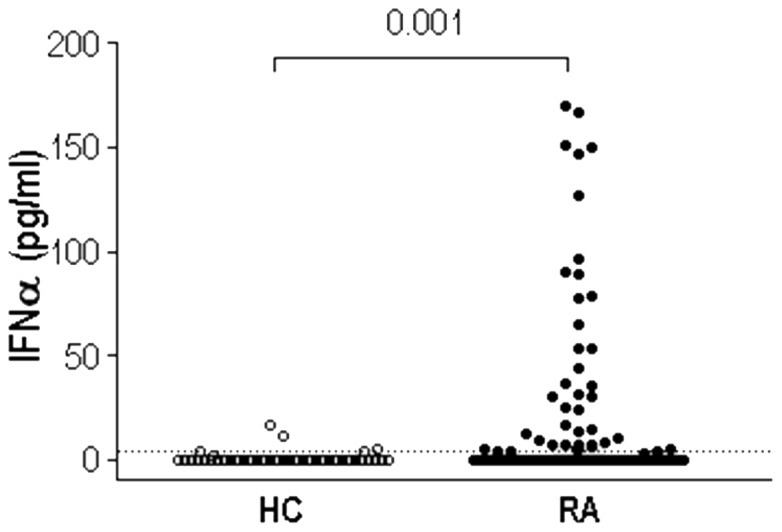
IFNα was quantified in 52 HC and 120 RA patients by CBA immunoassay. Dotted line represents HC 90th Percentile (4.092 pg/ml), used to classify RA patients in IFNlow (n = 80, 66%) or IFNhigh (n = 40, 33%). Differences were measured by Mann-Withney U-test.
EPC Populations Differ According to IFNα Levels
In view of these results, we analyzed EPC populations in RA patients according to IFNα levels and disease duration, using as controls healthy donors (HC) and patients with SLE, a disease presenting altered levels of IFNα and EPCs [24]. Figure 3 shows that among ERA patients, those with normal IFNα levels (IFNlow) displayed similar pre-EPC, EPC and mEPC counts to HC, whereas IFNhigh ERA patients exhibited higher levels of these populations compared with both HC and IFNlow, but similar to SLE patients. However, the most remarkable results were detected in LRA patients, since those with normal IFNα levels showed significantly lower pre-EPC and EPC counts than HC, thus highlighting a significant depletion that was missing in IFNhigh patients. Therefore, EPC depletion seems to be a characteristic of RA patients unless the presence of high IFNα levels hides this effect. In fact, no significant differences in EPC populations were present between SLE and IFNhigh RA patients, independently of disease duration. In any case, it is important to note that although IFNhigh RA patients displayed enhanced EPC populations, the mEPC/EPC ratio, indicative of the endothelial repair capability [5], [6], was increased in this group compared with their IFNlow counterparts (1.22(1.40) vs. 0.56(1.71), p = 0.013), thus suggesting an endothelial repair failure in these patients. Interestingly, no significant differences in mEPC/EPC ratio were found between ERA and LRA patients (p = 0.090), neither by treatments (all p>0.050), so there is no evidence that disease duration or longer exposure to treatment drugs could modify the mEPC/EPC ratio in RA patients.
Figure 3. Long-standing RA patients with low IFNα levels exhibited a depletion of all endothelial progenitor populations.

Pre-EPC, EPC and mEPC counts in early (ERA) and long-standing (LRA) Rheumatoid Arthritis patients were analyzed according to their IFNα serum levels. Healthy donors (HC) and SLE patients were included as both healthy and disease controls. Differences were assessed by Kruskal-Wallis test and Dunn’s multiple comparisons post hoc test. *p<0.05, **p<0.01.
IFNα is Associated with a Higher Rate of Cardiovascular Events
Taking into account the reported role of IFNα in endothelial damage and vascular repair, we aimed to evaluate the relevance of IFNα serum levels as a CV risk factor for RA patients. To this end, we analyzed the CV events suffered by RA patients in relation to both IFNα groups, demographic and clinical variables. The frequency of RA patients who had suffered CV events was higher in the IFNhigh than in the IFNlow group (37.5 vs 15.0%, p = 0.005), although no significant differences between groups were detected in traditional CV risk factors (Table 1), thus supporting the role of IFNα as an independent CV risk factor. Univariate logistic regression analysis (Table 2) revealed that high IFNα levels, male sex, age at diagnosis, hypertension and diabetes were associated with the risk of CV events. After multivariate analysis by logistic regression adjusted by age at diagnosis, sex, disease activity (DAS28) and traditional CV risk factors, only the association with IFNα and age at diagnosis remained significant.
Table 2. Association between presence of IFNα serum marker and CV events in RA patients.
| Cardiovascular events | Univariate Analysis | Multivariate Analysis* | ||||
| Absent (n = 93) | Present (n = 27) | OR [95% CI] | p | OR [95% CI] | p | |
| IFNα | ||||||
| IFNlow | 68 (73.1) | 12 (44.4) | 1 | 1 | ||
| IFNhigh | 25 (26.9) | 15 (55.6) | 3.400 [1.401–8.253] | 0.007 | 4.816 [1.254–18.488] | 0.022 |
| Sex | ||||||
| Women | 83 (89.2) | 18 (66.6) | 1 | |||
| Men | 10 (10.8) | 9 (33.6) | 4.150 [1.475–11.680] | 0.007 | ||
| Age at diagnosis | 51.00 (17.00) | 56.00 (23.00) | 1.050 [1.008–1.093] | 0.019 | 1.038 [1.015–1.084] | 0.021 |
| DAS28 score | 3.94 (2.66) | 3.26 (2.09) | 0.814 [0.580–1.143] | 0.235 | ||
| HTA | ||||||
| Normotensive | 72 (77.4) | 15 (55.5) | 1 | |||
| Hypertensive | 21 (22.6) | 12 (44.4) | 3.032 [1.218–7.547] | 0.017 | ||
| DM | ||||||
| Non-diabetic | 84 (91.3) | 20 (74.1) | 1 | |||
| Diabetic | 9 (8.7) | 7 (25.9) | 3.675 [1.192–11.326] | 0.023 | ||
| Smoking habit | ||||||
| Non-smoker | 50 (53.7) | 19 (70.3) | 1 | |||
| Smoker | 43 (46.3) | 8 (29.6) | 1.947 [0.773–4.904] | 0.157 | ||
| Hypercholesterolemia | ||||||
| Normocholesterolemic | 83 (89.2) | 26 (96.2) | 1 | |||
| Hypercholesterolemic | 10 (10.7) | 1 (3.70) | 0.311 [0.038–2.559] | 0.278 | ||
| RF | ||||||
| Negative | 34 (36.5) | 8 (30.8) | 1 | |||
| Positive | 60 (64.5) | 19 (69.2) | 1.530 [0.598–3.916] | 0.375 | ||
| Anti-CCP | ||||||
| Negative | 33 (35.4) | 7 (25.9) | 1 | |||
| Positive | 50 (53.7) | 20 (74.1) | 1.791 [0.678–4.734] | 0.415 | ||
| ANA | ||||||
| Negative | 56 (60.2) | 18 (66.6) | 1 | |||
| Positive | 37 (47.3) | 9 (33.3) | 0.730 [0.296–1.800] | 0.499 | ||
Associations were evaluated by logistic regression analysis using the presence of CV events (ischemic heart disease, n = 10; cerebrovascular accidents, n = 4; heart failure, n = 14; peripheral arteriopathy, n = 1) as dependent variable. Associations that reached statistic significance in multivariate analyses are highlighted in bold.
Multivariate analysis adjusted by sex, age at diagnosis, disease activity, smoking habits and presence or absence of HTA, DM and hypercholesterolemia. Accuracy of prediction of the final model was 76.7%.
On the other hand, patients who had suffered CV events showed an increased mEPC/EPC ratio compared to those who had not experienced such complications (1.27(3.21) vs. 1.00(2.04), p = 0.010), as well as lower VEGF levels (55.84(90.69) vs. 122.34(150.39) pg/ml, p = 0.044), thus supporting the relevance of angiogenic cytokines and EPC balance in the endothelial repair maintenance. All these results support that high IFNα serum levels in RA patients could be associated with a higher rate of CV events, maybe by increasing the mEPC/EPC ratio and impairing endothelial repair.
IFNlow/high Groups Differ in their Cytokine Profiles
Finally, to analyze whether IFNα serum marker may influence cytokine profiles in RA patients, we studied IL-1β, IL-6, IL-8, IL-10, MIP-1α, VEGF-A165, TNFα and TGF-β levels in patients and controls. The whole RA population was characterized by increased levels of IL-6 (1.05(3.41) vs. 0.32(1.15) pg/ml, p = 0.004), IL-8 (17.12(18.08) vs. 10.18(14.06) pg/ml, p = 0.008), IL-10 (0.37(0.68) vs. 0.10(0.18) pg/ml), and TNFα (5.76(4.01) vs. 3.26(1.93) pg/ml, p = 0.015), whereas TGF-β was decreased (14.47(4.60) vs. 19.42(6.71) ng/ml, p<0.001). Regarding to disease duration, we observed that LRA patients showed lower amounts of IL-1β (p = 0.002), IL-6 (p = 0.001) and IL-10 (p = 0.028) and slightly higher of TGF-β (p = 0.060) than ERA, whereas TNFα levels were strikingly higher in the LRA group (p<0.001). Restoration of cytokine levels in LRA could probably be due to a successful response to the therapy, since almost all LRA patients were under treatment while half of the ERA patients were untreated. In fact, striking differences were observed between treated and untreated patients in IL-1β (p<0.001), IL-6 (p = 0.002) and IL-10 (p = 0.004), but not in TNFα.
Interestingly, it is remarkable that IFNhigh patients showed a cytokine profile more similar to SLE patients than those IFNlow, except for IL-1β (Figure 4A). In fact, IFNhigh RA patients displayed higher levels of IL-1β (0.47(1.48) vs. 0.13(0.06) pg/ml, p<0.001), IL-6 (1.93(13.76) vs. 0.75(2.27) pg/ml, p = 0.004), IL-10 (0.65(1.15) vs. 0.26(0.46) pg/ml, p<0.001), MIP-1α (3.29(4.47) vs. 0.00(4.28) pg/ml, p = 0.001) and lower of TGF-β (12.86(4.01) vs. (14.98(5.45) ng/ml, p = 0.025) than IFNlow patients. In addition, although no association between IFNα and TNFα levels was detected in the whole RA group (r = −0.028, p = 0.766), a positive correlation was found in the IFNhigh group (r = 0.407, p = 0.011). All these results indicate that both IFNα and treatment influence cytokine levels. In fact, Figure 4B shows that treatments seem to restore IL-1β, IL-6, IL-8, IL-10 and TGF-β levels to a greater extent in IFNlow patients than in IFNhigh ones. Moreover, several clinical markers suggest a better outcome of IFNlow-treated patients than in their IFNhigh counterparts (Figure 4C), thus suggesting a potential IFNα role in therapy outcomes.
Figure 4. Cytokine profiles and disease features in RA patients are dependent on IFNα serum levels.

(A) Proinflammatory cytokines are increased in IFNhigh patients compared with both HC and IFNlow patients. (B) Serum cytokines levels are restored in IFNlow-treated patients but not in IFNhigh ones. (C) Improvement in clinical parameters in IFNlow-treated patients compared with IFNhigh group. Differences between groups were assessed by Mann-Withney U-test. n. t.: non-treated; t.: treated. *p<0.05, **p<0.01, ***p<0.001.
Finally, we analyzed the clinical response to anti-TNF therapy among a 6-month period, since it has been proposed a role of the IFN/TNF cross-regulation in the response to this treatment [26]–[27]. Of note, within the IFNlow group (n = 14), 35% of the patients (n = 5) reached a good response, and a similar percentage was found for those with a moderate response. In contrast, any patient of the IFNhigh group (n = 12) reached a good response, whereas only 41.6% (n = 5) fulfilled the criteria for a moderate one and more than a half (n = 7) exhibited no response to anti-TNF treatment.
Discussion
Recent evidence suggests a role of type I IFNs in vascular damage and EPC disbalance, mainly in SLE patients [15]–[18], [24], probably due to the central involvement of IFNα in the SLE pathogenesis [14], [28]–[30]. However, whether IFNα levels could play a major role in the clinical outcome and/or vascular damage in RA patients remains unknown.
Although most of the previous works reported an EPC depletion in RA patients that could be associated with disease activity [31]–[33], recent studies show contradictory results [34]–[36]. Our data may explain these conflicting data, since we demonstrated that only a group of RA patients exhibited a significant EPC depletion. We have previously confirmed that EPC population decreases with disease duration, whereas at disease onset it was similar to healthy subjects [24]. This finding is in line with previous studies where CV risk in RA patients has been reported to be associated with disease duration, probably due to disease-specific factors [37]. In addition, we reported for the first time, that EPC and pre-EPC populations were significantly reduced in patients with low IFNα serum levels, whereas higher levels of this cytokine were associated with higher counts of EPC populations, which leads to an increase in the mEPC/EPC ratio, in a similar way to the results observed in SLE [24]. Moreover, IFNhigh patients displayed higher disease activity and an elevated prevalence of autoantibodies, as was reported in IFNhigh SLE patients [38]. Thus, we think that IFNα serum levels could be an important bias in EPC studies in autoimmune diseases and it could be taken into account in future works.
Recent genomic studies have reported the presence of type I IFN signature in around 25–50% of RA patients [28]–[30], [39], which is according to our IFNhigh subset size (30%) and using similar criteria as cut off (90th percentile), but no correlations had been detected between IFN signature and clinical or immunological disease parameters. However, in this study, we showed that serum IFNα is correlated with clinical parameters, in the same way that has been previously reported in SLE patients [40], thus supporting the feasibility of IFNα serum marker in autoimmunity.
Although EPC depletion has been linked to higher rates of CV disease, our data show that IFNhigh patients, with increased EPC counts, exhibit a higher occurrence of CV events, thus highlighting the role of IFNα levels as an independent CV risk biomarker. In fact, these results are in accordance with the reported role of IFNα in vascular damage and EPC dysfunction [15], [17], [18], and with the increased mEPC/EPC ratio found in these patients, suggestive of an impaired EPC function. Actually, recent studies have linked endothelial repair failure in autoimmunity with the IFNα pathway, probably by altering the balance between endothelial cell apoptosis and vascular repair mediated by EPC [15]. This effect has been proposed to be mediated, at least in part, through VEGF repression in EPC [16]. Accordingly, IFN signature in EPC-treated cultures has been associated with impaired functionality and endothelial dysfunction [18]. Moreover, type I IFNs have been linked to atherosclerosis progression and vascular damage in both murine models [17] and human patients [41]–[43], thus proposing a type I IFN-mediated pathogenic role in CV disease in autoimmune patients. In addition, IFNα pharmacological treatment in non-RA subjects has been associated with CV disease [44], [45]. The increased rate of CV events in IFNhigh patients reported in our study support these findings. Accordingly, Somers et al. [42] reported that type I IFNs were independently associated with atherosclerotic development after adjusting for Framingham (traditional) risk factors. Similarly, in SLE, high disease activity is considered a better CV disease predictor than traditional risk factors [46].
In view of our results, we hypothesize that a potential mechanism by which IFNα could increase CV risk may be by promoting a premature EPC differentiation, generating mEPC (CD133−) with little or no vasculogenic and/or repair capability [6], probably similar to the “non-angiogenic phenotype” reported in murine SLE models [16], consequently resulting in a defective EPC-mediated endothelial repair. That is, although counterintuitive, higher EPC counts are not associated with cardioprotection, but endothelial repair failure, because of the high IFNα levels, which are causing a shift towards the mEPC phenotype. In addition, we showed that patients who had experienced CV events exhibited a higher mEPC/EPC ratio, thus linking IFNα, EPC maturation and impaired EPC functionality.
Another interesting finding was the differences in the cytokine patterns of RA patients, which seem to be related to treatment and IFNα levels. In fact, IFNhigh RA patients showed cytokine disturbances closer to SLE patients, characterized by a proinflammatory profile and higher IL-10 levels, which are associated with disease activity and poor prognosis markers, suggesting that this cytokine could be acting as a proinflammatory mediator in these conditions, as some authors have reported [47], [48]. Moreover, the IFNhigh group exhibited a positive correlation between IFNα and TNFα serum levels, similar to previously reported in SLE patients [49], [50]. Although Palucka et al. [26] have been proposed a negative cross-regulation between these cytokines, many other associations have been published thereafter, highlighting the relevance of the disease, the experimental model, the sample origin and the characteristics of the patients. It seems that, in some autoimmune disorders, the negative TNF/IFN cross-regulation loop is missed, leading to high serum levels of both cytokines in patients in which they may exert a pathological effect [39], [49]. Moreover, different associations of these two mediators have been reported even in a single disease [51], as seen in our study.Actually, we think that IFNhigh RA patients might display an impaired endothelial repair partly due to their proinflammatory cytokine network, mainly represented by higher serum levels of IL-1β and IL-6 (a Th17 inducer cytokines) and low TGF-β, compared to their IFNlow counterparts. In fact, reported in vitro experiments showed that proinflammatory conditions are enough to impair EPC functionality [33], [52], [53]. Moreover, Mälarstig et al. [13] have showed that raised IL-10 levels are associated with poor outcomes and enhanced systemic inflammation in acute coronary syndrome, supporting, at least in part, our findings.
Finally, differences in cytokine levels between treated and untreated patients among IFNlow and IFNhigh groups suggest that IFNα could be a predictive factor for treatment outcomes, being IFNhigh patients associated with a poor response. This was especially clear for anti-TNF therapy, since clinical response among the previous 6 months was higher in those patients within the IFNlow group. Similar conclusions were published by other authors [29], [54]. Therefore, this result makes us hypothesize that the IFNhigh group could benefit from an anti-IFNα therapy [55] rather than traditional DMARDs. However, relatively short follow-up period, differences in treatment duration and the low numbers of patients included do not lead us to achieve consistent conclusions in this issue.
Conclusions
In summary, we show that high IFNα serum levels could identify a group of RA patients with increased disease activity, EPC imbalance, enhanced proinflammatory profile and higher cardiovascular risk, probably due, at least in part, to an impaired endothelial repair. In addition, IFNα could be not only a marker of poor prognosis, but also of poor response to therapy, thus highlighting the relevance of this cytokine as a potential therapeutic target in RA.
Funding Statement
This work was supported by European Union FEDER funds and the Fondo de Investigación Sanitaria (FIS, PI08/0570 and PI12/0053). JRC is a recipient of a FPU grant from the Ministerio de Educación. CP was supported by a fellowship from FIS and BdP was supported by a fellowship from FICYT. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Avina-Zubieta JA, Thomas J, Sadatsafavi M, Lehman AJ, Lacaille D (2012) Risk of incident cardiovascular events in patients with rheumatoid arthritis: a meta-analysis of observational studies. Ann Rheum Dis. annrheumdis-2011-200726 [pii];10.1136/annrheumdis-2011-200726 [doi]. [DOI] [PubMed]
- 2. Wolfe F, Mitchell DM, Sibley JT, Fries JF, Bloch DA, et al. (1994) The mortality of rheumatoid arthritis. Arthritis Rheum 37: 481–494. [DOI] [PubMed] [Google Scholar]
- 3.Hansel S, Lassig G, Pistrosch F, Passauer J (2003) Endothelial dysfunction in young patients with long-term rheumatoid arthritis and low disease activity. Atherosclerosis 170: 177–180. S0021915003002818 [pii]. [DOI] [PubMed]
- 4. Pompilio G, Capogrossi MC, Pesce M, Alamanni F, DiCampli C, et al. (2009) Endothelial progenitor cells and cardiovascular homeostasis: clinical implications. Int J Cardiol 131: 156–167 S0167-5273(08)00949-2 [pii];10.1016/j.ijcard.2008.08.033 [doi]. [DOI] [PubMed] [Google Scholar]
- 5. Gehling UM, Ergun S, Schumacher U, Wagener C, Pantel K, et al. (2000) In vitro differentiation of endothelial cells from AC133-positive progenitor cells. Blood 95: 3106–3112. [PubMed] [Google Scholar]
- 6. Peichev M, Naiyer AJ, Pereira D, Zhu Z, Lane WJ, et al. (2000) Expression of VEGFR-2 and AC133 by circulating human CD34(+) cells identifies a population of functional endothelial precursors. Blood 95: 952–958. [PubMed] [Google Scholar]
- 7. Friedrich EB, Walenta K, Scharlau J, Nickenig G, Werner N (2006) CD34−/CD133+/VEGFR-2+ endothelial progenitor cell subpopulation with potent vasoregenerative capacities. Circ Res 98: e20–e25 01.RES.0000205765.28940.93 [pii];10.1161/01.RES.0000205765.28940.93 [doi]. [DOI] [PubMed] [Google Scholar]
- 8. Hill JM, Zalos G, Halcox JP, Schenke WH, Waclawiw MA, et al. (2003) Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med 348: 593–600 10.1056/NEJMoa022287 [doi];348/7/593 [pii]. [DOI] [PubMed] [Google Scholar]
- 9. Werner N, Kosiol S, Schiegl T, Ahlers P, Walenta K, et al. (2005) Circulating endothelial progenitor cells and cardiovascular outcomes. N Engl J Med 353: 999–1007 353/10/999 [pii];10.1056/NEJMoa043814 [doi]. [DOI] [PubMed] [Google Scholar]
- 10. Full LE, Ruisanchez C, Monaco C (2009) The inextricable link between atherosclerosis and prototypical inflammatory diseases rheumatoid arthritis and systemic lupus erythematosus. Arthritis Res Ther 11: 217 ar2631 [pii];10.1186/ar2631 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sattar N, McCarey DW, Capell H, McInnes IB (2003) Explaining how “high-grade” systemic inflammation accelerates vascular risk in rheumatoid arthritis. Circulation 108: 2957–2963 10.1161/01.CIR.0000099844.31524.05 [doi];108/24/2957 [pii]. [DOI] [PubMed] [Google Scholar]
- 12. Dessein PH, Joffe BI, Singh S (2005) Biomarkers of endothelial dysfunction, cardiovascular risk factors and atherosclerosis in rheumatoid arthritis. Arthritis Res Ther 7: R634–R643 ar1717 [pii];10.1186/ar1717 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Malarstig A, Eriksson P, Hamsten A, Lindahl B, Wallentin L, et al. (2008) Raised interleukin-10 is an indicator of poor outcome and enhanced systemic inflammation in patients with acute coronary syndrome. Heart 94: 724–729 hrt.2007.119271 [pii];10.1136/hrt.2007.119271 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ronnblom L (2011) The type I interferon system in the etiopathogenesis of autoimmune diseases. Ups J Med Sci 116: 227–237 10.3109/03009734.2011.624649 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Denny MF, Thacker S, Mehta H, Somers EC, Dodick T, et al. (2007) Interferon-alpha promotes abnormal vasculogenesis in lupus: a potential pathway for premature atherosclerosis. Blood 110: 2907–2915 blood-2007-05-089086 [pii];10.1182/blood-2007-05-089086 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Thacker SG, Berthier CC, Mattinzoli D, Rastaldi MP, Kretzler M, et al. (2010) The detrimental effects of IFN-alpha on vasculogenesis in lupus are mediated by repression of IL-1 pathways: potential role in atherogenesis and renal vascular rarefaction. J Immunol 185: 4457–4469 jimmunol.1001782 [pii];10.4049/jimmunol.1001782 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Thacker SG, Zhao W, Smith CK, Luo W, Wang H, et al. (2012) Type I interferons modulate vascular function, repair, thrombosis, and plaque progression in murine models of lupus and atherosclerosis. Arthritis Rheum 64: 2975–2985 10.1002/art.34504 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lee PY, Li Y, Richards HB, Chan FS, Zhuang H, et al. (2007) Type I interferon as a novel risk factor for endothelial progenitor cell depletion and endothelial dysfunction in systemic lupus erythematosus. Arthritis Rheum 56: 3759–3769 10.1002/art.23035 [doi]. [DOI] [PubMed] [Google Scholar]
- 19. Kaplan MJ, Salmon JE (2011) How does interferon-alpha insult the vasculature? Let me count the ways. Arthritis Rheum 63: 334–336 10.1002/art.30161 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tan FK, Zhou X, Mayes MD, Gourh P, Guo X, et al. (2006) Signatures of differentially regulated interferon gene expression and vasculotrophism in the peripheral blood cells of systemic sclerosis patients. Rheumatology (Oxford) 45: 694–702 kei244 [pii];10.1093/rheumatology/kei244 [doi]. [DOI] [PubMed] [Google Scholar]
- 21. van Gestel AM, Anderson JJ, van Riel PL, Boers M, Haagsma CJ, et al. (1999) ACR and EULAR improvement criteria have comparable validity in rheumatoid arthritis trials. American College of Rheumatology European League of Associations for Rheumatology. J Rheumatol 26: 705–711. [PubMed] [Google Scholar]
- 22. Ho KK, Pinsky JL, Kannel WB, Levy D (1993) The epidemiology of heart failure: the Framingham Study. J Am Coll Cardiol 22: 6A–13A. [DOI] [PubMed] [Google Scholar]
- 23. Rodriguez-Rodriguez L, Gonzalez-Juanatey C, Palomino-Morales R, Vazquez-Rodriguez TR, Miranda-Filloy JA, et al. (2011) TNFA -308 (rs1800629) polymorphism is associated with a higher risk of cardiovascular disease in patients with rheumatoid arthritis. Atherosclerosis 216: 125–130 S0021-9150(11)00192-4 [pii];10.1016/j.atherosclerosis.2010.10.052 [doi]. [DOI] [PubMed] [Google Scholar]
- 24. Rodriguez-Carrio J, Prado C, de PB, Lopez P, Gomez J, et al. (2012) Circulating endothelial cells and their progenitors in systemic lupus erythematosus and early rheumatoid arthritis patients. Rheumatology (Oxford) 51: 1775–1784 kes152 [pii];10.1093/rheumatology/kes152 [doi]. [DOI] [PubMed] [Google Scholar]
- 25. Distler JH, Allanore Y, Avouac J, Giacomelli R, Guiducci S, et al. (2009) EULAR Scleroderma Trials and Research group statement and recommendations on endothelial precursor cells. Ann Rheum Dis 68: 163–168 ard.2008.091918 [pii];10.1136/ard.2008.091918 [doi]. [DOI] [PubMed] [Google Scholar]
- 26. Palucka AK, Blanck JP, Bennett L, Pascual V, Banchereau J (2005) Cross-regulation of TNF and IFN-alpha in autoimmune diseases. Proc Natl Acad Sci U S A 102: 3372–3377 0408506102 [pii];10.1073/pnas.0408506102 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cantaert T, Baeten D, Tak PP, van Baarsen LG (2010) Type I IFN and TNFalpha cross-regulation in immune-mediated inflammatory disease: basic concepts and clinical relevance. Arthritis Res Ther 12: 219 ar3150 [pii];10.1186/ar3150 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Higgs BW, Liu Z, White B, Zhu W, White WI, et al. (2011) Patients with systemic lupus erythematosus, myositis, rheumatoid arthritis and scleroderma share activation of a common type I interferon pathway. Ann Rheum Dis 70: 2029–2036 ard.2011.150326 [pii];10.1136/ard.2011.150326 [doi]. [DOI] [PubMed] [Google Scholar]
- 29. Reynier F, Petit F, Paye M, Turrel-Davin F, Imbert PE, et al. (2011) Importance of correlation between gene expression levels: application to the type I interferon signature in rheumatoid arthritis. PLoS One 6: e24828 10.1371/journal.pone.0024828 [doi];PONE-D-11-05657 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. van der Pouw Kraan TC, Wijbrandts CA, van Baarsen LG, Voskuyl AE, Rustenburg F, et al. (2007) Rheumatoid arthritis subtypes identified by genomic profiling of peripheral blood cells: assignment of a type I interferon signature in a subpopulation of patients. Ann Rheum Dis 66: 1008–1014 ard.2006.063412 [pii];10.1136/ard.2006.063412 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Egan CG, Caporali F, Garcia-Gonzalez E, Galeazzi M, Sorrentino V (2008) Endothelial progenitor cells and colony-forming units in rheumatoid arthritis: association with clinical characteristics. Rheumatology (Oxford) 47: 1484–1488 ken299 [pii];10.1093/rheumatology/ken299 [doi]. [DOI] [PubMed] [Google Scholar]
- 32. Grisar J, Aletaha D, Steiner CW, Kapral T, Steiner S, et al. (2005) Depletion of endothelial progenitor cells in the peripheral blood of patients with rheumatoid arthritis. Circulation 111: 204–211 01.CIR.0000151875.21836.AE [pii];10.1161/01.CIR.0000151875.21836.AE [doi]. [DOI] [PubMed] [Google Scholar]
- 33. Herbrig K, Haensel S, Oelschlaegel U, Pistrosch F, Foerster S, et al. (2006) Endothelial dysfunction in patients with rheumatoid arthritis is associated with a reduced number and impaired function of endothelial progenitor cells. Ann Rheum Dis 65: 157–163 ard.2005.035378 [pii];10.1136/ard.2005.035378 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Allanore Y, Batteux F, Avouac J, Assous N, Weill B, et al.. (2007) Levels of circulating endothelial progenitor cells in systemic sclerosis. Clin Exp Rheumatol 25: 60–66. 1982 [pii]. [PubMed]
- 35. Jodon dV, V, Avouac J, Ponceau A, Ruiz B, Kahan A, et al. (2010) Enhanced late-outgrowth circulating endothelial progenitor cell levels in rheumatoid arthritis and correlation with disease activity. Arthritis Res Ther 12: R27 ar2934 [pii];10.1186/ar2934 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yiu KH, Wang S, Mok MY, Ooi GC, Khong PL, et al. (2010) Role of circulating endothelial progenitor cells in patients with rheumatoid arthritis with coronary calcification. J Rheumatol 37: 529–535 jrheum.090782 [pii];10.3899/jrheum.090782 [doi]. [DOI] [PubMed] [Google Scholar]
- 37. Kerola AM, Kauppi MJ, Kerola T, Nieminen TV (2012) How early in the course of rheumatoid arthritis does the excess cardiovascular risk appear? Ann Rheum Dis 71: 1606–1615 annrheumdis-2012-201334 [pii];10.1136/annrheumdis-2012-201334 [doi]. [DOI] [PubMed] [Google Scholar]
- 38. Kirou KA, Lee C, George S, Louca K, Peterson MG, et al. (2005) Activation of the interferon-alpha pathway identifies a subgroup of systemic lupus erythematosus patients with distinct serologic features and active disease. Arthritis Rheum 52: 1491–1503 10.1002/art.21031 [doi]. [DOI] [PubMed] [Google Scholar]
- 39. Cantaert T, van Baarsen LG, Wijbrandts CA, Thurlings RM, van de Sande MG, et al. (2010) Type I interferons have no major influence on humoral autoimmunity in rheumatoid arthritis. Rheumatology (Oxford) 49: 156–166 kep345 [pii];10.1093/rheumatology/kep345 [doi]. [DOI] [PubMed] [Google Scholar]
- 40. Bauer JW, Baechler EC, Petri M, Batliwalla FM, Crawford D, et al. (2006) Elevated serum levels of interferon-regulated chemokines are biomarkers for active human systemic lupus erythematosus. PLoS Med 3: e491 05-PLME-RA-0506R3 [pii];10.1371/journal.pmed.0030491 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kirou KA, Cole P, Salmon JE, Roman MJ, Crow MK (2013) Identification of molecular pathways associated with progression of carotid atherosclerosis in systemic lupus erythematosus [abstract]. Arthritis Rheum 54 suppl:S807 [Google Scholar]
- 42. Somers EC, Zhao W, Lewis EE, Wang L, Wing JJ, et al. (2012) Type I interferons are associated with subclinical markers of cardiovascular disease in a cohort of systemic lupus erythematosus patients. PLoS One 7: e37000 10.1371/journal.pone.0037000 [doi];PONE-D-12-02237 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhao W, Somers EC, McCune WJ, Kaplan MJ (2009) Type I Interferon gene signatures are associated with vascular risk and atherosclerosis in systemic lupus erythematosus [abstract]. Arthritis Rheum 60 suppl 10582. [Google Scholar]
- 44. Kobayashi T, Sato Y, Hasegawa Y, Nagasawa T, Yoda Y, et al. (1992) Multiple myeloma complicated by congestive heart failure following first administration of recombinant alpha-interferon. Intern Med 31: 936–940. [DOI] [PubMed] [Google Scholar]
- 45. Kuwata A, Ohashi M, Sugiyama M, Ueda R, Dohi Y (2002) A case of reversible dilated cardiomyopathy after alpha-interferon therapy in a patient with renal cell carcinoma. Am J Med Sci 324: 331–334. [DOI] [PubMed] [Google Scholar]
- 46. Esdaile JM, Abrahamowicz M, Grodzicky T, Li Y, Panaritis C, et al. (2001) Traditional Framingham risk factors fail to fully account for accelerated atherosclerosis in systemic lupus erythematosus. Arthritis Rheum 44: 2331–2337. [DOI] [PubMed] [Google Scholar]
- 47. Cush JJ, Splawski JB, Thomas R, McFarlin JE, Schulze-Koops H, et al. (1995) Elevated interleukin-10 levels in patients with rheumatoid arthritis. Arthritis Rheum 38: 96–104. [DOI] [PubMed] [Google Scholar]
- 48. Lacki JK, Samborski W, Mackiewicz SH (1997) Interleukin-10 and interleukin-6 in lupus erythematosus and rheumatoid arthritis, correlations with acute phase proteins. Clin Rheumatol 16: 275–278. [DOI] [PubMed] [Google Scholar]
- 49.Lopez P, Gomez J, Prado C, Gutierrez C, Suarez A (2008) Influence of functional interleukin 10/tumor necrosis factor-alpha polymorphisms on interferon-alpha, IL-10, and regulatory T cell population in patients with systemic lupus erythematosus receiving antimalarial treatment. J Rheumatol 35: 1559–1566. 08/13/0621 [pii]. [PubMed]
- 50. Weckerle CE, Mangale D, Franek BS, Kelly JA, Kumabe M, et al. (2012) Large-scale analysis of tumor necrosis factor alpha levels in systemic lupus erythematosus. Arthritis Rheum 64: 2947–2952 10.1002/art.34483 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kariuki SN, Crow MK, Niewold TB (2008) The PTPN22 C1858T polymorphism is associated with skewing of cytokine profiles toward high interferon-alpha activity and low tumor necrosis factor alpha levels in patients with lupus. Arthritis Rheum 58: 2818–2823 10.1002/art.23728 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Deng XL, Li XX, Liu XY, Sun L, Liu R (2010) Comparative study on circulating endothelial progenitor cells in systemic lupus erythematosus patients at active stage. Rheumatol Int 30: 1429–1436 10.1007/s00296-009-1156-4 [doi]. [DOI] [PubMed] [Google Scholar]
- 53. Grisar J, Steiner CW, Bonelli M, Karonitsch T, Schwarzinger I, et al. (2008) Systemic lupus erythematosus patients exhibit functional deficiencies of endothelial progenitor cells. Rheumatology (Oxford) 47: 1476–1483 ken286 [pii];10.1093/rheumatology/ken286 [doi]. [DOI] [PubMed] [Google Scholar]
- 54. van Baarsen LG, Wijbrandts CA, Rustenburg F, Cantaert T, van der Pouw Kraan TC, et al. (2010) Regulation of IFN response gene activity during infliximab treatment in rheumatoid arthritis is associated with clinical response to treatment. Arthritis Res Ther 12: R11 ar2912 [pii];10.1186/ar2912 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yao Y, Richman L, Higgs BW, Morehouse CA, de los RM, et al. (2009) Neutralization of interferon-alpha/beta-inducible genes and downstream effect in a phase I trial of an anti-interferon-alpha monoclonal antibody in systemic lupus erythematosus. Arthritis Rheum 60: 1785–1796 10.1002/art.24557 [doi]. [DOI] [PubMed] [Google Scholar]