

Abstract
Parkinson's disease (PD) and related Lewy body diseases are characterized by deposition of α-synuclein aggregates in both the central nervous system and peripheral nervous system. Synucleinopathy lesions spread to larger brain areas as the disease progresses, and prion-like cell-to-cell transmission of aggregated α-synuclein is thought to be the underlying mechanism for this pathological spreading. LRRK2 is another protein linked to the pathogenesis of PD, and its presence in Lewy bodies has attracted much attention as to whether LRRK2 and α-synuclein interplay during the pathogenesis of PD. However, the relationship between these two crucial proteins still remains unclear. In this review article, we will discuss the current state of knowledge in terms of how these proteins cause the disease and provide the hypothetical mechanisms by which LRRK2 might modify the generation and progression of synucleinopathy.
Keywords: Parkinson's disease, LRRK2, alpha-synuclein, synucleinopathy, transmission, neurodegeneration
INTRODUCTION
Parkinson's disease (PD) is one of the most common neurode-generative diseases, with clinical symptoms of resting tremor, increased muscle tone, bradykinesia, and abnormal postural righting reflexes [1]. Pathologically, PD is characterized by loss of dopaminergic neurons in the Substantia nigra pars compacta, deposition of α-synuclein visualized in the forms of Lewy bodies and Lewy neurites, and neuroinflammation demonstrated by the activation of microglia [2]. Lewy body inclusions are primarily composed of misfolded/aggregated α-synuclein [3], and these pathological protein aggregates spread to different brain regions in a highly sequential way as the disease progresses [4]. Therefore, identifying factors that contribute to the formation and spreading of α-synuclein aggregates may be critical to the overall understanding of pathogenesis of PD.
α-Synuclein (SNCA) and LRRK2 genes harbor autosomal dominant mutations that cause familial PD with clinical characteristics similar to sporadic PD [5, 6]. Although some have argued that each factor operates independently [7], there might be interactions between the two genes in pathogenesis of PD. Here, we review the recent literature suggesting cooperation between α-synuclein and LRRK2 and propose potential mechanisms underlying this interaction.
GENETIC AND PATHOLOGICAL LINK OF LRRK2 AND α-SYNUCLEIN TO PARKINSON'S DISEASE
In 1996, researchers discovered genetic linkage to chromosome 4 markers in a large group of Greek/Italian descendants [8]. In this kindred and later in several others, SNCA gene (PARK1) displayed a missense mutation A53T [8] and other missense mutations A30P and E46K [9, 10] with largely similar phenotypes but seemingly worse presentation of dementia for E46K [10], as well as duplications [11] and triplications with varying penetrance [6, 12]. Several years after the first report of SNCA mutation, Funayama and colleagues found genetic linkage to chromosome 12 markers in a Japanese family with autosomal dominant parkinsonism [13]. And in 2004, the mutations were located in LRRK2 (PARK8) in chromosome 12 as a cause of PD and also detected in a number of other kindreds [14, 15]. Among the autosomal dominant mutations in LRRK2, G2019S is the most common mutation throughout ethnic groups [16], and is found most frequently in North-African Arabs and Ashkenazi Jews [5, 17].
Autosomal recessive causes of PD have also been linked to several genes: parkin (PARK2) [18], DJ-1 (PARK6) [19, 20], PINK1 (PARK7) [21], and ATP13A2 (PARK9) [22].
Recently, the genome-wide association studies have identified both SNCA and LRRK2 as strong genetic risk loci for sporadic PD, suggesting the roles of these two genes in the pathogenesis of idiopathic PD [6, 23-25].
PD patients with LRRK2 mutations have somewhat variable pathology. Most of these cases showed the typical Lewy body pathology, however, others showed a mixture of Lewy bodies and tau inclusions, and sometimes tau inclusions alone [26]. The fact that LRRK2 mutations are associated with both α-synuclein and tau pathologies raises the possibility of LRRK2 acting upstream of α-synuclein and tau aggregation.
PATHOGENIC MECHANISM OF α-SYNUCLEIN
α-synuclein is the primary component of the Lewy bodies and Lewy neurites [27], and is normally present in the presynaptic terminals [28, 29]. At the presynaptic terminal, α-synuclein promotes the formation of the SNARE complex, regulating the neurotransmitter release [30]. α-synuclein is also known to recognize defective bilayer membranes [31] and produce membrane curvature [32]. These abilities may be related to its function in vesicle dynamics and trafficking. It is still not clear whether the normal function of α-synuclein has relevant roles in PD pathogenesis.
Recombinant α-synuclein protein can generate amyloid fibrils that are indistinguishable to the ones found in Lewy bodies [33]. All the known mutations in SNCA linked to familial PD have common molecular phenotype of accelerated aggregation of α-synuclein (reviewed in [34]). Overproduction of α-synuclein in various animal models, including mice, rats, non-human primates, flies, and nematodes, resulted in aggregation of this protein followed by cell death [35, 36]. Reduction of α-synuclein aggregates by using pharmacological agents or overexpression of aggregation inhibitors, such as α-synuclein, prevented neuronal death and behavioral deficits caused by α-synuclein [37, 38]. Collectively, these findings strongly suggest that aggregation is necessary for the mechanism by which α-synuclein exerts its pathogenic actions.
Several mechanisms and cellular targets of α-synuclein have been suggested. These include vesicle trafficking [39], microtubule-dependent transport [40], Golgi fragmentation [41], synaptic vesicle cycling [42], autophagy [43], proteasomal activity [44], and mitochondrial function [45]. It is likely that α-synuclein aggregates have multiple targets, thereby leading to various types of impairment in cellular functions.
Spreading of α-synuclein aggregates
Lewy body pathology throughout the nervous system, both the CNS and PNS, and spreads within the CNS with a very specific topographical sequence during PD progression. Braak and colleagues suggested that Lewy body and Lewy neurite pathology first appears in the olfactory bulb and the dorsal motor nucleus of vagus, followed by the propagation through the mid brain and then to the mesocortex and the neocortex. This pathological propagation may explain the symptomatic progression of the disease. Therefore, although not all patients follow the same pattern of pathological propagation due to heterogeneity of the disease, it is important to note that protein aggregate pathology can spread to larger brain regions as the disease progresses, and understanding the mechanism of the aggregate propagation would provide critical insights into how disease progression occurs. Spreading of α-synuclein aggregates in human patients has been supported by recent observations that PD patients who had received transplantation of embryonic mesencephalic tissues displayed α-synuclein-positive Lewy bodies and Lewy neurites in grafted neurons [46, 47].
Mechanism of aggregate spreading
There is an increasing body of evidence that cell-to-cell transmission of α-synuclein aggregates is the underlying mechanism for spreading of Lewy pathology [48]. Our previous studies suggested that α-synuclein aggregates are released from neurons through the unconventional exocytosis [49]. Some suggested exosome-associated secretion [50], and others proposed exophagy of α-synuclein, a mechanism involves fusion of the autophagosomes and the plasma membrane [51]. Secretion of α-synuclein appears to be an ongoing process even in healthy neurons [49], however, when neurons were under stress conditions, such as oxidative stress and failure in protein quality control systems, vesicle translocation and secretion of α-synuclein were increased [49, 50, 52, 53].
Secreted α-synuclein aggregates are transferred to neighboring neurons through endocytosis [48, 54]. Endocytosed aggregates undergo trafficking through the endocytic pathway and were delivered to the lysosomes and degraded [55]. Impaired lysosomal function caused accumulation of internalized exogenous α-synuclein, thereby promoting cell-to-cell transmission [48]. Therefore, efficient trafficking and clearance of exogenous α-synuclein aggregates would prevent seeded aggregation of the endogenous α-synuclein. The mechanism of cell-to-cell transmission of α-synuclein aggregates and how the transferred seed of aggregates are cleared have become critical questions in understanding the mode of disease progression.
PATHOGENIC MECHANISM OF LRRK2
LRRK2 is a large, multi-domain GTPase/kinase protein [56], characterized by the presence of a ROC domain, a COR domain, and a MAPKKK domain [57-59]. These domains are known to control several cellular functions (Fig. 1) such as proliferation, differentiation, and survival [60]. Importantly, LRRK2 has been shown to play a significant role in regulating neural functions [61]. Mammalian LRRK2 has been discovered to control neurite outgrowth; mutant LRRK2 expression in neurons inhibited neurite outgrowth while knockdown of LRRK2 promotes it [61]. This has been further demonstrated in C. elegans model where over-expressed WT LRRK2, R1441C, and G2019S mutants displayed neurite impairment and loss of dopaminergic neurons with more severe loss of DA neurons in R1441C and G2019S mutant expressors than the WT expressor [62]. Over-expression of WT LRRK2, R1441C, and G2019S mutants also led to age-dependent behavioral deficits. Interestingly, LRRK2 has been observed not only in brain cells, but also in other peripheral organs such as kidney, lung, and spleen [63]. And since LRRK2 expression was notably high in immune cells like macrophagy and monocytes, it is suggested that LRRK2 might play a role in immune functions [64].
Fig. 1.
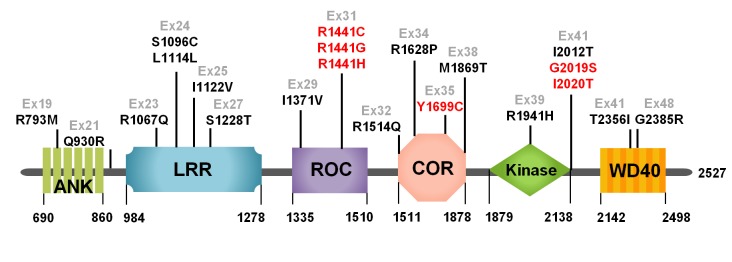
Domain structures of LRRK2 protein. ANK (ankyrin repeats), LRR (Leucine-rich repeats), ROC (Ras of Complex proteins), COR (C-terminal of ROC), Ex (Exon).
Role of kinase activity
LRRK2 protein contains a kinase and a GTPase domain and these two domains are known to harbor most of PD-linked mutations. Increased kinase activity associated with several mutations, such as G2019S, I2012T, R1441G/C, and Y1699C, has been shown to be responsible for increased neurotoxicity [65,66]. G2019S is the most common mutation of LRRK2 and exerts its pathogenic action by toxic gain-of-function through increased kinase activity [67].
Recent findings suggested that the major pathology associated with LRRK2 mutations was Lewy bodies, although some cases showed mixture of Lewy body and tau pathology, and rarely tau-only pathology [68,69]. However, the relationship between LRRK2 mutations and α-synuclein aggregation is not clearly understood. For example, it is not yet clear if LRRK2 directly phosphorylates SNCA [70]. One report showed that LRRK2 G2019S mutation may promote α-synuclein phosphorylation at S129 and increase α-synuclein aggregation [71]. However, other studies failed to show the relationship between LRRK2 G2019S mutation and increased levels of α-synuclein phosphorylation [72].
Some studies suggested that GTP binding in LRRK2 promoted the kinase activity [73, 74], and GTP-binding domain might be important for cytotoxicity of LRRK2 [75]. However, it is not quite clear how GTP binding and GTP hydrolysis regulate the kinase activity of LRRK2 and cause cytotoxicity.
Vesicle trafficking
Vesicle trafficking is among a number of pathways affected by LRRK2-dependent neuronal damage. Rab5b, one of the proteins known to interact with LRRK2 in regulating vesicle transport, resides in several different vesicle compartments and participates in membrane trafficking [76, 77]. LRRK2 has been shown to interact with Rab5b, and defects in synaptic vesicle endocytosis caused by either overexpression or knockdown of LRRK2 expression were rescued by expression of Rab5b in primary neurons [78]. Synaptic dysfunction is an early phenomenon presented in the pathogenesis of PD, and it was also reported that LRRK2 controls synaptic morphogenesis in Drosophila model [79]. Reduced LRRK2 expression led to synaptic overgrowth, while overexpression of WT LRRK2 resulted in synaptic depletion. Another study showed that LRRK2 had a significant effect on synaptic vesicle endocytosis by enhancing Endophilin A phosphorylation at S75 [80]. Perhaps, the most strong evidence for the involvement of LRRK2 in vesicle trafficking was provided by the study of MacLeod et al. [61]. This study showed that LRRK2 interacted with Rab7L1 (PARK16), and Rab7L deficiency resulted in neurodegeneration similar to the phenotype of LRRK2 mutant expression, whereas LRRK2-induced neurodegeneration was rescued by expression of Rab7L1. This study also showed defects of endolysosomal and Golgi-associated sorting and VPS35 defects in retromer complex by PD-associated defects in LRRK2 and Rab7L1. These defects were rescued by expression of WT VPS35. Therefore, LRRK2 collaborates with Rab7L1 and VPS35 in the endolysosomal pathway and the trafficking pathway that utilizes retromer complex, and defects in this system increase PD risk.
EVIDENCE FOR GENE-GENE INTERACTION BETWEEN LRRK2 AND α-SYNUCLEIN
Using tetracycline-inducible transgenic mice, Lin et al. [81] reported that overexpression of WT LRRK2 in A53T transgenic mice caused more rapid and robust neuropathological changes, including neurodegeneration, somatic α-synuclein accumulation, astrogliosis, and microglial activation, than the A53T single transgenic mice. The deteriorating effects of LRRK2 were expression level-dependent, and expression of the PD-linked G2019S LRRK2 resulted in even more severe neuropathology that of WT LRRK2 in the A53T mice. In contrast, neuropathological changes produced by transgenic expression of A53T α-synuclein were significantly reduced in LRRK2 knockout mice. This study clearly showed the pathogenic interplay between LRRK2 and α-synuclein. Since the mice expressing LRRK2 G2019S alone did not display neuropathological changes, LRRK2 may indirectly contribute to the disease progression. However, the role of LRRK2 in synucleinopathy has been challenged by other studies. These studies failed to show changes in α-synuclein and glial pathology, neurodegeneration, behavioral deficits, and timing of premature death in LRRK2 G2019S/α-synuclein A53T double transgenic mice, compared to the A53T single transgenic mice, casting a doubt in the role of LRRK2 mutations in α-synuclein-driven pathogenesis [7, 82].
IS LRRK2 A GENETIC MODIFIER OF SYNUCLEINOPATHIES?
Although mouse studies have produced mixed results, human pathology still suggests the role, albeit indirect, of LRRK2 in synucleinopathy. In this section, we propose a number of possible mechanisms by which LRRK2 plays roles in modulating synucleinopathy (Fig. 2).
Fig. 2.

Hypothetical mechanisms by which LRRK2 regulates α-synuclein aggregation. LRRK2 gene induction causes microglia activation to initiate inflammatory responses, which might enhance the efficiency of α-synuclein cell-to-cell transmission. LRRK2 in neurons might regulate endocytosis, lysosomal clearance, seeding, and exocytosis of extracellular α-synuclein (see text for details).
First, LRRK2 mutations may cause defects in protein degradation system, thereby leading to accumulation of α-synuclein. Homozygous deletion of LRRK2 gene resulted in reduced autophagy and age-dependent accumulation of α-synuclein and ubiquitinated proteins in mice [83]. LRRK2 WT and mutant proteins may also cause deficits in the degradation of α-synuclein by interfering with the chaperone-mediated autophagy [84].
Second, mutant LRRK2 expression caused proinflammatory responses from microglia [85]. Inhibition of LRRK2 kinase activity and knockdown of its expression resulted in attenuation of toll-like receptor 4-mediated microglia activation [86]. Considering that α-synuclein aggregation is sensitive to inflammatory environment [87], LRRK2 may modify synucleinopathy by altering inflammatory microenvironment surrounding neurons.
Finally, the most exciting possibility is the potential role of LRRK2 in cell-to-cell transmission of synucleinopathy. Several studies reported the role of LRRK2 in vesicle transport and autophagy (see above), which may be intimately related with the trafficking pathways of α-synuclein at multiple sites during cell-to-cell transmission. Secretion of α-synuclein is mediated by unconventional exocytosis, one possible mechanism is exophagy. LRRK2 might control α-synuclein secretion by modulating the autophagy pathway. After the transfer to the recipient cells, α-synuclein is transported through the endolysosomal pathway, where LRRK2 may control the rate of degradation of transferred α-synuclein by modulating the trafficking pathways. By either promoting or inhibiting the encounter of the exogenous and endogenous α-synuclein proteins within the endosomal system, LRRK2 may also regulate seeded aggregation of α-synuclein. Subsequent secretion of the seeded aggregates may be another site of control by LRRK2.
The possible roles of LRRK2 in development and progression of synucleinopathy described here are only speculations. However, investigations into these hypothetical mechanisms are likely to provide answers as to whether these two genes indeed interplay in generating synucleinopathy and potential targets for therapy for Lewy body diseases.
ACKNOWLEDGEMENTS
This work was supported by a National Research Foundation (NRF) grant, funded by the Korean Government (MEST) (No. 2010-0015188, to SJL), by the Korea Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (A111228, to SJL), and by grants from the National Research Foundation of Korea funded by the Korean Government (NRF-2012R1A1A2040840, to HJL).
References
- 1.Hoehn MM, Yahr MD. Parkinsonism: onset, progression and mortality. Neurology. 1967;17:427–442. doi: 10.1212/wnl.17.5.427. [DOI] [PubMed] [Google Scholar]
- 2.Dauer W, Przedborski S. Parkinson's disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- 3.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 4.Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 5.Lesage S, Dürr A, Tazir M, Lohmann E, Leutenegger AL, Janin S, Pollak P, Brice A French Parkinson's Disease Genetics Study Group. LRRK2 G2019S as a cause of Parkinson's disease in North African Arabs. N Engl J Med. 2006;354:422–423. doi: 10.1056/NEJMc055540. [DOI] [PubMed] [Google Scholar]
- 6.Simón-Sánchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, Scholz SW, Hernandez DG, Krüger R, Federoff M, Klein C, Goate A, Perlmutter J, Bonin M, Nalls MA, Illig T, Gieger C, Houlden H, Steffens M, Okun MS, Racette BA, Cookson MR, Foote KD, Fernandez HH, Traynor BJ, Schreiber S, Arepalli S, Zonozi R, Gwinn K, van der Brug M, Lopez G, Chanock SJ, Schatzkin A, Park Y, Hollenbeck A, Gao J, Huang X, Wood NW, Lorenz D, Deuschl G, Chen H, Riess O, Hardy JA, Singleton AB, Gasser T. Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat Genet. 2009;41:1308–1312. doi: 10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Daher JP, Pletnikova O, Biskup S, Musso A, Gellhaar S, Galter D, Troncoso JC, Lee MK, Dawson TM, Dawson VL, Moore DJ. Neurodegenerative phenotypes in an A53T alpha-synuclein transgenic mouse model are independent of LRRK2. Hum Mol Genet. 2012;21:2420–2431. doi: 10.1093/hmg/dds057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 9.Krüger R, Kuhn W, Müller T, Woitalla D, Graeber M, Kösel S, Przuntek H, Epplen JT, Schöls L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 10.Zarranz JJ, Alegre J, Gómez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atarés B, Llorens V, Gomez Tortosa E, del Ser T, Muñoz DG, de Yebenes JG. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- 11.Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, Waucquier N, Defebvre L, Amouyel P, Farrer M, Destée A. Alpha-synuclein locus duplication as a cause of familial Parkinson's disease. Lancet. 2004;364:1167–1169. doi: 10.1016/S0140-6736(04)17103-1. [DOI] [PubMed] [Google Scholar]
- 12.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. alpha-Synuclein locus triplication causes Parkinson's disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 13.Funayama M, Hasegawa K, Kowa H, Saito M, Tsuji S, Obata F. A new locus for Parkinson's disease (PARK8) maps to chromosome 12p11.2-q13.1. Ann Neurol. 2002;51:296–301. doi: 10.1002/ana.10113. [DOI] [PubMed] [Google Scholar]
- 14.Paisán-Ruíz C, Jain S, Evans EW, Gilks WP, Simón J, van der Brug M, López de, Aparicio S, Gil AM, Khan N, Johnson J, Martinez JR, Nicholl D, Carrera IM, Pena AS, de Silva R, Lees A, Martí-Massó JF, Pérez-Tur J, Wood NW, Singleton AB. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron. 2004;44:595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 15.Zimprich A, Müller-Myhsok B, Farrer M, Leitner P, Sharma M, Hulihan M, Lockhart P, Strongosky A, Kachergus J, Calne DB, Stoessl J, Uitti RJ, Pfeiffer RF, Trenkwalder C, Homann N, Ott E, Wenzel K, Asmus F, Hardy J, Wszolek Z, Gasser T. The PARK8 locus in autosomal dominant parkinsonism: confirmation of linkage and further delineation of the disease-containing interval. Am J Hum Genet. 2004;74:11–19. doi: 10.1086/380647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lesage S, Brice A. Parkinson's disease: from monogenic forms to genetic susceptibility factors. Hum Mol Genet. 2009;18:R48–R59. doi: 10.1093/hmg/ddp012. [DOI] [PubMed] [Google Scholar]
- 17.Ozelius LJ, Senthil G, Saunders-Pullman R, Ohmann E, Deligtisch A, Tagliati M, Hunt AL, Klein C, Henick B, Hailpern SM, Lipton RB, Soto-Valencia J, Risch N, Bressman SB. LRRK2 G2019S as a cause of Parkinson's disease in Ashkenazi Jews. N Engl J Med. 2006;354:424–425. doi: 10.1056/NEJMc055509. [DOI] [PubMed] [Google Scholar]
- 18.Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 19.Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, Dekker MC, Squitieri F, Ibanez P, Joosse M, van Dongen JW, Vanacore N, van Swieten JC, Brice A, Meco G, van Duijn CM, Oostra BA, Heutink P. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256–259. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- 20.Moore DJ, Zhang L, Dawson TM, Dawson VL. A missense mutation (L166P) in DJ-1, linked to familial Parkinson's disease, confers reduced protein stability and impairs homo-oligomerization. J Neurochem. 2003;87:1558–1567. doi: 10.1111/j.1471-4159.2003.02265.x. [DOI] [PubMed] [Google Scholar]
- 21.Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, González-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, Auburger G, Wood NW. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science. 2004;304:1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- 22.Ramirez A, Heimbach A, Gründemann J, Stiller B, Hampshire D, Cid LP, Goebel I, Mubaidin AF, Wriekat AL, Roeper J, Al-Din A, Hillmer AM, Karsak M, Liss B, Woods CG, Behrens MI, Kubisch C. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet. 2006;38:1184–1191. doi: 10.1038/ng1884. [DOI] [PubMed] [Google Scholar]
- 23.Pankratz N, Wilk JB, Latourelle JC, DeStefano AL, Halter C, Pugh EW, Doheny KF, Gusella JF, Nichols WC, Foroud T, Myers RH PSG-PROGENI and GenePD Investigators, Coordinators and Molecular Genetic Laboratories. Genomewide association study for susceptibility genes contributing to familial Parkinson disease. Hum Genet. 2009;124:593–605. doi: 10.1007/s00439-008-0582-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, Kawaguchi T, Tsunoda T, Watanabe M, Takeda A, Tomiyama H, Nakashima K, Hasegawa K, Obata F, Yoshikawa T, Kawakami H, Sakoda S, Yamamoto M, Hattori N, Murata M, Nakamura Y, Toda T. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson's disease. Nat Genet. 2009;41:1303–1307. doi: 10.1038/ng.485. [DOI] [PubMed] [Google Scholar]
- 25.Edwards TL, Scott WK, Almonte C, Burt A, Powell EH, Beecham GW, Wang L, Züchner S, Konidari I, Wang G, Singer C, Nahab F, Scott B, Stajich JM, Pericak-Vance M, Haines J, Vance JM, Martin ER. Genome-wide association study confirms SNPs in SNCA and the MAPT region as common risk factors for Parkinson disease. Ann Hum Genet. 2010;74:97–109. doi: 10.1111/j.1469-1809.2009.00560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wider C, Dickson DW, Wszolek ZK. Leucine-rich repeat kinase 2 gene-associated disease: redefining genotype-phenotype correlation. Neurodegener Dis. 2010;7:175–179. doi: 10.1159/000289232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with lewy bodies. Proc Natl Acad Sci U S A. 1998;95:6469–6473. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iwai A, Masliah E, Yoshimoto M, Ge N, Flanagan L, de Silva HA, Kittel A, Saitoh T. The precursor protein of non-A beta component of Alzheimer's disease amyloid is a presynaptic protein of the central nervous system. Neuron. 1995;14:467–475. doi: 10.1016/0896-6273(95)90302-x. [DOI] [PubMed] [Google Scholar]
- 29.Maroteaux L, Campanelli JT, Scheller RH. Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci. 1988;8:2804–2815. doi: 10.1523/JNEUROSCI.08-08-02804.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burré J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Südhof TC. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329:1663–1667. doi: 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nuscher B, Kamp F, Mehnert T, Odoy S, Haass C, Kahle PJ, Beyer K. Alpha-synuclein has a high affinity for packing defects in a bilayer membrane: a thermodynamics study. J Biol Chem. 2004;279:21966–21975. doi: 10.1074/jbc.M401076200. [DOI] [PubMed] [Google Scholar]
- 32.Varkey J, Isas JM, Mizuno N, Jensen MB, Bhatia VK, Jao CC, Petrlova J, Voss JC, Stamou DG, Steven AC, Langen R. Membrane curvature induction and tubulation are common features of synucleins and apolipoproteins. J Biol Chem. 2010;285:32486–32493. doi: 10.1074/jbc.M110.139576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Conway KA, Harper JD, Lansbury PT., Jr Fibrils formed in vitro from alpha-synuclein and two mutant forms linked to Parkinson's disease are typical amyloid. Biochemistry. 2000;39:2552–2563. doi: 10.1021/bi991447r. [DOI] [PubMed] [Google Scholar]
- 34.Kim C, Lee SJ. Controlling the mass action of alpha-synuclein in Parkinson's disease. J Neurochem. 2008;107:303–316. doi: 10.1111/j.1471-4159.2008.05612.x. [DOI] [PubMed] [Google Scholar]
- 35.Lashuel HA, Overk CR, Oueslati A, Masliah E. The many faces of alpha-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci. 2013;14:38–48. doi: 10.1038/nrn3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maries E, Dass B, Collier TJ, Kordower JH, Steece-Collier K. The role of alpha-synuclein in Parkinson's disease: insights from animal models. Nat Rev Neurosci. 2003;4:727–738. doi: 10.1038/nrn1199. [DOI] [PubMed] [Google Scholar]
- 37.Hashimoto M, Rockenstein E, Mante M, Mallory M, Masliah E. beta-Synuclein inhibits alpha-synuclein aggregation: a possible role as an anti-parkinsonian factor. Neuron. 2001;32:213–223. doi: 10.1016/s0896-6273(01)00462-7. [DOI] [PubMed] [Google Scholar]
- 38.Sarkar S, Perlstein EO, Imarisio S, Pineau S, Cordenier A, Maglathlin RL, Webster JA, Lewis TA, O'Kane CJ, Schreiber SL, Rubinsztein DC. Small molecules enhance autophagy and reduce toxicity in Huntington's disease models. Nat Chem Biol. 2007;3:331–338. doi: 10.1038/nchembio883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B, Liu K, Xu K, Strathearn KE, Liu F, Cao S, Caldwell KA, Caldwell GA, Marsischky G, Kolodner RD, Labaer J, Rochet JC, Bonini NM, Lindquist S. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson's models. Science. 2006;313:324–328. doi: 10.1126/science.1129462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee HJ, Khoshaghideh F, Lee S, Lee SJ. Impairment of microtubule-dependent trafficking by overexpression of alpha-synuclein. Eur J Neurosci. 2006;24:3153–3162. doi: 10.1111/j.1460-9568.2006.05210.x. [DOI] [PubMed] [Google Scholar]
- 41.Gosavi N, Lee HJ, Lee JS, Patel S, Lee SJ. Golgi fragmentation occurs in the cells with prefibrillar alpha-synuclein aggregates and precedes the formation of fibrillar inclusion. J Biol Chem. 2002;277:48984–48992. doi: 10.1074/jbc.M208194200. [DOI] [PubMed] [Google Scholar]
- 42.Nemani VM, Lu W, Berge V, Nakamura K, Onoa B, Lee MK, Chaudhry FA, Nicoll RA, Edwards RH. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron. 2010;65:66–79. doi: 10.1016/j.neuron.2009.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Winslow AR, Chen CW, Corrochano S, Acevedo-Arozena A, Gordon DE, Peden AA, Lichtenberg M, Menzies FM, Ravikumar B, Imarisio S, Brown S, O'Kane CJ, Rubinsztein DC. α-Synuclein impairs macroautophagy: implications for Parkinson's disease. J Cell Biol. 2010;190:1023–1037. doi: 10.1083/jcb.201003122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lindersson E, Beedholm R, Højrup P, Moos T, Gai W, Hendil KB, Jensen PH. Proteasomal inhibition by alpha-synuclein filaments and oligomers. J Biol Chem. 2004;279:12924–12934. doi: 10.1074/jbc.M306390200. [DOI] [PubMed] [Google Scholar]
- 45.Parihar MS, Parihar A, Fujita M, Hashimoto M, Ghafourifar P. Mitochondrial association of alpha-synuclein causes oxidative stress. Cell Mol Life Sci. 2008;65:1272–1284. doi: 10.1007/s00018-008-7589-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson's disease. Nat Med. 2008;14:504–506. doi: 10.1038/nm1747. [DOI] [PubMed] [Google Scholar]
- 47.Li JY, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ, Lashley T, Quinn NP, Rehncrona S, Björklund A, Widner H, Revesz T, Lindvall O, Brundin P. Lewy bodies in grafted neurons in subjects with Parkinson's disease suggest host-to-graft disease propagation. Nat Med. 2008;14:501–503. doi: 10.1038/nm1746. [DOI] [PubMed] [Google Scholar]
- 48.Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee SJ. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A. 2009;106:13010–13015. doi: 10.1073/pnas.0903691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee HJ, Patel S, Lee SJ. Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J Neurosci. 2005;25:6016–6024. doi: 10.1523/JNEUROSCI.0692-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH, Stefanis L, Vekrellis K. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J Neurosci. 2010;30:6838–6851. doi: 10.1523/JNEUROSCI.5699-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ejlerskov P, Rasmussen I, Nielsen TT, Bergström AL, Tohyama Y, Jensen PH, Vilhardt F. Tubulin polymerization-promoting protein (TPPP/p25α) promotes unconventional secretion of α-synuclein through exophagy by impairing autophagosome-lysosome fusion. J Biol Chem. 2013;288:17313–17335. doi: 10.1074/jbc.M112.401174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jang A, Lee HJ, Suk JE, Jung JW, Kim KP, Lee SJ. Non-classical exocytosis of alpha-synuclein is sensitive to folding states and promoted under stress conditions. J Neurochem. 2010;113:1263–1274. doi: 10.1111/j.1471-4159.2010.06695.x. [DOI] [PubMed] [Google Scholar]
- 53.Lee HJ, Baek SM, Ho DH, Suk JE, Cho ED, Lee SJ. Dopamine promotes formation and secretion of non-fibrillar alpha-synuclein oligomers. Exp Mol Med. 2011;43:216–222. doi: 10.3858/emm.2011.43.4.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hansen C, Angot E, Bergström AL, Steiner JA, Pieri L, Paul G, Outeiro TF, Melki R, Kallunki P, Fog K, Li JY, Brundin P. α-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J Clin Invest. 2011;121:715–725. doi: 10.1172/JCI43366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee HJ, Suk JE, Bae EJ, Lee JH, Paik SR, Lee SJ. Assembly-dependent endocytosis and clearance of extracellular alpha-synuclein. Int J Biochem Cell Biol. 2008;40:1835–1849. doi: 10.1016/j.biocel.2008.01.017. [DOI] [PubMed] [Google Scholar]
- 56.Bosgraaf L, Van Haastert PJ. Roc, a Ras/GTPase domain in complex proteins. Biochim Biophys Acta. 2003;1643:5–10. doi: 10.1016/j.bbamcr.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 57.Greggio E, Zambrano I, Kaganovich A, Beilina A, Taymans JM, Daniëls V, Lewis P, Jain S, Ding J, Syed A, Thomas KJ, Baekelandt V, Cookson MR. The Parkinson disease-associated leucine-rich repeat kinase 2 (LRRK2) is a dimer that undergoes intramolecular autophosphorylation. J Biol Chem. 2008;283:16906–16914. doi: 10.1074/jbc.M708718200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Klein CL, Rovelli G, Springer W, Schall C, Gasser T, Kahle PJ. Homo-and heterodimerization of ROCO kinases: LRRK2 kinase inhibition by the LRRK2 ROCO fragment. J Neurochem. 2009;111:703–715. doi: 10.1111/j.1471-4159.2009.06358.x. [DOI] [PubMed] [Google Scholar]
- 59.Mata IF, Wedemeyer WJ, Farrer MJ, Taylor JP, Gallo KA. LRRK2 in Parkinson's disease: protein domains and functional insights. Trends Neurosci. 2006;29:286–293. doi: 10.1016/j.tins.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 60.Milosevic J, Schwarz SC, Ogunlade V, Meyer AK, Storch A, Schwarz J. Emerging role of LRRK2 in human neural progenitor cell cycle progression, survival and differentiation. Mol Neurodegener. 2009;4:25. doi: 10.1186/1750-1326-4-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.MacLeod D, Dowman J, Hammond R, Leete T, Inoue K, Abeliovich A. The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron. 2006;52:587–593. doi: 10.1016/j.neuron.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 62.Yao C, El Khoury R, Wang W, Byrd TA, Pehek EA, Thacker C, Zhu X, Smith MA, Wilson-Delfosse AL, Chen SG. LRRK2-mediated neurodegeneration and dysfunction of dopaminergic neurons in a Caenorhabditis elegans model of Parkinson's disease. Neurobiol Dis. 2010;40:73–81. doi: 10.1016/j.nbd.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Maekawa T, Kubo M, Yokoyama I, Ohta E, Obata F. Age-dependent and cell-population-restricted LRRK2 expression in normal mouse spleen. Biochem Biophys Res Commun. 2010;392:431–435. doi: 10.1016/j.bbrc.2010.01.041. [DOI] [PubMed] [Google Scholar]
- 64.Thévenet J, Pescini Gobert R, Hooft van Huijsduijnen R, Wiessner C, Sagot YJ. Regulation of LRRK2 expression points to a functional role in human monocyte maturation. PLoS One. 2011;6:e21519. doi: 10.1371/journal.pone.0021519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.West AB, Moore DJ, Choi C, Andrabi SA, Li X, Dikeman D, Biskup S, Zhang Z, Lim KL, Dawson VL, Dawson TM. Parkinson's disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum Mol Genet. 2007;16:223–232. doi: 10.1093/hmg/ddl471. [DOI] [PubMed] [Google Scholar]
- 66.Di Fonzo A, Tassorelli C, De Mari M, Chien HF, Ferreira J, Rohé CF, Riboldazzi G, Antonini A, Albani G, Mauro A, Marconi R, Abbruzzese G, Lopiano L, Fincati E, Guidi M, Marini P, Stocchi F, Onofrj M, Toni V, Tinazzi M, Fabbrini G, Lamberti P, Vanacore N, Meco G, Leitner P, Uitti RJ, Wszolek ZK, Gasser T, Simons EJ, Breedveld GJ, Goldwurm S, Pezzoli G, Sampaio C, Barbosa E, Martignoni E, Oostra BA, Bonifati V Italian Parkinson's Genetics Network. Comprehensive analysis of the LRRK2 gene in sixty families with Parkinson's disease. Eur J Hum Genet. 2006;14:322–331. doi: 10.1038/sj.ejhg.5201539. [DOI] [PubMed] [Google Scholar]
- 67.Lee BD, Shin JH, VanKampen J, Petrucelli L, West AB, Ko HS, Lee YI, Maguire-Zeiss KA, Bowers WJ, Federoff HJ, Dawson VL, Dawson TM. Inhibitors of leucine-rich repeat kinase-2 protect against models of Parkinson's disease. Nat Med. 2010;16:998–1000. doi: 10.1038/nm.2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P, Asmus F, Müller-Myhsok B, Dickson DW, Meitinger T, Strom TM, Wszolek ZK, Gasser T. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 69.Rajput A, Dickson DW, Robinson CA, Ross OA, Dächsel JC, Lincoln SJ, Cobb SA, Rajput ML, Farrer MJ. Parkinsonism, Lrrk2 G2019S, and tau neuropathology. Neurology. 2006;67:1506–1508. doi: 10.1212/01.wnl.0000240220.33950.0c. [DOI] [PubMed] [Google Scholar]
- 70.Carballo-Carbajal I, Weber-Endress S, Rovelli G, Chan D, Wolozin B, Klein CL, Patenge N, Gasser T, Kahle PJ. Leucine-rich repeat kinase 2 induces alpha-synuclein expression via the extracellular signal-regulated kinase pathway. Cell Signal. 2010;22:821–827. doi: 10.1016/j.cellsig.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Qing H, Wong W, McGeer EG, McGeer PL. Lrrk2 phosphorylates alpha synuclein at serine 129: Parkinson disease implications. Biochem Biophys Res Commun. 2009;387:149–152. doi: 10.1016/j.bbrc.2009.06.142. [DOI] [PubMed] [Google Scholar]
- 72.Liu G, Aliaga L, Cai H. α-Synuclein, LRRK2 and their interplay in Parkinson's disease. Future Neurol. 2012;7:145–153. doi: 10.2217/fnl.12.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ito G, Okai T, Fujino G, Takeda K, Ichijo H, Katada T, Iwatsubo T. GTP binding is essential to the protein kinase activity of LRRK2, a causative gene product for familial Parkinson's disease. Biochemistry. 2007;46:1380–1388. doi: 10.1021/bi061960m. [DOI] [PubMed] [Google Scholar]
- 74.Guo L, Gandhi PN, Wang W, Petersen RB, Wilson-Delfosse AL, Chen SG. The Parkinson's disease-associated protein, leucine-rich repeat kinase 2 (LRRK2), is an authentic GTPase that stimulates kinase activity. Exp Cell Res. 2007;313:3658–3670. doi: 10.1016/j.yexcr.2007.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xiong Y, Coombes CE, Kilaru A, Li X, Gitler AD, Bowers WJ, Dawson VL, Dawson TM, Moore DJ. GTPase activity plays a key role in the pathobiology of LRRK2. PLoS Genet. 2010;6:e1000902. doi: 10.1371/journal.pgen.1000902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Seabra MC, Wasmeier C. Controlling the location and activation of Rab GTPases. Curr Opin Cell Biol. 2004;16:451–457. doi: 10.1016/j.ceb.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 77.Pfeffer S, Aivazian D. Targeting Rab GTPases to distinct membrane compartments. Nat Rev Mol Cell Biol. 2004;5:886–896. doi: 10.1038/nrm1500. [DOI] [PubMed] [Google Scholar]
- 78.Shin N, Jeong H, Kwon J, Heo HY, Kwon JJ, Yun HJ, Kim CH, Han BS, Tong Y, Shen J, Hatano T, Hattori N, Kim KS, Chang S, Seol W. LRRK2 regulates synaptic vesicle endocytosis. Exp Cell Res. 2008;314:2055–2065. doi: 10.1016/j.yexcr.2008.02.015. [DOI] [PubMed] [Google Scholar]
- 79.Lee S, Liu HP, Lin WY, Guo H, Lu B. LRRK2 kinase regulates synaptic morphology through distinct substrates at the presynaptic and postsynaptic compartments of the Drosophila neuromuscular junction. J Neurosci. 2010;30:16959–16969. doi: 10.1523/JNEUROSCI.1807-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Matta S, Van Kolen K, da Cunha R, van den Bogaart G, Mandemakers W, Miskiewicz K, De Bock PJ, Morais VA, Vilain S, Haddad D, Delbroek L, Swerts J, Chávez-Gutiérrez L, Esposito G, Daneels G, Karran E, Holt M, Gevaert K, Moechars DW, De Strooper B, Verstreken P. LRRK2 controls an EndoA phosphorylation cycle in synaptic endocytosis. Neuron. 2012;75:1008–1021. doi: 10.1016/j.neuron.2012.08.022. [DOI] [PubMed] [Google Scholar]
- 81.Lin X, Parisiadou L, Gu XL, Wang L, Shim H, Sun L, Xie C, Long CX, Yang WJ, Ding J, Chen ZZ, Gallant PE, Tao-Cheng JH, Rudow G, Troncoso JC, Liu Z, Li Z, Cai H. Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson's-disease-related mutant alpha-synuclein. Neuron. 2009;64:807–827. doi: 10.1016/j.neuron.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Herzig MC, Bidinosti M, Schweizer T, Hafner T, Stemmelen C, Weiss A, Danner S, Vidotto N, Stauffer D, Barske C, Mayer F, Schmid P, Rovelli G, van der Putten PH, Shimshek DR. High LRRK2 levels fail to induce or exacerbate neuronal alpha-synucleinopathy in mouse brain. PLoS One. 2012;7:e36581. doi: 10.1371/journal.pone.0036581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tong Y, Yamaguchi H, Giaime E, Boyle S, Kopan R, Kelleher RJ, 3rd, Shen J. Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of alpha-synuclein, and apoptotic cell death in aged mice. Proc Natl Acad Sci U S A. 2010;107:9879–9884. doi: 10.1073/pnas.1004676107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Orenstein SJ, Kuo SH, Tasset I, Arias E, Koga H, Fernandez-Carasa I, Cortes E, Honig LS, Dauer W, Consiglio A, Raya A, Sulzer D, Cuervo AM. Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci. 2013;16:394–406. doi: 10.1038/nn.3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gillardon F, Schmid R, Draheim H. Parkinson's disease-linked leucine-rich repeat kinase 2(R1441G) mutation increases proinflammatory cytokine release from activated primary microglial cells and resultant neurotoxicity. Neuroscience. 2012;208:41–48. doi: 10.1016/j.neuroscience.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 86.Moehle MS, Webber PJ, Tse T, Sukar N, Standaert DG, DeSilva TM, Cowell RM, West AB. LRRK2 inhibition attenuates microglial inflammatory responses. J Neurosci. 2012;32:1602–1611. doi: 10.1523/JNEUROSCI.5601-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gao HM, Kotzbauer PT, Uryu K, Leight S, Trojanowski JQ, Lee VM. Neuroinflammation and oxidation/nitration of alpha-synuclein linked to dopaminergic neurodegeneration. J Neurosci. 2008;28:7687–7698. doi: 10.1523/JNEUROSCI.0143-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]