

Abstract
Purpose The L216R mutation, seen in individuals of Polynesian descent, is considered one of the most severe mutations associated with holocarboxylase synthetase (HLCS) deficiency and is regarded as being unresponsive to biotin. This report describes the presentation and outcome in two surviving siblings, homozygous for this highly lethal mutation.
Methods and results Both cases had perinatal head imaging findings of brain hemorrhage and subependymal cysts. Both had metabolic decompensation within 24 h after birth consisting of metabolic acidosis, lactic acidosis, and thrombocytopenia. Biochemical profiles were consistent with HLCS deficiency, and genetic analysis confirmed homozygosity for the L216R mutation. After resolution of neonatal metabolic crisis, dosing of biotin was titrated on an outpatient basis to primarily control dermatitis. The eldest is currently on 1.2 g of oral biotin daily, well above any dose previously reported to treat HLCS deficiency. To date, neither patient has required hospital readmission for acute metabolic decompensation. At the age of 7, the eldest child is, to our knowledge, the oldest patient ever described in the literature who is homozygous for the L216R mutation. She has mild intellectual disability.
Conclusion This report contrasts previous reports of poor outcomes and neonatal deaths in homozygous L216R patients. We also provide data on the potential upper tolerable limit of biotin. These cases suggest that the outcome of HCLS deficiency due to a homozygous L216R mutation, when diagnosed and treated early with high-level neonatal care and biotin, may not be as severe as previously reported.
Introduction
Biotin is a water-soluble vitamin found in foods such as liver, milk, and egg yolk. Natural occurring D (+)-biotin is not synthesized by mammals and must be acquired by exogenous sources (Suormala et al. 1998). The adequate intake for adults is 30 ug/day. To date, sufficient data has not been collected to establish a tolerable upper intake level (Institute of Medicine (US) Standing Committee on the Scientific Evaluation of Dietary Reference Intakes and its Panel on Folate, Other B Vitamins, and Choline 1998). Biotin acts as a cofactor for five human carboxylase enzymes: acetyl-CoA carboxylase 1 and 2, pyruvate carboxylase, propionyl-CoA carboxylase, and 3-methylcrotonyl-CoA carboxylase (Bailey et al. 2008). If biotin metabolism is disturbed, these enzymes are affected leading to a disruption of normal cellular function (Mayende et al. 2012). The enzyme holocarboxylase synthetase (HLCS) is important for binding biotin to these enzymes.
Mutations in HLCS are responsible for HLCS deficiency (HLCS deficiency; OMIM #253270), a rare heritable disease transmitted in an autosomal recessive pattern. Many patients with HLCS deficiency present in the neonatal period with poor feeding, respiratory distress, lethargy, vomiting, hypotonia, seizures, and coma (Morrone et al. 2002). Skin findings can be particularly severe and include facial and full body erythrodermic dermatitis, hyperkeratosis, and diffuse non-scarring alopecia (Esparza et al. 2011). Laboratory abnormalities may include metabolic and/or lactic acidosis, organic acidemia, mild hyperammonemia, and variable hypoglycemia (Morrone et al. 2002). HLCS deficiency can lead to repeated bouts of metabolic decomposition, developmental delay, and is a potentially fatal condition if left untreated (Morrone et al. 2002, Dupuis et al. 1999).
With oral daily biotin supplementation (10–20 mg), most cases of HLCS deficiency can be successfully treated (Bailey et al. 2008; Dupuis et al. 1999). Untreated cases or patients who display an incomplete response to biotin supplementation, however, have a poor long-term prognosis (Bailey et al. 2008; Dupuis et al. 1999). Of the many discrete gene mutations, errors between amino acids 159 and 314 in the N terminal extension outside of the biotin-binding domain of HLCS are believed to be the most severe mutations and are considered biotin unresponsive (Mayende et al. 2012). The L216R mutation is seen in individuals of Polynesian ancestry. It causes one of the most severe forms of HLCS deficiency. Individuals homozygous for this mutation are considered biotin unresponsive, and the homozygous mutation is associated with high infant mortality (Bailey et al. 2008; Morrone et al. 2002; Wilson et al. 2005). In the first case report of a homozygous L216R patient, it was unclear if there was any clinical response on 100 mg/day of biotin (Morrone et al. 2002). At the age of 2, the patient had severe full body eczema with global developmental delay and deafness. This child was reported to have died at age 3 years in a later report (Wilson et al. 2005). Wilson et al. also describe another six homozygous L216R patients from a family of seven affected children. Four of these children died between the ages of 3 days and 3 years, one had a good recovery after birth, but had recurrent infections and metabolic acidosis at 18 months on 40–80 mg of biotin daily, and one was lost to follow-up at age 18 months (Wilson et al. 2005). Bailey et al. (2008) studied fibroblasts from two of the above patients. The fibroblast Km in their study could not be differentiated from negative controls and the mutant HLCS enzyme was found to have a faster turnover rate than that of the wild-type enzyme (Bailey et al. 2008). Other studies have also demonstrated that the L216R mutant has a biotin ligase activity near zero (Esaki et al. 2012).
In contrast to these patients with biotin unresponsiveness, patients heterozygous for a biotin-unresponsive allele and a biotin-responsive allele show good clinical response to biotin therapy between 20 and 40 mg/day (Dupuis et al. 1999). The favorable outcome is attributed to the presence of the biotin-responsive allele.
The purpose of this report is to present the clinical course of two siblings homozygous for the L216R mutation, a 7-year-old female and a 5-month-old male. The eldest is the longest surviving case for this severe mutation reported in the literature. Both are doing well on very large doses of biotin, much larger than any previously reported.
Subjects, Methods, and Results
Case 1
A female was born to an 18-year-old Samoan/Tongan primigravida and her 19-year-old Samoan/German partner. The pregnancy was complicated by maternal thrombocytopenia. The mother received a diagnosis of idiopathic thrombocytopenic purpura (ITP) at the age of 12 years and had been treated intermittently with corticosteroids, including during the pregnancy. The mother presented in labor at 34–6/7 weeks’ gestation after premature rupture of membranes. She had an uncomplicated spontaneous vaginal delivery. The birth weight was 2,262 g, the length was 44.5 cm, and the occipitofrontal circumference (OFC) was 36 cm. Apgar scores were 5 and 7, at 1 and 5 min, respectively. The infant had poor respiratory effort and tone. In the newborn intensive care unit (NICU), the infant’s initial course was complicated by a large subgaleal hematoma, thrombocytopenia, and coagulopathy with an initial platelet count of 36,000 × 109/L with elevated prothrombin time, partial thromboplastin time, and fibrin degradation products, and a low fibrinogen. Computed tomography (CT) of the head on day 1 of life revealed moderate bilateral intraventricular hemorrhages, left cerebellar hemorrhage, acute subdural hemorrhage of the posterior cerebellum and left occipital lobe, diffuse hypodensity in the cerebral white matter, and bilateral subgaleal hemorrhage. Brain magnetic resonance imaging (MRI) on day 3 confirmed the CT scan findings. A diffusion scan showed small cortical infarctions in the occipital, left temporal, and inferior right frontal lobes. Periventricular cysts were noted. She was treated with intravenous immunoglobulin (IVIG) for the first 4 days of life and required multiple platelet, fresh frozen plasma, and cryoprecipitate transfusions.
Persistent metabolic acidosis was noted. The serum lactic acid was 11.4 mmol/L on day of life (DOL) 10. Initial qualitative urine organic acids showed a complex pattern considered to be consistent with HLCS deficiency. Biotinidase activity could not be reported on the newborn screening due to the transfusions, but was eventually normal. Total and free carnitines were low (2.2 and 12.7 umol/L, respectively), with an elevated acyl/free ratio of 4.8. An acylcarnitine profile revealed an abnormal elevation of propionylcarnitine and 3-hydroxyisovalerylcarnitine, supporting the diagnosis of HLCS deficiency. Plasma amino acids on DOL 10 revealed numerous elevations including leucine (973 nmol/mL), isoleucine (413 nmol/mL), valine (980 nmol/mL), and the presence of alloisoleucine (24 nmol/mL), thought to be secondary derangements. Holocarboxylase synthetase activity was performed on lymphocytes and showed an elevated Km of 30.6 nmol/L (reference range 1.0–7.8), with maximum velocity (Vmax) of 187 fmol/mg/h compared to simultaneous control Km of 7.28 nmol/L and Vmax of 222 fmol/mg/h. HLCS gene sequencing documented homozygosity for a c.647T>G (L216R) mutation.
Biotin, at a dose of 10 mg/day, and carnitine were started on DOL 10. She developed a significant erythematous rash in the diaper area (Fig. 1a) that progressed over time to involve her intertriginous regions. Neutropenia was noted at 3 weeks of age with an absolute neutrophil count of 328, which gradually improved. Cholestasis was also noted at 3 weeks of age, associated with hepatomegaly and a liver palpable 4 cm below the right costal margin. Direct hyperbilirubinemia improved, but at 7–8 weeks of age, she had elevated liver transaminases and acholic stools. She was treated with ursodiol and vitamins A, D, E, and K without improvement. A liver biopsy was consistent with giant cell hepatitis with bile duct hyperplasia. A hepatobiliary iminodiacetic acid (HIDA) scan showed no excretion. The intraoperative cholangiogram, however, was normal, and her cholestasis gradually resolved. The postoperative course was complicated by possible seizures. She had no further recurrence of seizure-like activity and phenobarbital therapy was weaned over time and discontinued.
Fig. 1.
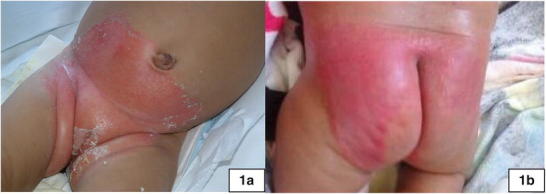
Images of neonatal dermatitis. (a) The dermatitis in Case 1 in early infancy, showing the significant skin denudation and erythematous rash. (b) The dermatitis in Case 2 in early infancy after neonatal discharge from the hospital
The biotin dose was gradually increased to control dermatitis. Her most recent daily maximum dose was 1.2 g/day (25 mg/kg/day) of a biotin powder compounded with carboxymethyl cellulose powder, simple syrup, and sterile water to make a 5 mg/mL oral solution; no specific solubility was sought by the pharmacy. She has had improvement in her rash on this higher dose of biotin, with only minimal chronic rash remaining around her eyes (Fig. 2). Carnitine at 50–100 mg/kg/day has been continued. No dietary restrictions were recommended, except avoidance of prolonged fasting. Propionylcarnitine (6.5–10 umole/L, reference range 0.08–1.77) and 3-hydroxyisovalerylcarnitine (0.36–0.82 umole/L, reference range 0.01–0.11), when checked at ages 6 and 7 years, remained elevated above clinical baseline. There was no persistence of elevated branch chain amino acids. Serious metabolic decompensation did not recur. She received early intervention services due to her high-risk status, although her early motor and language milestones were within normal limits. Speech delay was noted at age 5 years. She repeated kindergarten and qualified for special education services upon entering grade 1, however, neurodevelopmental testing results are unavailable. Formal audiology testing at age 5 revealed only conductive hearing loss, secondary to cerumen impaction. When last seen at 7½ years of age, her height was at the 75th centile for age, with a weight of 44.5 kg (>97th centile for age), and an OFC of 55 cm (>97th centile).
Fig. 2.
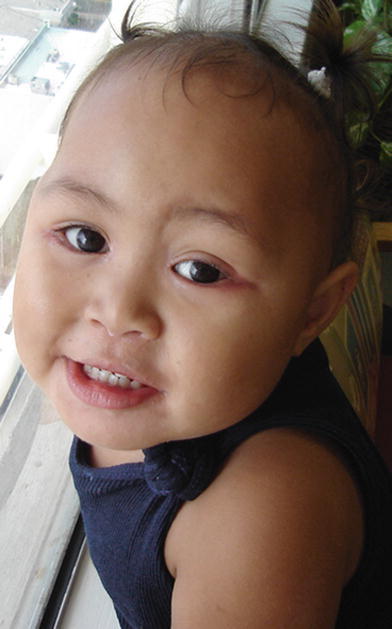
Case 1 at age 2 years. Note the persistent rash at the outer canthi bilaterally
Case 2
The prenatal course was complicated by abnormal ultrasound findings including fetal ventriculomegaly and possible brain hemorrhage first noted at 27 weeks. Subependymal cysts were additionally noted on follow-up fetal MRI. During this pregnancy, the mother had normal platelet counts and no evidence of ITP. Prenatal diagnosis by molecular testing for the L216R mutation was declined. Due to the risk of recurrent HCLS deficiency, and based on the limited available information on biotin use for at-risk pregnancies (Suormala et al. 1998;Thuy et al. 1999), it was recommended that the mother take 10 mg/day of biotin, although she only took it during the last week prior to delivery. A male infant was born at 40 weeks gestation by normal vaginal delivery. The infant initially had poor respiratory effort but responded well to continuous positive airway pressure. Apgar scores were 7 and 9.
In the first few hours of life, the infant began having respiratory distress and petechiae were noted on the face, torso, and limbs. Complete blood count was obtained revealing a platelet count of 26,000 × 109/L. The infant was transferred to the NICU, where initial blood pH was 7.1. Initial lactate level was 18 mmol/L, which slowly decreased over the first week of life. Given the family history of HLCS, oral biotin was started on DOL 1 at 25 mg/kg/day divided three times daily. A carnitine profile collected on DOL 2 showed an elevated propionylcarnitine level of 20.56 umol/L (reference range 0.07–14.85) as well as an elevated 3-hydroxyisovalerylcarnitine of 1.23 umol/L (reference range 0.01–0.07). Free carnitine was depleted with an elevation in acyl/free ratio. Oral levocarnitine was started at 50 mg/kg/day.
A mild intertriginous rash, without skin breakdown, was noted throughout the NICU stay. The thrombocytopenia persisted and IVIG was given two times requiring multiple platelet transfusions (5 total). Platelet counts and biochemical status improved and the infant was discharged on DOL 34 on biotin 40 mg/kg of compounded (as above) 5 mg/ml oral solution divided twice daily, and levocarnitine 50 mg/kg/day. Molecular analysis confirmed L216R homozygosity. Enzyme assay was not performed.
As an outpatient, biotin was increased to 60 mg/kg/day divided twice daily to control dermatitis (Fig. 1b), he also remained on carnitine. No dietary restrictions were recommended except avoidance of prolonged fasting. At 5 months, no major developmental delays were noted.
Discussion
These siblings, affected with a severe form of HLCS deficiency due to a homozygous L216R mutation, presented within hours of birth with thrombocytopenia and metabolic acidosis. Both cases had perinatal CNS findings and brain hemorrhage. Ventriculomegaly and subependymal cysts have been reported previously in cases homozygous for the L216R mutation (Wilson et al. 2005). Hemorrhage may have been a result of fetal thrombocytopenia. The generalized acidosis and accumulation of organic acids during episodes of severe metabolic imbalance have been postulated to account for the neonatal thrombocytopenia sometimes seen in HLCS (Roth et al. 1980; Briones et al. 1989). However, the thrombocytopenia in this family is also complicated by the maternal history of ITP and possible bleeding disorder that has never been fully evaluated. Although, there was no maternal thrombocytopenia or significant bleeding disorder noted during the second pregnancy, this could have been a confounding factor in both cases, especially since the IVIG seemed to stabilize the platelet counts.
These siblings have required very large doses of biotin but are doing well with normal growth and only mild cognitive delays in the oldest girl. The dose of 25 mg/kg/day (1.2 g/day) of biotin in the eldest child, to control dermatitis, is sixfold above the daily reported maximum dosing of up to 200 mg/day previously reported in the literature to treat biotin deficiency due to any underlying cause (Institute of Medicine (US) Standing Committee on the Scientific Evaluation of Dietary Reference Intakes and its Panel on Folate, Other B Vitamins, and Choline 1998; Bailey et al. 2008; Morrone et al. 2002; Wilson et al. 2005). The risk of human toxicity from the usual dietary intake of biotin or from supplements appears to be low and no tolerable upper intake level has been determined (Institute of Medicine (US) Standing Committee on the Scientific Evaluation of Dietary Reference Intakes and its Panel on Folate, Other B Vitamins, and Choline 1998). In our experience, high-dose biotin titration clearly improves the dermatitis. Anecdotally, when the eldest sibling goes days without biotin, her periocular rash significantly worsens. Although no pharmacokinetic data was collected on the siblings, it was postulated that at least four times per day dosing would be desirable based on the rapid clearance studies and estimated 1 h and 50 min half-life of biotin (Bitsch et al. 1989). However, two times daily dosing was chosen to improve compliance.
This report provides documentation about the natural history and the potential positive outcomes in patients with HLCS deficiency due a homozygous L216R mutation when treated with high-dose oral biotin, avoidance of fasting, and carnitine supplementation. More research will need to be completed on HLCS deficiency with special attention to prenatal diagnosis and biotin therapy. Pregnancies at risk for severe HLCS deficiency can be monitored by prenatal ultrasound with close attention given to CNS findings. Prenatal diagnosis and maternal biotin therapy can be considered. Evaluation of the affected or at-risk neonate may need to include special attention to the platelet count, cranial imaging studies, and prompt treatment of any metabolic derangements. The dose of biotin should be minimally optimized to prevent metabolic decompensation and control dermatitis, but ideally should try to maximize the long-term developmental outcome.
Acknowledgments
We would like to thank the family and all of the laboratory and medical personnel involved in the care of these children. We would like to particularly thank Dr. Wade Kyono, the hematologist involved in their care.
Synopsis
This report provides documentation about the natural history and potential positive outcomes in two patients with HLCS deficiency due a homozygous L216R mutation when treated with high-dose oral biotin.
Compliance with Ethics Guidelines
Conflict of Interest
Thomas Slavin, Syed Zaidi, Charles Neal, Brenda Nishikawa, and Laurie Seaver declare that they have no conflict of interest.
Contributions by Authors
Thomas Slavin and Laurie Seaver were the metabolic clinicians involved in the care of the siblings and primary writers of the report. Charles Neal provided neonatal care and assisted with report editing and the discussion of IVIG therapy. Syed Zaidi helped care for Case 2 as a neonate and assisted with the background research. Brenda Nishikawa was the children’s pediatrician and helped edit this report for overall accuracy.
Informed Consent
Patients described in this case report were treated with standard medical care. Proper consent was obtained for inclusion of the patients in this case report. This report was reviewed by Hawai‘i Pacific Health and was found to meet the definition of a case report and is in compliance with applicable research and patient privacy regulations. Additional informed consent was obtained for all patients for whom identifying information is included in this report.
Footnotes
Competing interests: None declared
Contributor Information
Collaborators: Johannes Zschocke and K Michael Gibson
References
- Bailey LM, Ivanov RA, Jitrapakdee S, Wilson CJ, Wallace JC, Polyak SW. Reduced half-life of holocarboxylase synthetase from patients with severe multiple carboxylase deficiency. Hum Mutat. 2008;29:E47–57. doi: 10.1002/humu.20766. [DOI] [PubMed] [Google Scholar]
- Bitsch R, Salz I, Hotzel D. Studies on bioavailability of oral biotin doses for humans. Int J Vitam Nutr Res. 1989;59:65–71. [PubMed] [Google Scholar]
- Briones P, Ribes A, Vilaseca MA, Rodriguez-Valcarcel G, Thuy LP, Sweetman L. A new case of holocarboxylase synthetase deficiency. J Inherit Metab Dis. 1989;12:329–330. doi: 10.1007/BF01799228. [DOI] [PubMed] [Google Scholar]
- Dupuis L, Campeau E, Leclerc D, Gravel RA. Mechanism of biotin responsiveness in biotin-responsive multiple carboxylase deficiency. Mol Genet Metab. 1999;66:80–90. doi: 10.1006/mgme.1998.2785. [DOI] [PubMed] [Google Scholar]
- Esaki S, Malkaram SA, Zempleni J. Effects of single-nucleotide polymorphisms in the human holocarboxylase synthetase gene on enzyme catalysis. Eur J Hum Genet. 2012;20:428–433. doi: 10.1038/ejhg.2011.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esparza EM, Golden AS, Hahn SH, Patterson K, Brandling-Bennett HA. What syndrome is this? Infantile periorificial and intertriginous dermatitis preceding sepsis-like respiratory failure. Pediatr Dermatol. 2011;28:333–334. doi: 10.1111/j.1525-1470.2011.01439.x. [DOI] [PubMed] [Google Scholar]
- Institute of Medicine (US) Standing Committee on the Scientific Evaluation of Dietary Reference Intakes and its Panel on Folate, Other B Vitamins, and Choline (1998) Dietary reference intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline. National Academies Press (US), Washington (DC) [PubMed]
- Mayende L, Swift RD, Bailey LM, Soares da Costa TP, Wallace JC, Booker GW, Polyak SW. A novel molecular mechanism to explain biotin-unresponsive holocarboxylase synthetase deficiency. J Mol Med (Berl) 2012;90:81–88. doi: 10.1007/s00109-011-0811-x. [DOI] [PubMed] [Google Scholar]
- Morrone A, Malvagia S, Donati MA, et al. Clinical findings and biochemical and molecular analysis of four patients with holocarboxylase synthetase deficiency. Am J Med Genet. 2002;111:10–18. doi: 10.1002/ajmg.10532. [DOI] [PubMed] [Google Scholar]
- Roth KS, Yang W, Foremann JW, Rothman R, Segal S. Holocarboxylase synthetase deficiency: a biotin-responsive organic acidemia. J Pediatr. 1980;96:845–849. doi: 10.1016/S0022-3476(80)80554-3. [DOI] [PubMed] [Google Scholar]
- Suormala T, Fowler B, Jakobs C, et al. Late-onset holocarboxylase synthetase-deficiency: pre- and post-natal diagnosis and evaluation of effectiveness of antenatal biotin therapy. Eur J Pediatr. 1998;157:570–575. doi: 10.1007/s004310050881. [DOI] [PubMed] [Google Scholar]
- Thuy LP, Belmont J, Nyhan WL. Prenatal diagnosis and treatment of holocarboxylase synthetase deficiency. Prenat Diagn. 1999;19:108–112. doi: 10.1002/(SICI)1097-0223(199902)19:2<108::AID-PD476>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Wilson CJ, Myer M, Darlow BA, et al. Severe holocarboxylase synthetase deficiency with incomplete biotin responsiveness resulting in antenatal insult in samoan neonates. J Pediatr. 2005;147:115–118. doi: 10.1016/j.jpeds.2005.03.006. [DOI] [PubMed] [Google Scholar]