

Abstract
Here we report a patient with a new pathogenic mutation in ACAD9. Shortly after birth she presented with respiratory insufficiency and a high lactate level. At age 7 weeks, she was diagnosed with severe hypertrophic cardiomyopathy and she suffered from muscle weakness and hypotonia. Her condition deteriorated during intercurrent illnesses and she died at 6 months of age in cardiogenic shock. Analysis of respiratory chain activities in muscle and fibroblasts revealed an isolated complex I deficiency. A genome-wide screen for homozygosity revealed several homozygous regions. Four candidate genes were found and sequencing revealed a homozygous missense mutation in ACAD9. The mutation results in an Ala220Val amino acid substitution located near the catalytic core of ACAD9. SDS and BN-PAGE analysis showed severely decreased ACAD9 and complex I protein levels, and lentiviral complementation of patient fibroblasts partially rescued the complex I deficiency. Riboflavin supplementation did not ameliorate the complex I deficiency in patient fibroblasts. More than a dozen ACAD9 patients with complex I deficiency have been identified in the last 3 years, indicating that ACAD9 is important for complex I assembly, and that ACAD9 mutations are a relatively frequent cause of complex I deficiency.
Introduction
Inborn errors of metabolism (IEM) is a large group of disorders, with a wide spectrum of symptoms, and this also holds true for an IEM subgroup: the oxidative phosphorylation disorders. One in every 5,000 newborns suffers from such a disorder (Skladal et al. 2003), and 20–25% of these cases are isolated complex I deficiencies (Bugiani et al. 2004; Loeffen et al. 2000; Scaglia et al. 2004; Thorburn 2004). Complex I deficiency can be caused by mutations in genes encoding the structural components of the complex, either in mtDNA or nuclear DNA, or by mutations in genes encoding proteins aiding the assembly of the 45 subunits of the complex. Defects in assembly factors form a new class of disease (Nouws et al. 2012), which are probably responsible for 50% of the isolated complex I deficiencies. In 2010, we identified a new complex I assembly factor, ACAD9, and to date 14 patients in 8 families with ACAD9 mutations have been reported (Gerards et al. 2011; Haack et al. 2010; Haack et al. 2012; Nouws et al. 2010). The patients were identified within a period of 3 years, indicating that ACAD9 is a crucial protein in the assembly process, and that mutations in this gene are a relatively frequent cause of complex I deficiency. Most patients with ACAD9 mutations present with exercise intolerance in combination with hypertrophic cardiomyopathy, a thickening of the heart muscle with various presentations, and, when diagnosed in (pre-)adolescent children, a poor prognosis. Hypertrophic cardiomyopathy may be a consequence of mitochondrial dysfunction, caused by mutations in proteins that are involved in fatty acid oxidation, mitochondrial translation, or oxidative phosphorylation. Here we report a new pathogenic mutation in ACAD9, causing hypertrophic cardiomyopathy and early death. In contrast to a previous report (Gerards et al. 2011) of the beneficial effect of riboflavin in ACAD9-deficient patients, this patient did not respond to riboflavin treatment.
Materials and Methods
Clinical Data
The patient was a girl, the first child of healthy first-cousin parents of Turkish origin. The pregnancy was uncomplicated, apart from a maternal febrile urinary infection shortly before birth. The patient was born in gestation week 41+4 by cesarean section, with no clinical signs of asphyxia.
Shortly after birth, she had respiratory insufficiency requiring nasal CPAP therapy with high oxygen supplementation, and she developed a compensated metabolic acidosis with a plasma lactate up to 9 mmol/l. A chest X-ray showed bilateral dense lungs and antibiotic therapy was initiated. At the age of 23 postnatal hours, her condition deteriorated into cardiorespiratory collapse with severe pulseless bradycardia: she was given cardiac massage, intubated, and mechanically ventilated. Plasma lactate rose to a maximum of 20 mmol/l. Echocardiography demonstrated a structurally normal heart with signs of severe pulmonary hypertension. The liver was enlarged, and paralytic ileus was suspected on abdominal X-ray, but this resolved over the following week. She was extubated after 5 days, requiring nasal CPAP for another week. At 3 weeks of age, her condition had improved and she was discharged to a local hospital. Plasma lactate levels had stabilized to 2.2–3.3 mmol/l.
At age 7 weeks, she presented with tachypnea, paradoxical respiration, muscle weakness, and hypotonia. Echocardiography showed severe concentric left ventricle hypertrophy, which progressed over the next month, despite propranolol therapy. At age 3 months, treatment with 100 mg riboflavin per day was initiated. Her condition deteriorated during intercurrent infections, and she died at age 6 months in cardiogenic failure.
Urine organic acids measured three times showed excretion of lactate, citric acid cycle metabolites and a consistent slight elevation of 3-methylglutaconic acid, in the range of 21–65 μmol/mmol creatinine (ref < 9). Plasma amino acids showed elevated alanine (1221 μmol/l, ref 148–475) and low citrulline (7 μmol/l, ref. 8–47).
Enzyme Measurements
Respiratory chain enzyme analysis in muscle and fibroblasts was performed as described before (Cooperstein and Lazarow 1951; Janssen et al. 2007; Wibrand et al. 2010) Values are expressed relative to the mitochondrial reference enzyme citrate synthase (Srere 1969).
SNP Array Analysis and Homozygosity Mapping
DNA was extracted from peripheral blood according to standard procedures and used for a genome-wide search for homozygosity with the Affymetrix Genomewide Human SNP Array 6.0 (Affymetrix Inc., Santa Clara, CA). In brief, DNA was digested with the restriction enzymes StyI and NspI, mixed with StyI and NspI adapters and ligated with the T4 DNA ligase. The ligated DNA was PCR-amplified, pooled, and purified. The purified PCR product was fragmented with DNase I and end-labeled with biotin. The samples were hybridized to an array for 18 h in a hybridization oven. The array was washed, stained, and scanned with an Affymetrix GeneChip scanner 3000. Affymetrix software was used to analyze the data, and homozygositymapper (homozygositymapper.org) was used to identify the homozygous regions.
Mutation Analysis
PCR of genomic DNA was performed with the Promega GoTaq PCR system and the following conditions: 0.2 mM dNTPs, 1 x buffer, 1.5 mM MgCl2, 0.5 mM of each primer, 10 ng template and 1.5U polymerase in a total volume of 50 μl. The PCR program was: 94° C for 2 min, 35 cycles of denaturing at 94° C for 30 s, annealing at 60° C for 30 s, and extension at 72° C for 30 s, and a final extension step of 72° C for 7 min.
Sequencing was performed with the ABI Big Dye Terminator v. 1.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, USA). The sequencing reactions were vacuum-purified with the Montage Seq96 Sequencing Reaction Cleanup Kit (Millipore SA, SaintQuentin, France) and analyzed on an ABI 3130xl Gene Analyzer (Applied Biosystems). The primer sequences are available upon request.
The data were analyzed with Sequencing Analysis Software Version 5.2 from Applied Biosystems, and mutation screening was performed using Mutation Surveyor v.3.1 Software (SoftGenetics). Verified variations were evaluated through the bioinformatice tools of Alamut (interactive Biosoftware). Primer sequences and PCR conditions are available on request.
Blue Native, SDS-PAGE, and In-Gel Activity Assays
Eighty micrograms of solubilized mitochondrial protein was loaded on 5–15% blue native gradient gels or on one-dimensional 10% sodium dodecyl sulfate polyacrylamide gels (SDS-PAGE) as described previously (Calvaruso et al. 2008; Ugalde et al. 2004). After electrophoresis, the gels were processed for either complex I in-gel activity (CI-IGA) analysis or immunoblotting. For blotting, proteins were transferred to a PROTRAN nitrocellulose membrane (Schleicher & Schuell).
Antibodies and ECL Detection
Immunodetection was performed by the use of the following primary antibodies: CI-NDUFA9, CII-SDHA, CV-ATPase-α, and V5 (invitrogen), CII-SDHB (Abcam), Porin (VWR international), ACAD9 (a gift from J. Vockley, University of Pittsburgh, USA). Secondary detection was performed using peroxidase conjugated anti-mouse or anti-rabbit IgGs (Invitrogen). The signal was generated using ECL (Pierce, Rockford, USA).
Lentiviral Complementation of Patient Fibroblasts
ACAD9WT and the Cox 8 leader sequence were cloned into pDONR201 as described before (Hoefs et al. 2008; Nouws et al. 2010). To construct the lentiviral compatible vector containing a C-terminally V5-tagged ACAD9WT or Cox 8 leader sequence, the pDONR201-ACAD9WT and pDONR201-cox8 vectors were recombined with pLenti6.2⁄V5-DEST Gateway Vector using the Gateway LR Clonase II enzyme mix (Invitrogen). HEK293FT cells were transfected to produce lentivirus with pLP1, pLP2, pVSV/G, and one of the two expression vectors according to the manufacturer’s protocol (Invitrogen). The virus was harvested after 72 h and added to the human fibroblasts overnight. The following day, the virus was removed and the medium refreshed. After 48 h, 2.5 μg/ml blasticidin was added to the medium to select the transduced cells. Cells were selected for 14 days, in which time the mock-infected cells (without virus) died. Blasticidin-resistant cells were used for biochemical analysis within six passages after transduction.
Riboflavin Treatment of Patient Cells
Human skin fibroblasts were cultured in medium 199 (Gibco) supplemented with 10% fetal bovine serum (FBS v/v) (PAA), 1% penicillin/streptomycin (Gibco), 1mM sodium pyruvate (Sigma), and 1 μg/ml riboflavin (Sigma) for one week, and the medium was refreshed every 2 days.
Modeling
The ACAD9 modeling was executed as described in (Nouws et al. 2010). Modeling details can be found online at http://www.cmbi.ru.nl/_hvensela/ACAD9/
Results
Enzyme Measurements and Gel Analyses
Analysis of respiratory chain enzyme activity in muscle showed a severe isolated complex I deficiency, with a CI/CS ratio of ≤0.01 (ref. 0.19–1.54). In fibroblasts, the CI/CS ratio was decreased to 0.16 (ref 0.24–0.37). Western blot analysis showed a severe decrease of ACAD9 protein, suggesting that the mutation leads to instability of the protein (Fig. 1a). To compare with other ACAD9 mutations, we included fibroblasts from two previously identified patients in both the SDS and the blue native analysis. Our patient had a severe decrease of complex I in-gel activity and of the total amount of complex I by immunoblot analysis, compared both to controls and to the two other ACAD9 patients (Fig. 1b).
Fig. 1.
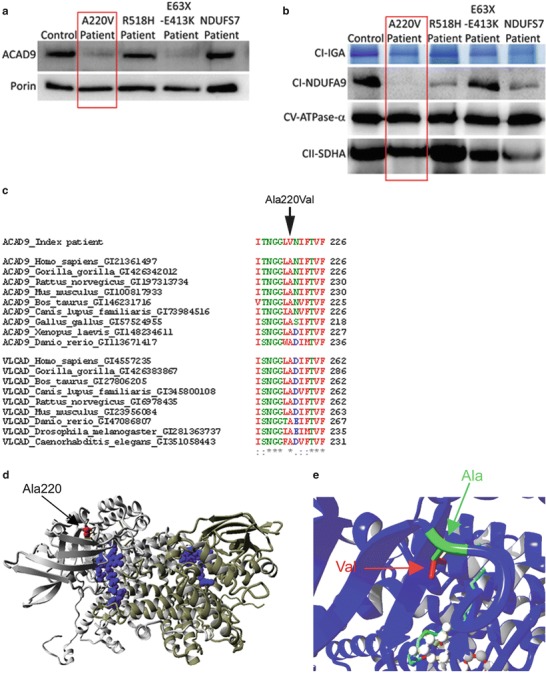
The ACAD9 p.Ala220Val substitution leads to instability of the protein. (a, b) ACAD9 expression level and complex I expression level in control, ACAD9 and NDUFS7 patient fibroblasts. The p.Ala220Val mutation is described in this chapter, whereas the two other ACAD9 patients (R518H and E63X-E413K) and the NDUFS7 patient were described previously (Nouws et al. 2010; Triepels et al. 1999). (a) SDS-PAGE immunodetections of ACAD9 and porin, showing a decreased amount of ACAD9 in the patient. (b) BN-PAGE analysis of complex I in-gel activity (CI-IGA) and western blot immunodetection of the complex I subunit NDUFA9, showing a decreased activity and amount of fully assembled complex I in the patient. Complex V-CV-ATPase-α and complex II-SDHA were used as loading controls. (c) Conservation of the altered amino acids is shown via clustalW alignments. Asterisks (*) indicate identical amino acids, colons (:) indicate conserved substitutions, and periods (.) indicate semiconserved substitutions. (d) Model of ACAD9 (PDB code 3b96) and the p.Ala220Val mutation, showing the two ACAD9 monomers (grey and yellow), the location of the FAD cofactor (blue), and the arrow indicates the location of the mutation (red). (e) The mutation shown in closer detail; the replacement of alanine (in green and indicated by green arrow) by valine leads to an addition of two methylgroups (indicated in red and by a red arrow). Moreover, the mutation is in proximity to the fatty acid (light blue stretch) and FAD (white balls) binding sites in the core of the protein
Gene and Conservation Analyses
A genome-wide search for homozygosity revealed several homozygous regions, altogether comprising 298.5 Mb. No mutations were found in the structural complex I genes NDUFA4, NDUFB4, or NDUFB9 that were located in the homozygous regions. Sequencing of ACAD9 located in a homozygous region on chromosome 3 showed a homozygous missense mutation, c.659C>T (p.Ala220Val). The variant was absent from the dbSNP, ESP, and the 1,000 genomes databases (Altshuler et al. 2010), and in silico prediction by PolyPhen2, SIFT, and Mutation Taster all pointed to a possible pathogenic effect. Both parents were heterozygous carriers of the mutation. The affected amino acid, Ala220, is highly conserved among organisms expressing ACAD9 (Fig. 1c). In addition, this amino acid is also conserved in other ACAD family members like VLCAD (Fig. 1c) and SCAD and MCAD (not shown).
Modeling of the p.Ala220Val Mutation
The affected alanine is located in proximity of the fatty acid and FAD (flavin adenine dinucleotide) binding sites in the core of the protein (Fig 1d–e). The mutation changes alanine to valine, which are both hydrophobic and small amino acids.
Functional Complementation
To confirm that the ACAD9 p.Ala220Val amino acid change caused the complex I deficiency in this patient, we complemented patient fibroblasts with C-terminally V5-tagged wild-type ACAD9. The protein was introduced in the cells by stable lentiviral transduction, and we could demonstrate the presence of V5-tagged ACAD9 on SDS western blot (Fig. 2a). Both complex I amount and activity (CI-IGA) increased, indicating that the wild-type ACAD9 indeed rescued the complex I deficiency in the patient cells (Fig. 2b). Consistent with these results, also spectrophotometric analysis demonstrated an increase in CI/CS ratio from 0.17 to 0.23 after complementation of the patient cells, whereas this did not change in control cells.
Fig. 2.
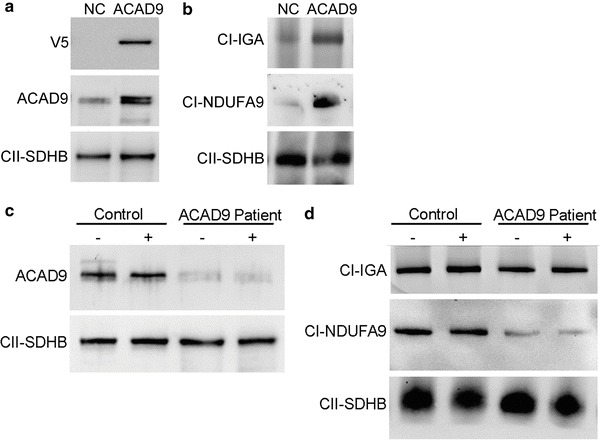
Complementation and riboflavin treatment of patient fibroblasts. (a-b) Lentiviral complementation of patient fibroblasts with negative control V5-tagged COX8 leader sequence, indicated as NC and V5-tagged wild-type ACAD9 as ACAD9. (a) SDS-PAGE immunodetections of V5 and ACAD9 showing the presence of V5-tagged ACAD9. SDHB was used as a loading control. (b) BN-PAGE analysis of complex I in-gel activity (CI-IGA) and western blot immunodetection of the complex I subunit NDUFA9 showing an increase in complex I in-gel activity and amount by complementation with WT ACAD9. Complex II-SDHB was used as a loading control. (c, d) Riboflavin treatment of control and patient fibroblasts. (c) SDS-PAGE immunodetections of ACAD9 and SDHB in control and ACAD9 patient fibroblasts, showing no differences in ACAD9 amounts between fibroblasts cultured without (−) and with (+) riboflavin supplementation. (d) BN-PAGE analysis of complex I in-gel activity (CI-IGA) and western blot immunodetection of complex I subunit NDUFA9 showing no differences in complex I activity or amount between fibroblasts cultured without (−) and with (+) riboflavin supplementation. Complex II-SDHB was used as a loading control
Riboflavin Treatment of Fibroblasts
Since riboflavin has been reported to have a beneficial effect in several cases of complex I deficiency caused by ACAD9 mutations in patients and patient fibroblasts, we decided to test riboflavin treatment in this case as well. The patient did not show clinical improvement after riboflavin administration. We also assayed whether riboflavin had a beneficial effect on the patient fibroblasts, but treatment of the p.Ala220Val patient cell line with riboflavin did not have any effect; it did not stabilize the ACAD9 protein nor did it increase complex I activity or amount (Fig. 2c–d).
Discussion
We report a new pathogenic mutation in the complex I assembly factor ACAD9. The mutation results in a p.Ala220Val substitution that affects a highly conserved amino acid. The ACAD9 protein level was decreased, leading to a pronounced reduction of both complex I amount and activity, and functional complementation with wild-type ACAD9 led to an increase in complex I activity and amount of fully assembled complex I. The affected amino acid is located in the proximity of the fatty acid and FAD binding sites in the core of the protein, which may interfere with their binding. The mutation leads to the substitution of alanine with valine, and although both amino acids are hydrophobic and small, valine may fit less well in the protein structure and possibly pose a sterical hindrance for the side chains of other amino acids. Taken together, these results indicate that the mutation is disease-causing.
Including this patient, 15 patients have been reported up to date with complex I deficiency due to mutations in ACAD9, Tables 1 and 2 show an overview of the patients, the biochemical data, and the clinical findings. The major clinical presentation of this patient was hypertrophic cardiomyopathy, as reported in 10 out of 14 complex I–deficient patients with ACAD9 mutations (Nouws et al. 2010; Haack et al. 2012; Gerards et al. 2011; Haack et al. 2012). Additional symptoms among these ten patients included lactic acidosis, failure to thrive, hypotonia, exercise intolerance, encephalomyopathy, cardiorespiratory depression, mild hearing loss, and short stature. We found a consistent slightly elevated excretion of 3-methylglutaconic acid, which can be an unspecific marker of a respiratory chain disorder. Excretion of 3-methylglutaconic acid was not reported in any of the other ACAD9 patients. The variability in the severity of the disorder was wide, with one patient dying at age 46 days, and one patient being alive at age 18 years (Nouws et al. 2010). The four remaining ACAD9 patients presented with a primarily muscular phenotype with fatiguability, exercise intolerance, and lactic acidosis in childhood, whereas cardiac function was normal (Gerards et al. 2011). Moreover, these patients had a relatively late onset of their symptoms. Three of the patients with the muscular phenotype were homozygous for a p.Arg532Trp mutation, which was also found homozygous in three patients with the cardiomyopathic phenotype (Gerards et al. 2011; Haack et al. 2010). Since the mutation is found in both groups, there does not seem to be an apparent genotype-phenotype correlation; a possible explanation for the clinical differences could be modifying variants in other genes.
Table 1.
Biochemical data on patients with complex I deficiency due to ACAD9 mutations
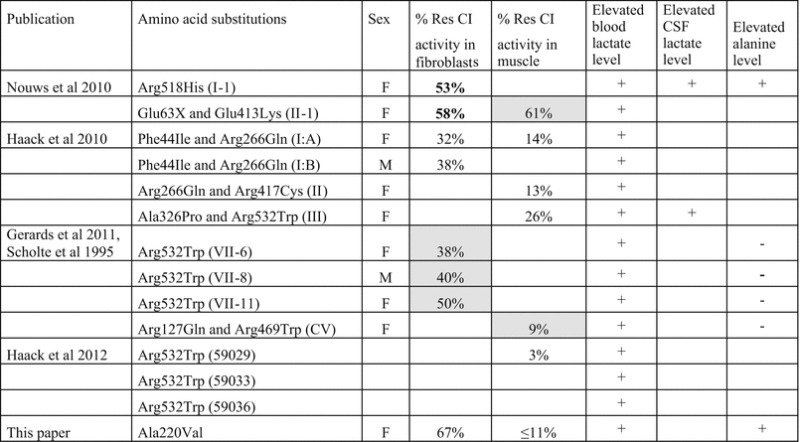
Residual CI activity in fibroblasts = complex I activity normalized for citrate synthase, percentage of lowest control value (values in bold are normalized to COX activity); grey box = percentage of control mean. Residual CI activity in muscle = complex I activity normalized for citrate synthase activity for percentage of lowest control value; grey box = percentage of control mean. + symptom present, – symptom absent, Empty box if information is not available
Table 2.
Clinical findings in patients with complex I deficiency due to mutations in ACAD9
| Publication | Amino acid substitutions | Pregnancy duration | Age on onset | Age of death | Alive at | Riboflavin therapy | Neurological symptoms | Brain abnormalities | Seizures | Psychiomotor dev. delay | Hypotonia | Hearing loss | Respiratory problems | Tachypnea | Dyspnoea | Respiratory disturbance | Other organ failure | Hyperthrophic cardiomyopathy | Hepatomology | Other | Failure to thrive | Exercise intolerance | Short stature |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nouws et al. 2010 | Arg518His (I-1) | Term | 8 m | 18 y | – | – | – | – | + | + | – | – | + | + | + | + | + | ||||||
| Glu63X and Glu413Lys (II-1) | 35 | 4 m | 6 m | + | – | + | + | ||||||||||||||||
| Haack et al. 2010 | Phe44Ile and Arg266Gln (I:A) | 39 w + 6 d | 24 h | 46 d | + | + | – | + | + | + | – | – | |||||||||||
| Phe44Ile and Arg266Gln (I:B) | Birth | 5 y | + | + | – | – | + | – | + | ||||||||||||||
| Arg266Gln and Arg417Cys (II) | Birth | 12 y | – | – | + | – | – | ||||||||||||||||
| Ala326Pro and Arg532Trp (III) | Birth | 2 y | + | – | + | + | – | ||||||||||||||||
| Gerards et al. 2011, Scholte et al. 1995 |
Arg532Trp (VII-6) | 4 y | 31 y | + | – | – | – | – | – | – | + | + | |||||||||||
| Arg532Trp (VII-8) | 4 y | 38 y | + | – | + | v | – | – | – | – | + | ||||||||||||
| Arg532Trp (VII-11) | 4y | 40 y | + | – | + | – | – | – | – | – | + | ||||||||||||
| Arg127Gln and Arg469Trp (CV) | Early CH | 27 y | + | – | – | – | – | + | – | – | – | + | |||||||||||
| Haack et al. 2012 | Arg532Trp (59029) | + | + | + | |||||||||||||||||||
| Arg532Trp (59033) | + | + | + | ||||||||||||||||||||
| Arg532Trp (59036) | + | + | + | ||||||||||||||||||||
| This report | Ala220Val | 41 w + 4 d | Birth | 6 m | – | – | – | + | + | + | + | + | + | + | – |
Dev. developmental, + symptom present, – symptom absent, empty box if information is not available, CH childhood, d days, w weeks, m months, y years
In addition to the aforementioned patients, He and coworkers described two other patients with deficiency of ACAD9 who had a liver phenotype and one patient displaying cardiomyopathy. However, these patients did not have isolated complex I deficiency, and no pathogenic mutations were identified, hence the exact contribution of the ACAD9 mutations to the phenotype of these patients is unclear (He et al. 2007).
Riboflavin treatment has been reported to alleviate symptoms in a few patients with the muscular phenotype (Scholte et al. 1995). The patients reported increased endurance power and disappearance of exercise-related nausea and pain after riboflavin therapy was started. Moreover, one of the patients had had one stroke-like episode at age 13 years, and had no more episodes on riboflavin therapy. After 1–2 years of riboflavin therapy, the patients’ resting lactate decreased and exercise tolerance assessed by ergometry increased.
In vitro treatment of two ACAD9-deficient fibroblast cell lines, which were compound heterozygous for the Phe44Ile and Arg266Gln substitutions resulted in 2.1- and 1.7-fold increases in complex I activity, respectively (Haack et al. 2010). We did not find any increase in ACAD9 protein level or complex I activity on riboflavin treatment of fibroblasts of our patient, in accordance with the lack of a clinical response to treatment with 100 mg riboflavin per day. None of the other patients with the cardiomyopathic phenotype were reported to receive riboflavin treatment.
Riboflavin is a precursor of flavin mononucleotide (FMN) and FAD, which are cofactors in complex I. The mode of action for riboflavin remains elusive, but it has been suggested to increase mitochondrial FAD concentration, which may support FAD binding. This binding could be necessary for the catalytic activity of ACAD9, however, also its folding or stability may improve (Henriques et al. 2010). Riboflavin might also function as an electron acceptor, since fibroblasts from patients with deficiency of NDUFS2, a subunit without FAD or FMN binding sites, responded to riboflavin treatment (Bar-Meir et al. 2001). An explanation for the lack of response to riboflavin treatment in our patient could be the specific location of the mutation, although both Ala220Val and Arg532Trp, found in patients who apparently responded to riboflavin treatment, are located near the FAD binding site. The scarce and contrasting results of riboflavin administration suggest that further studies, both in vivo and in vitro, are needed on riboflavin treatment in patients with complex I deficiency due to ACAD9 mutations to establish the effect of the treatment.
Take-Home Message
ACAD9 mutations are a major cause of complex I deficiency and ACAD9 patients do not always respond to riboflavin treatment.
Footnotes
Competing interests: None declared
Contributor Information
Leo Nijtmans, Email: l.nijtmans@cukz.umcn.nl.
Collaborators: Johannes Zschocke and K Michael Gibson
References
- Altshuler DL, Durbin RM, Abecasis GR, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467(7319):1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Meir M, Elpeleg ON, Saada A. Effect of various agents on adenosine triphosphate synthesis in mitochondrial complex I deficiency. J Pediatr. 2001;139(6):868–870. doi: 10.1067/mpd.2001.118885. [DOI] [PubMed] [Google Scholar]
- Bugiani M, Invernizzi F, Alberio S, et al. Clinical and molecular findings in children with complex I deficiency. Biochim Biophys Acta. 2004;1659(2–3):136–147. doi: 10.1016/j.bbabio.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Calvaruso MA, Smeitink J, Nijtmans L. Electrophoresis techniques to investigate defects in oxidative phosphorylation. Methods. 2008;46(4):281–287. doi: 10.1016/j.ymeth.2008.09.023. [DOI] [PubMed] [Google Scholar]
- Cooperstein SJ, Lazarow A. A microspectrophotometric method for the determination of cytochrome oxidase. J Biol Chem. 1951;189(2):665–670. [PubMed] [Google Scholar]
- Gerards M, van den Bosch BJ, Danhauser K, et al. Riboflavin-responsive oxidative phosphorylation complex I deficiency caused by defective ACAD9: new function for an old gene. Brain. 2011;134(Pt 1):210–219. doi: 10.1093/brain/awq273. [DOI] [PubMed] [Google Scholar]
- Haack TB, Danhauser K, Haberberger B, et al. Exome sequencing identifies ACAD9 mutations as a cause of complex I deficiency. Nat Genet. 2010;42(12):1131–1134. doi: 10.1038/ng.706. [DOI] [PubMed] [Google Scholar]
- Haack TB, Haberberger B, Frisch EM, et al. Molecular diagnosis in mitochondrial complex I deficiency using exome sequencing. J Med Genet. 2012;49(4):277–283. doi: 10.1136/jmedgenet-2012-100846. [DOI] [PubMed] [Google Scholar]
- He M, Rutledge SL, Kelly DR, et al. A new genetic disorder in mitochondrial fatty acid beta-oxidation: ACAD9 deficiency. Am J Hum Genet. 2007;81(1):87–103. doi: 10.1086/519219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriques BJ, Olsen RK, Bross P, Gomes CM. Emerging roles for riboflavin in functional rescue of mitochondrial beta-oxidation flavoenzymes. Curr Med Chem. 2010;17(32):3842–3854. doi: 10.2174/092986710793205462. [DOI] [PubMed] [Google Scholar]
- Hoefs SJ, Dieteren CE, Distelmaier F, et al. NDUFA2 complex I mutation leads to Leigh disease. Am J Hum Genet. 2008;82(6):1306–1315. doi: 10.1016/j.ajhg.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen AJ, Trijbels FJ, Sengers RC, et al. Spectrophotometric assay for complex I of the respiratory chain in tissue samples and cultured fibroblasts. Clin Chem. 2007;53(4):729–734. doi: 10.1373/clinchem.2006.078873. [DOI] [PubMed] [Google Scholar]
- Loeffen JL, Smeitink JA, Trijbels JM, et al. Isolated complex I deficiency in children: clinical, biochemical and genetic aspects. Hum Mutat. 2000;15(2):123–134. doi: 10.1002/(SICI)1098-1004(200002)15:2<123::AID-HUMU1>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Nouws J, Nijtmans L, Houten SM, et al. Acyl-CoA dehydrogenase 9 is required for the biogenesis of oxidative phosphorylation complex I. Cell Metab. 2010;12(3):283–294. doi: 10.1016/j.cmet.2010.08.002. [DOI] [PubMed] [Google Scholar]
- Nouws J, Nijtmans LG, Smeitink JA, Vogel RO. Assembly factors as a new class of disease genes for mitochondrial complex I deficiency: cause, pathology and treatment options. Brain. 2012;135(Pt 1):12–22. doi: 10.1093/brain/awr261. [DOI] [PubMed] [Google Scholar]
- Scaglia F, Towbin JA, Craigen WJ, et al. Clinical spectrum, morbidity, and mortality in 113 pediatric patients with mitochondrial disease. Pediatrics. 2004;114(4):925–931. doi: 10.1542/peds.2004-0718. [DOI] [PubMed] [Google Scholar]
- Scholte HR, Busch HF, Bakker HD, et al. Riboflavin-responsive complex I deficiency. Biochim Biophys Acta. 1995;1271(1):75–83. doi: 10.1016/0925-4439(95)00013-T. [DOI] [PubMed] [Google Scholar]
- Skladal D, Halliday J, Thorburn DR. Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain. 2003;126:1905–1912. doi: 10.1093/brain/awg170. [DOI] [PubMed] [Google Scholar]
- Srere PA. Citrate synthase : [EC 4.1.3.7. Citrate oxaloacetate-421 lyase (CoA-acetylating)] In: John ML, editor. Methods in enzymology. Citric acid cycle. London: Academic Press; 1969. pp. 3–11. [Google Scholar]
- Thorburn DR. Mitochondrial disorders: prevalence, myths and advances. J Inherit Metab Dis. 2004;27(3):349–362. doi: 10.1023/B:BOLI.0000031098.41409.55. [DOI] [PubMed] [Google Scholar]
- Triepels RH, van den Heuvel LP, Loeffen JL, et al. Leigh syndrome associated with a mutation in the NDUFS7 (PSST) nuclear encoded subunit of complex I. Ann Neurol. 1999;45(6):787–790. doi: 10.1002/1531-8249(199906)45:6<787::AID-ANA13>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Ugalde C, Vogel R, Huijbens R, van den Heuvel LP, Smeitink J, Nijtmans L. Human mitochondrial complex I assembles through the combination of evolutionary conserved modules: a framework to interpret complex I deficiencies. Hum Mol Genet. 2004;13(20):2461–2472. doi: 10.1093/hmg/ddh262. [DOI] [PubMed] [Google Scholar]
- Wibrand F, Jeppesen TD, Frederiksen AL, et al. Limited diagnostic value of enzyme analysis in patients with mitochondrial tRNA mutations. Muscle Nerve. 2010;41(5):607–613. doi: 10.1002/mus.21541. [DOI] [PubMed] [Google Scholar]