

Abstract
We evaluated a family with a 16-month-old boy with cirrhosis and hepatocellular carcinoma and his 30-month-old brother with cirrhosis. After failing to identify a diagnosis after routine metabolic evaluation, we utilized a combination of RNA-Seq and whole exome sequencing to identify a novel homozygous p.Ser171Phe Transaldolase (TALDO1) variant in the proband, his brother with cirrhosis, as well as a clinically asymptomatic older 8-year-old brother. Metabolite analysis and enzymatic testing of TALDO1 demonstrated elevated ribitol, sedoheptitol, and sedoheptulose-7P, and lack of activity of TALDO1 in the three children homozygous for the p.Ser171Phe mutation. Our findings expand the phenotype of transaldolase deficiency to include early onset hepatocellular carcinoma in humans and demonstrate that, even within the same family, individuals with the same homozygous mutation demonstrate a wide range of phenotypes.
Introduction
With the advent of next-generation sequencing (NGS), we now have the tools to identify the etiology of even the rarest of familial diseases in a less biased manner (Chung et al. 2009). As we identify the etiology of these diseases by this method, we often redefine the spectrum of the phenotype of rare diseases. For diseases associated with cancer, identifying an underlying hereditary cause has significant implications for treatment, long-term cancer surveillance, and risk stratification for other family members.
We describe a 16-month-old male with hepatocellular carcinoma and a history of hepatomegaly and liver dysfunction with a family history significant for a brother with cirrhosis. The use of genomic analysis of the hepatocellular carcinoma identified a transaldolase (taldo) deficiency, a rare inborn error of metabolism in the pentose phosphate pathway (PPP). Taldo is a reversible enzyme of the non-oxidative branch of the PPP, catalyzing the transfer of a three-carbon keto unit, corresponding to dihydroxyacetone (DHA), from sedoheptulose-7-phosphate (S7P) to glyceraldehyde-3-phosphate (G3P) generating erythrose-4-phosphate (E4P) and fructose-6-phosphate (F6P). The first transaldolase-deficient patient was described by Verhoeven with liver cirrhosis, hepatosplenomegaly, thrombocytopenia, and dysmorphic features (Verhoeven et al. 2001). Since then, 22 additional cases have been described in the literature, but the condition continues to be rare (Verhoeven et al. 2005; Valayannopoulos et al. 2006; Fung et al. 2007; Wamelink et al. 2008b; Tylki-Szymanska et al. 2009; Balasubramaniam et al. 2011; Eyaid et al. 2013). The transaldolase knockout mouse is associated with a significant risk of hepatocellular carcinoma observed in 46% of the mice (Hanczko et al. 2009).
Methods
All individuals provided informed consent, and all studies were approved by the Columbia University Institutional Review Board. Blood and urine specimens were collected. Liver tissue from the proband (II.4) was collected at the time of liver resection and immediately flash frozen and stored at −80 °C.
Homozygosity mapping was performed by genotyping the four children and both parents using an Affymetrix 500k NspI genotyping array according to the manufacturer’s instructions (Affymetrix, Santa Clara, CA) and homozygosity mapper (http://www.homozygositymapper.org/) to identify regions of homozygosity in the 30-month-old brother of the proband (II.3) and the proband (II.4).
RNA was extracted from normal liver by homogenizing tissue and adding to Qiazol (Qiagen) (Chomczynski and Sacchi 1987). Total RNA was purified using RNeasy kit with DNase treatment (Qiagen) and mRNA isolation using a poly-A pulldown (Wang et al. 2009) and reverse transcription to generate cDNA. The cDNA was sequenced using 14 million (normal liver) and 21 million (tumor) 100 bp single-end sequencing reads on a HiSeq2000 according to manufacturer’s recommendations (Illumina; San Diego, CA). The pass filter (PF) reads were mapped to human reference genome hg19 using TopHat (version 2.0.4) (Trapnell et al. 2009). For each read, we allowed up to three mismatches during the mapping. For variant calling, we used SAMTools (Li et al. 2009) combined with additional false-positive filters to call single nucleotide variants (SNVs) and short indels. For each candidate variant site, we allowed the maximal read depth = 1,000,000, minimum mapping quality = 5, minimum base quality = 17. We removed the variant calls in which the fraction of reads carrying the non-reference allele was less than a 10 % threshold. For exome sequencing, genomic DNA was extracted from blood, fragmented and captured with the Agilent SureSelect XT Human All Exon v.2 44Mb capture kit. Libraries were sequenced on an Illumina HiSeq2000 with 100bp paired end reads at average on-target depth of coverage of 60×.
Urine polyols, heptuloses, and sugar-phosphates (sugar-P) were measured using liquid chromatography-tandem mass spectrometry (LC-MS/MS) and gas chromatography with flame ionization detection (Jansen et al. 1986, Wamelink et al. 2005a, 2007).
Transaldolase enzyme activity in lymphoblasts was measured by incubating the cells for 2 h with ribose-5P and measuring the sugar-P formed by LC-MS/MS (Wamelink et al. 2005b).
Results
Clinical Description
The proband (II.4 in Fig. 1) presented at 7 months of age with tachypnea and was found on physical examination to have hepatomegaly, which was confirmed on ultrasound. He was also found to have liver dysfunction with elevated aspartate aminotransferase (AST) of 228 (normal 12–36 U/L), alanine aminotransferase (ALT) of 104 (normal 7–41 U/L), and alpha-fetoprotein (AFP) of 2,108 (a marker for hepatocellular carcinoma, normal 0–9 ng/mL). Serum acylcarnitines, amino acids, very long chain fatty acids, copper, ceruloplasmin, iron, urine organic acids, and alpha 1 antitrypsin genotype were all normal. He was the product of a full-term pregnancy without prenatal or neonatal complications and had a normal New York State newborn screen. His growth and development were normal. His family history was significant for a 30-month-old brother (II.3) who had a history of hepatomegaly and elevated AST and ALT that progressed to cirrhosis. Both of his parents are from the Gambia, and there is no known history of consanguinity. All three pregnancies were uncomplicated by intercurrent illness, diabetes, or hypertension. The mother used no prescription or recreational drugs, alcohol or tobacco during any pregnancy. Routine prenatal testing was unremarkable. Birth weights were unremarkable, ranging from 5 lbs to 6 lbs 4 oz (Table 1). Each was discharged within 3 days of birth without complications. The proband had an abdominal MRI at 13 months of age that showed a 1.7 cm × 1.6 cm × 2.1 cm mass in segment 5 and another 1.3 cm × 1 × 1.4 cm mass in segment 6. The masses were biopsied, and both cores demonstrated well-differentiated hepatocellular carcinoma (Fig. 2) within a background of cirrhosis with mild inflammation. There was no evidence of storage material. The proband received a cadaveric liver transplant at 16 months of age at which time liver specimens were obtained for genomic analysis. The proband had normal cardiac structure and function with an ejection fraction of 68% and normal cardiac dimensions, normal renal function with blood urea nitrogen (BUN) of 8–19 mg/dL (7–20 mg/dL), creatinine of 0.2 mg/dL (0.6–1.2 mg/dL), and normal platelet counts ranging from 281 to 402 (165–415). There was no succinylacetone in the urine of the proband to suggest tyrosinemia.
Fig. 1.
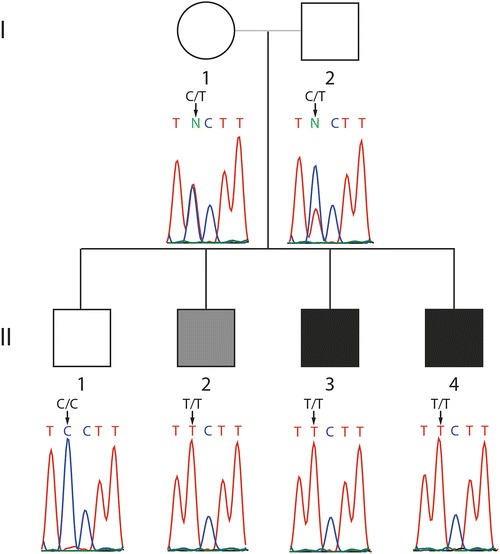
Pedigree with clinically affected individuals indicated in black and biochemically affected but asymptomatic individual indicated in gray
Table 1.
Metabolic and genetic results of four siblings. Urinary excretion of polyols, heptuloses, and sugar-phosphates in mmol/mol creatinine. Urine of II.4 collected after liver transplant
| Phenotype | II.4 | II.3 | II.2 | II.1 | |
|---|---|---|---|---|---|
| Age at time of sample collection | 1 year | 3 years | 9 years | 13 years | Control values |
| Clinical status | Proband with hepatocellular carcinoma and cirrhosis | Cirrhosis | Asymptomatic | Unaffected | |
| Birth weight | 6 lbs 4 oz | 5 lbs | 5 lbs 4 oz | ||
| Genetic status at TALDO1 | Homozygous Phe171 | Homozygous Phe171 | Homozygous Phe171 | Homozygous Ser171 | |
| ALT | 228 | 57 | 16 | 19 | 7–41 U/L |
| AST | 104 | 70 | 20 | 23 | 12–36 U/L |
| GGT | 70 | 20 | 21 | 15 | 9–58 U/L |
| AFP | 2,108 | 15.2 | 4.6 | 0.8 | 0–9 ng/mL |
| Platelet count | 281–402 | 171–325 | 273–275 | 165–415 | |
| BUN | 8–19 | 7–18 | 14 | 7–20 mg/dL | |
| Creatine | 0.2 | 0.3 | 0.47 | 0.6–1.2 mg/dL | |
| Erythritola | 203 | 117 | 103 | 22 | 1–2 years: 76–192 2–6 years: 55–105 6–18 years: 35–179 |
| Ribitola | 54 | 60 | 54 | 5 | 1–2 years: 9–24 2–6 years: 8–11 6–18 years: 4–11 |
| Arabitola | 152 | 124 | 77 | 17 | <89c |
| Sedoheptitolb | 2 | 2 | 1.1 | <1 | <1 |
| Perseitolb | <1 | 2 | 3 | <1 | <1 |
| Sedoheptuloseb | 600 | 300 | 270 | 2.7 | <9 |
| Mannoheptuloseb | Disturbedd | Disturbedd | Disturbedd | 2.8 | <3 |
| Sedoheptulose-7Pb | 0.93 | 0.96 | 1.7 | n.d. | <0.07c |
| Ribose-5Pb | 0.76 | 0.23 | 0.26 | n.d. | <0.13c |
| Ribulose-5P + xylulose-5Pb | 0.84 | 0.53 | 0.46 | 0.09 | <0.44c |
| Transaldolase activity | Undetectable | Undetectable | Undetectable | Normal |
Fig. 2.
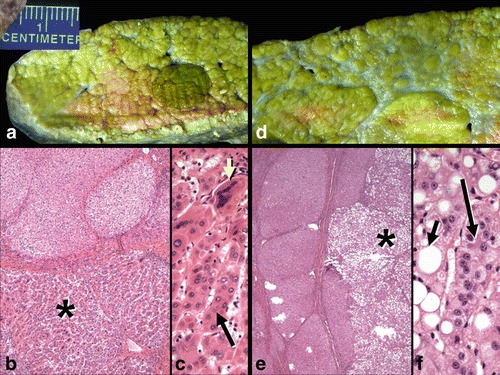
Explanted liver specimen from II.4 showing mixed micro-macronodular cirrhosis and several well-differentiated hepatocellular carcinomas. (a) A well-circumscribed carcinoma (dark green nodule) is present. (b) Microscopic section of the lesion seen in panel A shows an inactive cirrhosis at top and the hepatocellular carcinoma (*) below. (c) High magnification of the tumor in panel B shows partial microtrabecular pattern (long arrow) and many giant, multinucleated tumor cells (short yellow arrow). (d) Another nodule is grossly pale and is a steatotic hepatocellular carcinoma. (e) The underlying inactive cirrhosis (left) contrasts to the multilobulated fatty carcinoma at right (*). (f) The steatotic carcinoma shows many neoplastic hepatocytes with large fat vacuoles (short arrow) as well as focal microtrabecular growth (long arrow). (Hematoxylin and eosin stains; b: x 40; c: x 200; e: x 40; f: x 200)
The affected child has three older brothers (Fig. 1). One brother (II.3), as mentioned above, had a history of hepatomegaly and cirrhosis associated with increased AST and ALT diagnosed at 1 year of age. Acylcarnitines, urine organic acids, and serum amino acids were normal. A liver biopsy demonstrated cirrhosis. II.3 has normal platelet counts ranging from 171 to 325 (165–415) and normal renal function with BUN of 7–18 mg/dL (7–20 mg/dL), creatinine of 0.3 mg/dL (0.6–1.2 mg/dL).
The oldest two brothers, ages 12 and 8 years old (II.1 and II.2), have normal liver size and liver function studies. II.2 has normal platelet counts ranging from 273 to 275 (165–415) and normal renal function with BUN of 14 mg/dL (7–20 mg/dL), creatinine of 0.47 mg/dl (0.6–1.2 mg/dL).
Both parents are clinically asymptomatic but have not undergone clinical investigation.
Genomic Analysis
Based upon the family history, we assumed either an autosomal-recessive or X-linked mode of inheritance. There were no stretches of homozygosity > 1 Mb shared by II.3 and II.4.
We identified 1,901 homozygous rare/novel variants in the RNA-Seq data from the liver of the proband. Of these variants, 121 were either novel or appeared in the HGMD database of known mutations. Of these, 21 were either in the HGMD database or called damaging by SIFT. These variants were filtered to remove variants in the EVS database (http://evs.gs.washington.edu/EVS/) to eliminate those variants with an allele frequency in African Americans of greater than 5 %, leaving only five variants. One of these five variants (rs59947000) was a common indel. Another variant is in a repeat region that presumably is an alignment error. Two variants were not confirmed by Sanger sequencing. The remaining variant (aligned to 22 alternate and no reference alleles), confirmed to be homozygous in the proband, was c.512C>T (p.Ser171Phe) at TALDO1 (chr11: 763394 in hg19).
We identified 24,967 variants in the coding regions by whole exome sequencing in blood from the proband. As expected, many more variants were identified by exome sequencing compared to RNA-Seq. Of these variants, 8,584 were homozygous, and 233 were novel and homozygous; 146 were non-synonymous; 26 were predicted to be damaging by SIFT. After filtering out common SNPs found in the EVS dataset for African Americans (allele frequency >5 %), 11 variants remained. Of these 11 variants, 5 were confirmed by Sanger sequencing as homozygous in the proband. However, only the p.Ser171Phe TALDO1 variant was also homozygous in the other affected brother, II.3.
Genotyping of all the family members for c.512C>T TALDO1 identified that both parents were carriers, the two clinically affected children (II.3 and II.4) were homozygous mutant (T/T), and unaffected brother (II.1) was homozygous wild type (C/C), and, surprisingly, a clinically asymptomatic brother (II.2) was homozygous for the mutation (T/T).
To functionally evaluate the transaldolase activity, urine samples of the four siblings (collected after liver transplant in II.4) were analyzed for polyols, heptuloses, and sugar-P (Table 1). All three boys that were homozygous 171Phe TALDO1 had elevated excretion of ribitol, sedoheptitol, perseitol, sedoheptulose, and sedoheptulose-7P (Table 1) consistent with transaldolase deficiency. Erythritol and arabitol were only mildly elevated in the two youngest children (II.3 and II.4) and normal in II.2. Mannoheptulose could not be separately measured due to co-elution with sedoheptulose. The sugar-P ribose-5-P and xylulose-5P + ribulose-5P were also highly elevated in urine of II.3 and II.4 and mildly elevated in II.2. II.1 had a normal metabolite profile consistent with his normal genotype.
We directly tested the transaldolase activity in lymphoblasts. After incubating lymphoblasts with ribose-5P, there was normal formation of glucose-6P + fructose-6P in the controls and II.1 while there was no formation of glucose-6P + fructose-6P in II.2, II.3, and II.4, confirming Taldo deficiency (Table 1).
Discussion
We have identified a family with three children with a novel homozygous p.Ser171Phe mutation in TALDO1 associated with complete enzymatic deficiency of TALDO1 and a clinical course of hepatomegaly progressing to cirrhosis and hepatocellular carcinoma by 16 months of age. Intriguingly, within the same family, another sibling has cirrhosis but no hepatocellular carcinoma by age 30 months, and one 8-year-old child is clinically asymptomatic with normal liver size, function, and histology. None of the affected children have evidence of renal impairment, thrombocytopenia, or cardiac disease. It is unclear what is responsible for the clinical variability between individuals with the same TALDO1 genotype and whether there might have previously been a phenotype in the oldest affected brother. In two other families, a deletion of Ser171 was found in the TALDO1 gene as the cause of TALDO deficiency (10–11). This amino acid is part of a highly conserved region (Thorell et al. 2000). These data, the fact that serine 171 is highly conserved, and that this variant was not detected in a cohort of 13,005 alleles (EVS), indicate that this variant is pathogenic. Taldo deficiency in humans has been associated with a range of phenotypes from intrauterine lethality associated with fetal multimalformation syndrome and hydrops fetalis to the more common presentation of cirrhosis, liver failure, hepatosplenomegaly, anemia, thrombocytopenia, dysmorphia, congenital heart defects, and tubulopathy (Verhoeven et al. 2001, 2005; Valayannopoulos et al. 2006; Fung et al. 2007; Wamelink et al. 2008b; Tylki-Szymanska et al. 2009; Balasubramaniam et al. 2011; Eyaid et al. 2013). Our family fits into the range of phenotypes most commonly observed; however, only the liver is affected and without extrahepatic involvement. This is the first association of transaldolase deficiency with hepatocellular carcinoma and an asymptomatic presentation to our knowledge.
Lower concentrations of the polyols erythritol and arabitol could be relevant for the asymptomatic phenotype in II.2, but could also be related to the older age, since in other patients the polyol concentrations tend to be highest in the neonatal period and accumulate less when they are older (Wamelink et al. 2008a).
Taldo1+/− and Taldo1−/− mice are 27- and 79-fold more likely to spontaneously develop liver cirrhosis and nodular dysplasia, respectively, than their wild-type Taldo1+/+ littermates (Hanczko et al. 2009). Seventeen percent of Taldo1+/− and 46% of Taldo1−/− mice spontaneously developed hepatocellular carcinoma, and there was evidence of significant oxidative stress in the animal model with accumulation of sedoheptulose-7P, failure to recycle ribose 5-phosphate, depleted NADPH and glutathione, and increased production of lipid hydroperoxides. When Taldo1−/− mice were administered the antioxidant N-acetylcysteine, there was a marked reduction in the incidence of hepatocellular carcinoma (Hanczko et al. 2009), supporting the hypothesis that oxidative stress is responsible for tumorigenesis and possibly suggesting a strategy to prevent hepatocellular carcinoma in these patients. Because of the effectiveness of the antioxidant strategy in the mice, we are treating the three children with antioxidants.
Taldo deficiency is particularly challenging to diagnose with routine clinical tests. Routine metabolic tests including acylcarnitines, urine organic acids, serum amino acids, very long chain fatty acids, copper, ceruloplasmin, iron, alpha 1 antitrypsin genotype were all normal in this patient, and there was no identifiable storage material in hepatocytes or unique pathology. Lack of routine measurement of urinary polyols may account for the small number of recognized cases. In this case, homozygosity mapping did not identify genomic regions upon which to focus. Only with the advent of NGS were we able to make a diagnosis because transaldolase deficiency had not previously been associated with hepatocellular carcinoma in humans and was therefore not considered in the original differential diagnosis.
Notably, II.4 continues to have an abnormal metabolic profile in the urine, characteristic of transaldolase deficiency even after a liver transplant, suggesting that other tissues also contribute significantly to pentose phosphate metabolism. Having a definitive diagnosis for the proband and his siblings will now allow for surveillance of liver and kidney functions and hepatocellular carcinoma in the future.
We identified the disease mutation using both RNA-Seq and exome sequencing. However, the reduced cost of the experiment and the significantly smaller number of variants (~5x less) generated from the RNA-Seq data suggest that this can offer a significant advantage to genomic analysis when the appropriate tissue for analysis is available since the analysis will focus only on genes expressed within the tissue of interest. Mutations that lead to nonsense-mediated decay would, however, be expected to pose a challenge if only the sequence itself were analyzed, and RNA expression levels should also be included in the analysis. The availability of RNA-seq data from many primary tissues (such as GTEx projects) in healthy individuals will provide a background model for such analysis.
Transaldolase can now be added to the list of other inborn errors associated with increased risk of hepatocellular carcinomas including tyrosinemia and glycogen storage disease IV. It would be interesting to compare our RNA-Seq results with those of other inborn errors of metabolism to better understand if there are common drivers of carcinogenesis in inborn errors of metabolism and better define the mechanism of carcinogenesis.
Acknowledgments
Birthe Roos and Erwin Jansen are kindly acknowledged for their analytical contributions.
One-Sentence Summary
We utilized a combination of RNA-Seq and whole exome sequencing to identify a novel homozygous p.Ser171Phe Transaldolase (TALDO1) mutation in a 16-month-old child to develop cirrhosis and hepatocellular carcinoma and biochemically confirmed that three affected siblings demonstrate a wide range of phenotypes from being asymptomatic to cirrhosis to hepatocellular carcinoma.
Conflict of Interest
Charles A. LeDuc, Elizabeth E. Crouch, Ashley Wilson, Jay Lefkowitch, Mirjam M.C. Wamelink, Cornelis Jakobs, Gajja S Salomons, Xiaoyun Sun, Yufeng Shen, and Wendy K. Chung declare that they have no conflict of interest.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000 (5). Informed consent was obtained from all patients for being included in the study.
Details of the Contributions of Individual Authors
CAL and WKC wrote the manuscript with assistance from EEC, YS, MW, CJ, and GS. JL did the histological analysis. MW, CJ, and GS did the metabolic analysis. YS and XS did the alignment and variant calling. CAL and EEC tested and analyzed the variants. AW and WKC did the clinical workup on the patient. The study was designed by WKC.
Footnotes
Competing interests: None declared
Contributor Information
Wendy K. Chung, Email: Wkc15@columbia.edu
Collaborators: Johannes Zschocke and K Michael Gibson
References
- Balasubramaniam S, Wamelink MM, Ngu LH, et al. Novel heterozygous mutations in TALDO1 gene causing transaldolase deficiency and early infantile liver failure. J Pediatr Gastroenterol Nutr. 2011;52(1):113–116. doi: 10.1097/MPG.0b013e3181f50388. [DOI] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162(1):156–159. doi: 10.1016/0003-2697(87)90021-2. [DOI] [PubMed] [Google Scholar]
- Chung WK, Shin M, Jaramillo TC, et al. Absence epilepsy in apathetic, a spontaneous mutant mouse lacking the h channel subunit, HCN2. Neurobiol Dis. 2009;33(3):499–508. doi: 10.1016/j.nbd.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyaid W, Al Harbi T, Anazi S et al (Jan 12 2013) Transaldolase deficiency: report of 12 new cases and further delineation of the phenotype. J Inherit Metab Dis (Epub ahead of print) [DOI] [PubMed]
- Fung CW, Siu S, Mak C, Poon G, et al. A rare cause of hepatosplenomegaly-Transaldolase deficiency. J Inherit Metab Dis. 2007;30(Suppl 1):62. [Google Scholar]
- Hanczko R, Fernandez DR, Doherty E, et al. Prevention of hepatocarcinogenesis and increased susceptibility to acetaminophen-induced liver failure in transaldolase-deficient mice by N-acetylcysteine. J Clin Invest. 2009;119(6):1546–1557. doi: 10.1172/JCI35722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen G, Muskiet FA, Schierbeek H, Berger R, van der Slik W. Capillary gas chromatographic profiling of urinary, plasma and erythrocyte sugars and polyols as their trimethylsilyl derivatives, preceded by a simple and rapid prepurification method. Clin Chim Acta. 1986;157(3):277–293. doi: 10.1016/0009-8981(86)90303-7. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, et al. The sequence alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorell S, Gergely P, Jr, Banki K, Perl A, Schneider G. The three-dimensional structure of human transaldolase. FEBS Lett. 2000;475(3):205–208. doi: 10.1016/S0014-5793(00)01658-6. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tylki-Szymanska A, Stradomska TJ, Wamelink MM, et al. Transaldolase deficiency in two new patients with a relative mild phenotype. Mol Genet Metab. 2009;97(1):15–17. doi: 10.1016/j.ymgme.2009.01.016. [DOI] [PubMed] [Google Scholar]
- Valayannopoulos V, Verhoeven NM, Mention K, et al. Transaldolase deficiency: a new cause of hydrops fetalis and neonatal multi-organ disease. J Pediatr. 2006;149(5):713–717. doi: 10.1016/j.jpeds.2006.08.016. [DOI] [PubMed] [Google Scholar]
- Verhoeven NM, Huck JH, Roos B, et al. Transaldolase deficiency: liver cirrhosis associated with a new inborn error in the pentose phosphate pathway. Am J Hum Genet. 2001;68(5):1086–1092. doi: 10.1086/320108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhoeven NM, Wallot M, Huck JH, et al. A newborn with severe liver failure, cardiomyopathy and transaldolase deficiency. J Inherit Metab Dis. 2005;28(2):169–179. doi: 10.1007/s10545-005-5261-6. [DOI] [PubMed] [Google Scholar]
- Wamelink MM, Smith DE, Jakobs C, Verhoeven NM. Analysis of polyols in urine by liquid chromatography-tandem mass spectrometry: a useful tool for recognition of inborn errors affecting polyol metabolism. J Inherit Metab Dis. 2005;28(6):951–963. doi: 10.1007/s10545-005-0233-4. [DOI] [PubMed] [Google Scholar]
- Wamelink MM, Struys EA, Huck JH, et al. Quantification of sugar phosphate intermediates of the pentose phosphate pathway by LC-MS/MS: application to two new inherited defects of metabolism. J Chromatogr B Analyt Technol Biomed Life Sci. 2005;823(1):18–25. doi: 10.1016/j.jchromb.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Wamelink MM, Smith DE, Jansen EE, Verhoeven NM, Struys EA, Jakobs C. Detection of transaldolase deficiency by quantification of novel seven-carbon chain carbohydrate biomarkers in urine. J Inherit Metab Dis. 2007;30(5):735–742. doi: 10.1007/s10545-007-0590-2. [DOI] [PubMed] [Google Scholar]
- Wamelink MM, Struys EA, Jakobs C. The biochemistry, metabolism and inherited defects of the pentose phosphate pathway: a review. J Inherit Metab Dis. 2008;31(6):703–717. doi: 10.1007/s10545-008-1015-6. [DOI] [PubMed] [Google Scholar]
- Wamelink MM, Struys EA, Salomons GS, Fowler D, Jakobs C, Clayton PT. Transaldolase deficiency in a two-year-old boy with cirrhosis. Mol Genet Metab. 2008;94(2):255–258. doi: 10.1016/j.ymgme.2008.01.011. [DOI] [PubMed] [Google Scholar]
- Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10(1):57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]