

Abstract
Infantile sialic acid storage disease (ISSD) is a lysosomal storage disease characterized by accumulation of covalently unlinked (free) sialic acid in multiple tissues. ISSD and Salla disease (a predominantly neurological disorder) are allelic disorders caused by recessive mutations of a lysosomal anionic monosaccharide transporter, SLC17A5. While Salla disease is common in Finland due to a founder-effect mutation (p.Arg39Cys), ISSD is comparatively rare in all populations studied.
Here, we describe the clinical and molecular features of two unrelated Canadian Inuit neonates with a virtually identical presentation of ISSD. Both individuals presented antenatally with fetal hydrops, dying shortly following delivery. Urinary free sialic acid excretion was markedly increased in the one case in which urine could be obtained for testing; postmortem examination showed a picture of widespread lysosomal storage in both. Both children were homozygous for a novel splice site mutation (NM_012434:c.526-2A>G) resulting in skipping of exon 4 and an ensuing frameshift. Analysis of a further 129 pan-Arctic Inuit controls demonstrated a heterozygous carrier rate of 1/129 (~0.4 %) in our sample. Interestingly, lysosomal enzyme studies showed an unexplained ninefold increase in neuraminidase activity, with lesser elevations in the activities of several other lysosomal enzymes. Our results raise the possibility of a common founder mutation presenting as hydrops in this population. Furthermore, if confirmed in subsequent cases, the marked induction of neuraminidase activity seen here may prove useful in the clinical diagnosis of ISSD.
Introduction
Defects of the sialic acid transporter SLC17A5 cause a lysosomal storage disease characterized by systemic accumulation of free sialic acid in a wide range of tissues (Verheijen et al. 1999). SLC17A5-related conditions include infantile sialic acid storage disease (ISSD; OMIM #269920, a lethal multisystem disorder), Salla disease (OMIM #604369, a slowly progressive, predominantly neurological condition), and intermediate phenotypes (Aula et al. 2000; Verheijen et al. 1999). Recently, SLC17A5 mutations have also been identified in children and adults with cerebral palsy-like chronic encephalopathy; thus, the recognized spectrum of clinical phenotypes continues to expand (Debray et al. 2011; Mochel et al. 2009, 2010). ISSD is situated at the opposite (severe) end of the SLC17A5 disease spectrum; cardinal features include profound developmental delay, failure to thrive, hepatosplenomegaly, coarse facies, hypopigmentation, and (typically) death in infancy (Lemyre et al. 1999). Findings seen in a subset of cases include dysostosis multiplex, cardiomegaly, heart failure, and prematurity. A majority of ISSD cases demonstrate fetal hydrops and/or ascites; hence, this can be considered a prenatal-onset condition (Froissart et al. 2005). General genotype-phenotype correspondences have been established, and (with exceptions) most Salla disease patients have at least one copy of the common Finnish founder allele, p.Arg39Cys (NM_012434.4:c.115C>T) bearing some residual transporter activity in vitro (Aula et al. 2000; Morin et al. 2004; Verheijen et al. 1999; Wreden et al. 2005; Mochel et al. 2009). In contrast, a wide range of mutation types are seen in ISSD; those studied in vitro appear to be functionally null (Aula et al. 2000; Morin et al. 2004; Myall et al. 2007; Ruivo et al. 2008; Wreden et al. 2005).
Previous reports have described SLC17A5 mutations in Finnish, Swedish, Danish, Italian, French, Polish, Yugoslav, Turkish, Bedouin, Japanese, and North American (including French Canadian) patients (Biancheri et al. 2002, 2005; Coker et al. 2009; Erikson et al. 2002; Landau et al. 2004; Nakano et al. 1996; Sonderby Christensen et al. 2003; Tylki-Szymanska et al. 2003; Verheijen et al. 1999). Apart from the common p.Arg39Cys allele, no other founder alleles have been described. Here, we describe the clinical features of two unrelated affected individuals of non-consanguineous Canadian Inuit descent. SLC17A5 sequencing identified both probands to be homozygous for a novel splice site mutation, making this the first report of ISSD in the Inuit, and raising the possibility of a common ancestral mutation in this population.
Patient 1
Patient 1 was the product of a spontaneous pregnancy to healthy parents; no infectious or teratogenic risks were disclosed. Fetal hepatomegaly and ascites were first noted at 20 weeks’ gestational age; karyotype was normal and maternal TORCH screen was negative. Increasing abdominal girth, cardiomegaly, and facial edema were apparent by 29 weeks. Fetal paracentesis yielded 700 cc of yellow, non-chylous fluid. Spontaneous preterm vaginal delivery of a male weighing 1,769 g (+1–+2SD) ensued at 31+ 4 weeks. Apgar scores were 4 at 1 min and 7 at 5 min; the child was intubated, received surfactant, and was ventilated with high-frequency oscillator in 100 % oxygen. Despite these measures, the patient was critically unstable for the first several hours of life, with a persistent mixed acidosis (pH < 6.90) and poor oxygen saturations. The clinical picture stabilized by 12 h of age, with resolution of acidosis and improved oxygenation.
Newborn examination was significant for dysmorphic facies, massive hepatomegaly, significant peripheral edema, and bilateral talipes. CBC and Coombs’ test were negative, and echocardiograph showed a structurally normal heart. The child’s general condition remained poor, with marked hypotonia, absence of spontaneous movement, and complete reliance on ventilator support. The NICU course was punctuated by two abrupt episodes of decompensation, during which laboratory studies showed a nonspecific biochemical picture of hypoglycemia, hyponatremia, and mild lactic acidosis; these were attributed to suspected ventilator-associated pneumonia, and responded to empiric antibiotic treatment. Repeated red cell and platelet transfusions were required due to persistent anemia and thrombocytopenia. There was evidence of an ongoing acute-phase response, with C-reactive protein levels ranging between 44 and 222 mg/L (upper normal limit = 8 mg/L), a marked left shift, and ongoing significant bandemia (0.41 − 8.8 × 109/L). The patient died on day 26 of life due to respiratory failure despite maximal ventilation. On autopsy, deposition of foamy, clear cytoplasmic material was seen in neural cell bodies throughout the central and peripheral nervous systems, as well as in the pituitary gland, liver, lymph nodes, and pancreatic islet cells (Fig. 1).
Fig. 1.
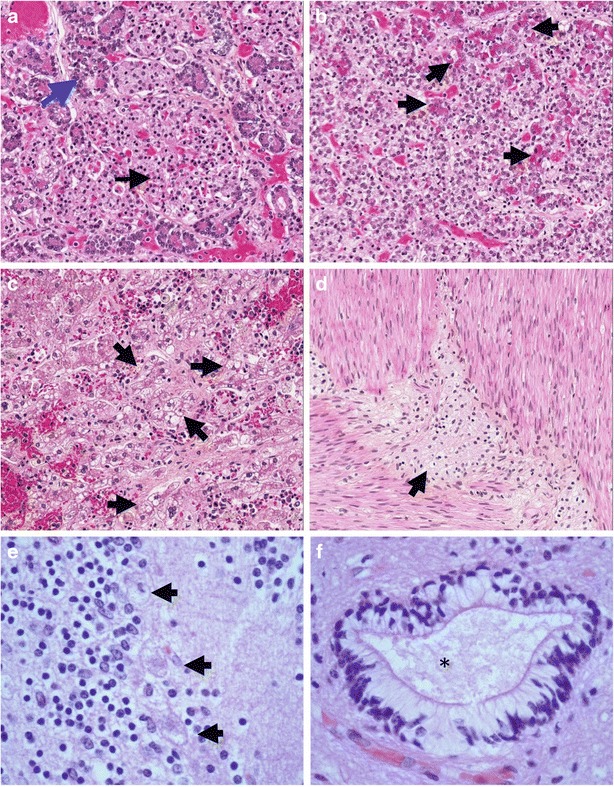
Pathologic findings in Patient 1. (a) Section through pancreas showing enlarged, vacuolated islet cells (black arrow), with relatively normal-appearing acinar cells (blue arrow) at periphery (b) Pituitary gland contains a majority of pale, vacuolated cells with a foamy appearance; some acidophilic cells with a superimposed foamy quality (arrows) are seen (c) Sections of liver show granular, swollen, pale, foamy cells throughout (some clusters indicated by arrows). The stored material does not stain by Oil Red-O or periodic acid-Schiff histochemical stains (data not shown) (d) Ganglion cells of the myenteric plexus (arrow) have enlarged, pale cytoplasm with a foamy quality (e) Cerebellar cortex: three Purkinje cells are swollen, with cytoplasmic accumulation of foamy storage material and loss of Nissl substance (arrows) (f) Ependymal canal, spinal cord: ependymal cells have a clear cytoplasm due to presence of storage material (asterisk indicates center of canal)
Biochemical investigations in this patient showed the following: Urine oligosaccharides showed increased excretion of free sialic acid, providing the major clue to diagnosis. Interestingly, repeat urine oligosaccharide on day 19 showed a normal pattern. Mucopolysaccharide screen was initially too dilute for analysis; repeat on day 19 was negative. Urine organic acids were unremarkable apart from elevated lactate and pyruvate. Plasma amino acids were normal. Lactate was high (8.5 mmol/L) immediately following birth, but normalized rapidly over the following 4 h. Ammonia and liver enzymes were normal. The most striking finding on enzymology of cultured skin fibroblasts was a nearly tenfold increase in neuraminidase activity, with less marked elevations of several other lysosomal enzyme activities (Table 1). Plasma hexosaminidases were not consistent with I-cell disease. NPC1 and NPC2 sequencing were normal.
Table 1.
Lysosomal enzyme activities in ISSD
| Cultured fibroblast enzyme activities (nmol/h/mg) | |||
|---|---|---|---|
| Enzyme | Reference interval | Patient 1 | Patient 2 |
| Neuraminidase | 11.9–20.1 | 181 | 199 |
| β-Galactosidase | 335–435 | 629 | 789 |
| β-Glucuronidase | 105–233 | 95 (93) | 141 |
| β-Glucocerebrosidase | 71–108 | 237 | NA |
| Sphingomyelinase | 56–113 | 158 | NA |
| Arylsulfatase A | 22–50 | 70 | 78 |
| Acid lipase | NA | 801 | 1,108 |
| α-Mannosidase | 64–184 | 50 | 146 |
| Total hexosaminidase | 8160–11,500 | 18,329 | 20,018 |
| Hexosaminidase A | 390–750 | 1,925 | 2,155 |
| Serum hexosaminidases (nmol/h/mL) | ||
|---|---|---|
| Enzyme | Reference interval | Patient 1 |
| Total serum hexosaminidase | 439–1,300 | 572 |
| Serum hexosaminidase A | 30–45 % of total | 249 (43.5 % of total) |
Lysosomal enzyme activities were measured from cultured skin fibroblast extracts in the laboratory of Dr. T. Rupar according to standard methods, in triplicate with simultaneous triplicate measurement of a same-day control specimen. Skin biopsy for fibroblast culture was obtained antemortem in Patient 1, and postmortem in Patient 2. Serum hexosaminidases were tested at the Hospital for Sick Children, Toronto, Canada, according to standard methods
NA Not available
Given our clinical suspicion of ISSD, SLC17A5 sequencing was performed. This showed a homozygous change, NM_012434:c.526-2A>G, situated within the exon 4 splice acceptor site. The effect of this sequence change on splicing was examined by reverse transcriptase PCR using primers situated in exons 3 and 5 (Fig. 2a,b). While a control sample produced a band with the expected size of 420 bp, the patient’s sample showed a smaller band of approximately 350 bp. Direct sequence analysis of this smaller fragment confirmed skipping of exon 4 (r.526_613del) (Fig. 2c). This change is predicted to result in deletion of 28 amino acid residues, a frameshift, and consequently in a prematurely truncated protein (p.Gly176_Ala204delinsfs*7). This mutation is not represented in any of the following catalogues of variation: dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP/), 1,000 genomes (http://www.1000genomes.org/), or the NHLBI Exome Variant Server (http://evs.gs.washington.edu/EVS/). Genotyping of 129 circumpolar Inuit samples from a variety of locales identified a single individual heterozygous for the same mutation (sample minor allele frequency: 1/258 = 0.4 %; 95 % confidence interval = [0.01–2.2 %]).
Fig. 2.
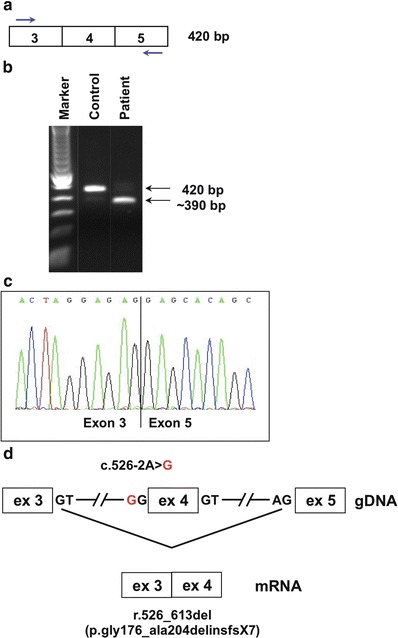
Aberrant splicing due to SLC17A5 mutation. Mutation analysis was performed on genomic DNA from whole blood by direct sequencing of all exons and exon/intron boundaries of SLC17A5 (NM_012434.4), according to standard protocols. For SLC17A5 mRNA studies, cDNA was prepared from cultured skin fibroblasts according to standard protocols: (a) Strategy for RT-PCR amplification of a region containing exon 4 (primer sequences and PCR protocol are available upon request). (b) The amplified product visualized on an agarose gel. The expected size of 420 bp is seen in an unaffected control, whereas in Patient 1, an additional band of approximately 350 bp was seen. (c) Direct sequence analysis of the ~350 bp fragment reveals an 88 bp deletion corresponding to exon 4. (d) A schematic overview of the gDNA with the acceptor splice site mutation c.526-2A>G. Disruption of the exon 4 splice acceptor site results in exon skipping, with exon 3 spliced directly onto exon 5 (r.526_613del). This is predicted to result in a frameshift and premature stop codon at the seventh downstream position (p.Gly176_Ala204delinsfs*7)
Patient 2
Patient 2 was referred to our pathology department for postmortem review and was identified retrospectively based on the strikingly similar neuropathologic findings versus those of Patient 1. Patient 2 was the product of a spontaneous singleton pregnancy to healthy non-consanguineous Inuit parents. At 25 weeks’ gestation, the mother presented with threatened preterm labor, and the fetus was discovered to have severe hydrops, polyhydramnios, and mild cardiomegaly, as well as bilateral cerebral ventriculomegaly and bilateral talipes. Amniocentesis showed a normal male karyotype; TORCH screen was negative. Fetal echo was structurally normal apart from cardiomegaly. Serial ultrasounds showed a picture of progressively worsening fetal hydrops. By 27+ 6 weeks, the measured fetal abdominal circumference was in keeping with 41 weeks, due to hepatomegaly and massive ascites. Variable decelerations were noted, and presentation being footling breach, an emergency Caesarean section was performed. A male infant was born weighing 2,314 g (+4SD). The child died shortly after birth following an unsuccessful attempt at resuscitation.
At autopsy, note was made of massive serous ascites, bilateral pleural effusions, and moderate diffuse subcutaneous edema. Pulmonary hypoplasia, cardiomegaly, and hepatosplenomegaly were evident. Histological findings were essentially identical to those of Patient 1, with foamy vacuolated cells in the central nervous system, peripheral nerve ganglia, pituitary chromophobe cells, hepatocytes, renal tubular epithelium, thyroid follicular epithelium, macrophages, adrenal glands, and epithelial cells of the renal glomeruli. Placental accumulation of storage material was also seen. As for Patient 1, neuraminidase activity in cultured skin fibroblasts was shown to be markedly increased (Table 1). SLC17A5 sequencing showed this individual to be homozygous for the same mutation previously identified in Patient 1.
Discussion
Both of the children described here followed a severe clinical course, characterized by antenatal development of fetal hydrops, multisystem involvement, and early demise despite maximal supportive treatment. This is essentially in keeping with the reported phenotype of other patients with truncating SLC17A5 mutations, suggesting a poor prognosis in patients lacking residual transporter activity (Reviewed in Froissart et al. 2005). From a molecular standpoint, both patients were homozygous for the same SLC17A5 mutation (c.526-2A>G). As far as we are aware, this constitutes the first report of ISSD among the Inuit; as our two patients are unrelated and from geographically remote reaches of the Arctic, the question of an ancestral founder mutation in SLC17A5 is raised. Determining whether this is the case will require analysis of a larger ethnic cohort and/or use of SNP-based linkage disequilibrium methods, beyond the scope of this report.
The distribution of the observed storage material in our patients is similar to that reported previously (Lemyre et al. 1999), and corresponds in a general sense to the expression pattern of sialin in the mouse (Yarovaya et al. 2005).
The cause of the nearly tenfold increase in fibroblast neuraminidase activity seen in our patients is unclear. We are aware of one earlier report of two children with free sialic acid storage and increased neuraminidase activity in lymphocytes, although activity was normal in fibroblasts in these same patients (Ylitalo et al. 1986). In contrast, several authors have reported neuraminidase activity in ISSD patients as “normal” or “not decreased” (Baumkötter et al. 1985; Paschke et al. 1986; Pueschel et al. 1988; Fois et al. 1987; Nakano et al. 1996; Lemyre et al. 1999). Whether this apparent inconsistency represents true biological variation, differences in technique, or a chance observation due to the influence of other genetic modifiers is unclear at present. Neuraminidases are known to be inducible in response to various external stimuli, for instance during immune cell differentiation and activation (Stamatos et al. 2005; Wang et al. 2004), although the basal regulation of “housekeeping” neuraminidase expression is poorly understood. In general, mechanisms that could conceivably lead to increased neuraminidase activity include (1) bulk expansion of the lysosomal compartment, (2) dysregulation of neuraminidase gene expression, and (3) allosteric effects on neuraminidase activity at the enzyme level, or some combination thereof. When present, the presence of this highly unusual finding should therefore prompt the clinician to consider testing for ISSD via conventional means, such as urine oligosaccharides analysis and/or SLC17A5 sequencing.
Synopsis
The sequential ascertainment of two unrelated Canadian Inuit patients homozygous for the same SLC17A5 mutation raises the possibility of a founder mutation for infantile sialic acid storage disease (ISSD) in this population.
Author Contributions
Matthew Lines: Primary authorship of manuscript
Tony Rupar and Jack Rip: Enzymatic analyses and assistance with writing
Berivan Baskin and Peter Ray: Molecular analyses
David Grynspan and Jean Michaud: Description of pathological findings and preparation of histologic images
Michael Geraghty: Corresponding author, study design, critical appraisal, assistance with writing
Guarantor
Michael Geraghty
Compliance with Ethics Guidelines
Matthew Lines, Tony Rupar, Jack Rip, Berivan Baskin, Peter Ray, Robert Hegele, David Grynspan, Jean Michaud, and Michael Geraghty each declare that they have no conflict of interest.
This manuscript is a retrospective case series containing no identifying patient information, and data were collected in the course of provisioning the routine standard of clinical care (rather than as part of a research study). All procedures followed were in accordance with the Helsinki Declaration of 1975, as revised in 2000.
Footnotes
Competing interests: None declared
Contributor Information
Michael T. Geraghty, Email: mgeraghty@cheo.on.ca
Collaborators: Johannes Zschocke and K Michael Gibson
References
- Aula N, Salomaki P, Timonen R, Verheijen F, Mancini G, Mansson JE, Aula P, Peltonen L. The spectrum of SLC17A5-gene mutations resulting in free sialic acid-storage diseases indicates some genotype-phenotype correlation. Am J Hum Genet. 2000;67(4):832–840. doi: 10.1086/303077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumkötter J, Cantz M, Mendla K, Baumann W, Friebolin H, Gehler J, Spranger J. N-Acetylneuraminic acid storage disease. Hum Genet. 1985;71(2):155–159. doi: 10.1007/BF00283373. [DOI] [PubMed] [Google Scholar]
- Biancheri R, Verbeek E, Rossi A, Gaggero R, Roccatagliata L, Gatti R, van Diggelen O, Verheijen FW, Mancini GM. An Italian severe Salla disease variant associated with a SLC17A5 mutation earlier described in infantile sialic acid storage disease. Clin Genet. 2002;61(6):443–447. doi: 10.1034/j.1399-0004.2002.610608.x. [DOI] [PubMed] [Google Scholar]
- Biancheri R, Rossi A, Verbeek HA, Schot R, Corsolini F, Assereto S, Mancini GM, Verheijen FW, Minetti C, Filocamo M. Homozygosity for the p.K136E mutation in the SLC17A5 gene as cause of an Italian severe Salla disease. Neurogenetics. 2005;6(4):195–199. doi: 10.1007/s10048-005-0011-3. [DOI] [PubMed] [Google Scholar]
- Coker M, Kalkan-Ucar S, Kitis O, Ucar H, Goksen-Simsek D, Darcan S, Gokben S. Salla disease in Turkish children: severe and conventional type. Turk J Pediatr. 2009;51(6):605–609. [PubMed] [Google Scholar]
- Debray FG, Lefebvre C, Colinet S, Segers K, Stevens R. Free sialic acid storage disease mimicking cerebral palsy and revealed by blood smear examination. J Pediatr. 2011;158(1):165. doi: 10.1016/j.jpeds.2010.06.057. [DOI] [PubMed] [Google Scholar]
- Erikson A, Aula N, Aula P, Mansson JE. Free sialic acid storage (Salla) disease in Sweden. Acta Paediatr. 2002;91(12):1324–1327. doi: 10.1111/j.1651-2227.2002.tb02828.x. [DOI] [PubMed] [Google Scholar]
- Fois A, Balestri P, Farnetani MA, Mancini GM, Borgogni P, Margollicci MA, Molinelli M, Alessandrini C, Gerli R. Free sialic acid storage disease. A new Italian case. Eur J Pediatr. 1987;146(2):195–198. doi: 10.1007/BF02343235. [DOI] [PubMed] [Google Scholar]
- Froissart R, Cheillan D, Bouvier R, Tourret S, Bonnet V, Piraud M, Maire I. Clinical, morphological, and molecular aspects of sialic acid storage disease manifesting in utero. J Med Genet. 2005;42(11):829–836. doi: 10.1136/jmg.2004.029744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau D, Cohen D, Shalev H, Pinsk V, Yerushalmi B, Zeigler M, Birk OS. A novel mutation in the SLC17A5 gene causing both severe and mild phenotypes of free sialic acid storage disease in one inbred Bedouin kindred. Mol Genet Metab. 2004;82(2):167–172. doi: 10.1016/j.ymgme.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Lemyre E, Russo P, Melancon SB, Gagne R, Potier M, Lambert M. Clinical spectrum of infantile free sialic acid storage disease. Am J Med Genet. 1999;82(5):385–391. doi: 10.1002/(SICI)1096-8628(19990219)82:5<385::AID-AJMG6>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Mochel F, Yang B, Barritault J, Thompson JN, Engelke UF, McNeill NH, Benko WS, Kaneski CR, Adams DR, Tsokos M, et al. Free sialic acid storage disease without sialuria. Ann Neurol. 2009;65(6):753–757. doi: 10.1002/ana.21624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochel F, Engelke UF, Barritault J, Yang B, McNeill NH, Thompson JN, Vanderver A, Wolf NI, Willemsen MA, Verheijen FW, Seguin F, Wevers RA, Schiffmann R. Elevated CSF N-acetylaspartylglutamate in patients with free sialic acid storage diseases. Neurology. 2010;74(4):302–305. doi: 10.1212/WNL.0b013e3181cbcdc4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin P, Sagne C, Gasnier B. Functional characterization of wild-type and mutant human sialin. EMBO J. 2004;23(23):4560–4570. doi: 10.1038/sj.emboj.7600464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myall NJ, Wreden CC, Wlizla M, Reimer RJ. G328E and G409E sialin missense mutations similarly impair transport activity, but differentially affect trafficking. Mol Genet Metab. 2007;92(4):371–374. doi: 10.1016/j.ymgme.2007.08.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano C, Hirabayashi Y, Ohno K, Yano T, Mito T, Sakurai M. A Japanese case of infantile sialic acid storage disease. Brain Dev. 1996;18(2):153–156. doi: 10.1016/0387-7604(95)00142-5. [DOI] [PubMed] [Google Scholar]
- Paschke E, Trinkl G, Erwa W, Pavelka M, Mutz I, Roscher A. Infantile type of sialic acid storage disease with sialuria. Clin Genet. 1986;29(5):417–424. doi: 10.1111/j.1399-0004.1986.tb00514.x. [DOI] [PubMed] [Google Scholar]
- Pueschel SM, O’Shea PA, Alroy J, Ambler MW, Dangond F, Daniel PF, Kolodny EH. Infantile sialic acid storage disease associated with renal disease. Pediatr Neurol. 1988;4(4):207–212. doi: 10.1016/0887-8994(88)90032-X. [DOI] [PubMed] [Google Scholar]
- Ruivo R, Sharifi A, Boubekeur S, Morin P, Anne C, Debacker C, Graziano JC, Sagne C, Gasnier B. Molecular pathogenesis of sialic acid storage diseases: insight gained from four missense mutations and a putative polymorphism of human sialin. Biol Cell. 2008;100(9):551–559. doi: 10.1042/BC20070166. [DOI] [PubMed] [Google Scholar]
- Sonderby Christensen P, Kaad PH, Ostergaard JR. Two cases of Salla disease in Danish children. Acta Paediatr. 2003;92(11):1357–1358. doi: 10.1111/j.1651-2227.2003.tb00514.x. [DOI] [PubMed] [Google Scholar]
- Stamatos NM, Liang F, Nan X, Landry K, Cross AS, Wang LX, Pshezhetsky AV. Differential expression of endogenous sialidases of human monocytes during cellular differentiation into macrophages. FEBS J. 2005;272(10):2545–2556. doi: 10.1111/j.1742-4658.2005.04679.x. [DOI] [PubMed] [Google Scholar]
- Tylki-Szymanska A, Czartoryska B, Lugowska A, Verheijen FW, Mancini GM, Rokicki D, Taybert J, Chmielinska E. Infantile sialic acid storage disease (ISSD): report of the first case detected in Poland. Pediatr Int. 2003;45(2):199–200. doi: 10.1046/j.1442-200X.2003.01693.x. [DOI] [PubMed] [Google Scholar]
- Verheijen FW, Verbeek E, Aula N, Beerens CE, Havelaar AC, Joosse M, Peltonen L, Aula P, Galjaard H, van der Spek PJ, Mancini GM. A new gene, encoding an anion transporter, is mutated in sialic acid storage diseases. Nat Genet. 1999;23(4):462–465. doi: 10.1038/70585. [DOI] [PubMed] [Google Scholar]
- Wang P, Zhang J, Bian H, Wu P, Kuvelkar R, Kung TT, Crawley Y, Egan RW, Billah MM. Induction of lysosomal and plasma membrane-bound sialidases in human T-cells via T-cell receptor. Biochem J. 2004;380(Part 2):425–433. doi: 10.1042/BJ20031896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wreden CC, Wlizla M, Reimer RJ. Varied mechanisms underlie the free sialic acid storage disorders. J Biol Chem. 2005;280(2):1408–1416. doi: 10.1074/jbc.M411295200. [DOI] [PubMed] [Google Scholar]
- Yarovaya N, Schot R, Fodero L, McMahon M, Mahoney A, Williams R, Verbeek E, de Bondt A, Hampson M, van der Spek P, Stubbs A, Masters CL, Verheijen FW, Mancini GM, Venter DJ. Sialin, an anion transporter defective in sialic acid storage diseases, shows highly variable expression in adult mouse brain, and is developmentally regulated. Neurobiol Dis. 2005;19(3):351–365. doi: 10.1016/j.nbd.2004.12.020. [DOI] [PubMed] [Google Scholar]
- Ylitalo V, Hagberg B, Rapola J, Månsson JE, Svennerholm L, Sanner G, Tonnby B. Salla disease variants. Sialoylaciduric encephalopathy with increased sialidase activity in two non-Finnish children. Neuropediatrics. 1986;17(1):44–47. doi: 10.1055/s-2008-1052498. [DOI] [PubMed] [Google Scholar]