

Abstract
We report an adult male with classic propionic acidemia (PA) who developed chronic kidney disease in the third decade of his life. This diagnosis was recognized by an increasing serum creatinine and confirmed by reduced glomerular filtration on a 99mTc-diethylenetriamine pentaacetate (DTPA) scan. Histopathology of the kidney showed moderate glomerulo- and tubulointerstitial fibrosis with very segmental mesangial IgA deposits. This is the second reported case of kidney disease in an individual with propionic acidemia possibly indicating that chronic kidney disease may be a late-stage complication of propionic acidemia. Additionally, this is the first description of the histopathology of kidney disease in an individual with propionic acidemia. As more cases emerge, the clinical course and spectrum of renal pathology in this disorder will be better defined.
Introduction
Propionic acidemia (PA) is a chronic, life-threatening organic acidemia that presents in childhood due to deficiency of the enzyme propionyl-CoA carboxylase (PCC), which catalyzes the conversion of propionyl-CoA to methylmalonyl-CoA (Fenton et al. 2001; de Baulny et al. 2012). Biochemical findings in affected patients include an elevation of the plasma C3 acylcarnitine species and elevated urine organic acids such as 3-hydroxypropionate, methylcitrate, tiglylglycine, and propionylglycine. Plasma amino acids demonstrate elevated glycine. The diagnosis can be confirmed by showing deficient enzyme activity in leukocytes or fibroblasts, or mutations in both alleles of either PCCA or PCCB, the genes that encode the subunits of PCC (Desviat et al. 2004; Ugarte et al. 1999).
Well-recognized morbidities of PA include acute episodes of hyperammonemia and metabolic acidosis. Affected individuals are susceptible to acute basal ganglia infarctions, pancreatitis, and bone marrow suppression. Chronic issues include intellectual disability, poor growth, cardiomyopathy, prolonged QTc, and immune defects (de Baulny et al. 2012; Lee et al. 2009; Lücke et al. 2004; Surtees et al. 1992; Grünert et al. 2012; Sutton et al. 2012; Carillo-Carrasco and Venditti 2012; Baumgartner et al. 2007). As individuals with this disorder live longer, new complications potentially related to the disorder are being recognized. Acute-onset optic neuropathy is one of the more recently identified complications of this disorder (Williams et al. 2009; Ianchulev et al. 2003).
To date, only one case report has attempted to associate PA with chronic kidney disease (CKD) (Lam et al. 2011). Herein, we report an adult male with classic propionic acidemia who developed CKD in the third decade of life. We propose that CKD is a late complication of PA.
Methods and Results
Clinical Summary
The patient, now 29 years of age, first came to medical attention at 8 days of life. He presented with dehydration, lethargy, metabolic acidosis with total CO2 19 mEq/L and anion gap 24 mEq/L, hyperammonemia at 114 μmol/L (nl. 0–32 μmol/L), and a plasma glycine level of 900 μmol/L (nl. 87–323 μmol/L). The diagnosis of PA was supported by increased urinary excretion of hydroxypropionic acid and methylcitrate. The diagnosis was subsequently confirmed by absence of propionyl-CoA carboxylase and normal 3-methylcrotonyl carboxylase activities in peripheral leukocytes. Due to refractory hyperammonemia at 11 days of life (peak 690 μmol/L), he received peritoneal dialysis for approximately 1 month but has not required renal replacement therapy in the ensuing years.
The patient was recognized to have developmental delay at 13 months of age, when his development was estimated to be at around 8–9 months globally. Obsessive compulsive symptoms and anxiety symptoms were noted early in the second decade of life. Currently, he is considered to have mild intellectual disability with diagnoses of autistic disorder, anxiety disorder, and obsessive compulsive disorder. He is followed regularly by psychiatry, lives with his parents, and attends a 5-day-a-week day program.
Later in the second decade, he developed debilitating bowel dysmotility and was discovered to have intestinal malrotation. He underwent a Ladd procedure at 18 years of age, but continues to have severely delayed gastric emptying with intermittent bilious vomiting. He receives gastrostomy tube feedings of PediaSure®, Polycose, valine, and carnitine to provide 33 kcal/kg/day and 0.8 g/kg/day of protein.
At 15 years of life, mild to moderate left ventricular dysfunction was noted by echocardiography with prolonged QTc interval. At 21 years of life, his left ventricular fractional shortening had decreased from baseline 25% to 18%, and he was initiated on lisinopril 5 mg daily. Hypertension was not present, and echocardiography at 29 years of life demonstrated mild to moderate global hypokinesis of the left ventricle with an ejection fraction of 40%. Additional past medical history was notable for osteoporosis with a femur fracture at 6 years of life and hypothyroidism diagnosed at 23 years of life.
Renal Evaluation
The patient was referred to nephrology at 29 years of age due to an elevated serum creatinine which had slowly risen over the preceding 7 years from 0.8 mg/dL to 1.5 mg/dL. The estimated glomerular filtration rate (eGFR) by the 4-variable Modification of Diet in Renal Disease equation was 51 mL/min/1.73 m2, consistent with stage 3 CKD. At the time of referral to nephrology, the only urinary symptom was nocturnal enuresis attributed to 20-h enteral feedings. His medication regimen had not changed in several years and consisted of lisinopril 5 mg daily, risperidone, domperidone, ondansetron, levothyroxine, cetirizine, mirtazapine, omeprazole, sodium bicarbonate, and the above-listed enteral feedings. Family history was significant for the absence of kidney disease. Physical examination was normal, with blood pressure 90/60 mmHg, weight 66.2 kg, and height 163.8 cm. Additional laboratory evaluation (Table 1) was notable only for mild anemia and normal urinary protein excretion in the setting of angiotensin-converting enzyme inhibitor use. Liver function testing was normal. Computed tomography imaging of the abdomen with intravenous and oral contrast 5 years earlier had demonstrated structurally normal kidneys, as did renal ultrasonography at the time of kidney biopsy. A 99mTc-diethylenetriamine pentaacetate (DTPA) scan calculated a GFR of 38.5 mL/min/1.73 m2, confirming reduced glomerular filtration reasonably consistent with the creatinine-based eGFR.
Table 1.
Selected laboratory values at the time of nephrology evaluation
| Variable | Value | Reference |
|---|---|---|
| Blood urea nitrogen (mg/dL) | 18 | 7–22 |
| Creatinine (mg/dL) | 1.7 | 0.6–1.3 |
| Sodium (mEq/L) | 136 | 135–148 |
| Total CO2 (mEq/L) | 24 | 21–31 |
| Calcium (mg/dL) | 9.2 | 8.4–10.5 |
| Phosphorous (mg/dL) | 4.1 | 2.5–4.5 |
| Uric acid (mg/dL) | 4.2 | 3.7–8.6 |
| Albumin (g/dL) | 3.8 | 3.5–5.3 |
| Ammonia (μ/dL) | 100 | 27–102 |
| Hemoglobin (g/dL) | 11.3 | 13.9–16.3 |
| Platelets (cells/mm3) | 162,000 | 150,000–350,000 |
| Urinalysis | ||
| pH | 5 | 4.6–8.0 |
| Protein | negative | negative |
| Red blood cells (per HPF) | 1 | 0–5 |
| White blood cells (per HPF) | 0 | 0–5 |
| Urine microalbumin/creatinine (mg/g) | 3.2 | 0.0–30.0 |
| 25-vitamin D (ng/mL) | 36.7 | 30–100 |
| Complement C3 (mg/dL) | 130 | 90–180 |
| Complement C4 (mg/dL) | 26 | 9–36 |
| Antinuclear antibody | Negative | Negative |
| Free plasma carnitine (μmol/L) | 12.6 | 22–66 |
| Total plasma carnitine (μmol/L) | 122.8 | 28–84 |
| Urine citrate* (μg/mg creatinine) | 12 | 12–96 |
| Urine methylcitrate* (μg/mg creatinine) | 105 | 0 |
*Semi-quantitative values derived from urine organic acid analysis. methylcitrate/citrate ratio is 8.75
An ultrasound-guided percutaneous kidney biopsy was performed for further evaluation. Light microscopy revealed up to 31 glomeruli per section, of which up to 9 were globally sclerosed. One subcapsular glomerulus had a focal scar, and the remaining glomeruli were unremarkable (Fig. 1a). Immunofluorescence was performed on a frozen, non-sclerosed glomerulus, demonstrating fine granular, very segmental mesangial staining for IgG 2+, IgA 3-4+, IgM 1-2+, C3 2-3+ very sparse, kappa light chain 2+, and lambda light chain 2+. Glomerular staining was negative for C1q, with nonspecific linear staining for albumin 2+ in capillary loops and tubular basement membrane and fibrinogen 1-2+ in the interstitium (Fig. 1b, c). Electron microscopy was performed on one normal appearing glomerulus. Ultrastructurally, there was mild expansion of the mesangial matrix with a segmentally wrinkled but otherwise unremarkable glomerular basement membrane. There were very sparse small electron-dense deposits at the periphery of the mesangium (Fig. 1d). Podocytes demonstrated occasional vacuolizations and mild focal effacement of the foot processes estimated to involve less than 10% of the capillary surface. With such sparse glomerular electron-dense deposits and lack of mesangial proliferation, the possibility of a secondary nature of IgA deposition could not be excluded.
Fig. 1.
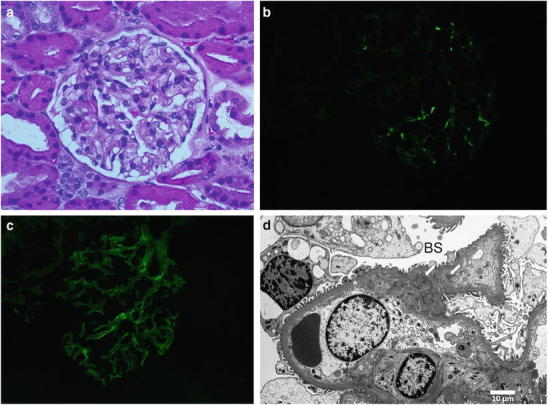
(a) Histologically normal glomerulus by light microscopy (400x); (b) sparse IgA and (c) IgG mesangial deposits by immunofluorescence; (d) sparse electron-dense mesangial deposits (arrows) on electron microscopy. Glomerular basement membrane (*) and Bowman’s space (BS)
The tubules showed focal injury with cell blebbing and vacuolization, focal dilatation, and flattened epithelium. Ultrastructurally, the tubular cells showed focally enlarged mitochondria with disorganized cristae (Fig. 2a and b).
Fig. 2.
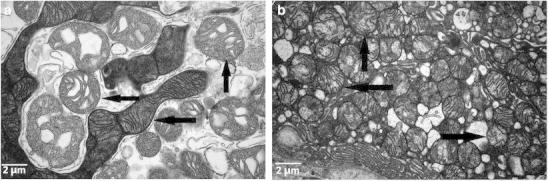
(a and b) Enlarged abnormal mitochondria in tubular cells with disruption of cristae (indicated by arrows)
The interstitium showed mild, lymphocytic inflammatory infiltrate with rare eosinophils mostly associated with areas of fibrosis. Moderate tubular atrophy and interstitial fibrosis was present, involving approximately 25–30% of the cortical parenchyma. Three arteries up to interlobular size were present and appeared unremarkable, as did the arterioles.
Summary and Discussion
We report a patient with classical PA who presented with progressive CKD in the third decade of life. The clinical history did not reveal a likely etiology for CKD, apart from a low-normal blood pressure in the context of lisinopril administration at the time of referral. The kidney biopsy demonstrated moderate glomerular and tubulointerstitial scarring, evidence of significant chronicity although nonspecific for an etiology.
While the glomerular immunofluorescence demonstrated fine mesangial IgA deposits with small electron-dense deposits on electron microscopy, the absence of both mesangial proliferative changes and clinically significant proteinuria and hematuria render a primary diagnosis of autoimmune IgA nephropathy less likely. Autoimmune IgA nephropathy is the most common glomerulonephritis worldwide, estimated to account for 10% of all glomerulonephritis in the United States (Tumlin et al. 2007). Secondary forms of IgA nephropathy have been associated with multiple disorders, including cirrhosis, celiac disease, and inflammatory bowel disease. These are commonly due to impaired IgA clearance in the setting of cirrhosis or the formation of alternative immune complexes such as gliadin/anti-gliadin IgA with celiac disease (Pouria and Barratt 2008). In autoimmune IgA nephropathy, at least mild mesangial hypercellularity is typically present, with absent glomerular changes potentially suggesting a secondary etiology (Haas 1997). Deposition of complement in the mesangium is also characteristic of autoimmune IgA nephropathy, and this patient had only sparse C3 by immunofluorescence perhaps more consistent with nonspecific immunoglobulin trapping or a secondary etiology. It is not known if IgA metabolism is altered in the setting of PA.
Renal pathology in humans affected with PA has not been previously described. In a mouse model of PA (PCCA knockout), all mice died within 24–36 h of birth due to dehydration and severe ketoacidosis (Miyazaki et al. 2001). Kidney histology revealed enlarged collecting ducts with protein resorption droplets. It was possible these findings stemmed from dehydration and malnutrition rather than an intrinsic renal process specific to PA. The early death of these animals precludes the observation of long-term renal sequelae of PA.
Renal pathology in the related disorder, methylmalonic acidemia (MMA), has been well described in both affected humans and in mouse models. Kidney biopsies from individuals with MMA classically show tubulointerstitial inflammation with mononuclear cell infiltrates and interstitial fibrosis with tubular atrophy (Rutledge et al. 1993; Walter et al. 1989; D’Angio et al. 1991; Molteni et al. 1991; Hörster and Hoffmann 2004). Kidneys from affected mice demonstrate dysmorphic mitochondria with abnormal cristae in the proximal tubular cells of the kidney, beginning at the end of the first week of life. Kidneys from older mice have severe and widespread tubulointerstitial changes similar to those reported in patients with MMA (Chandler et al. 2009). As noted, these tubulointerstitial changes are nonspecific but are consistent with those seen in our patient. Tubular cell mitochondria in our patient also appeared mildly dysmorphic, further suggesting a role for altered cellular metabolism in the pathology of PA-associated CKD.
We identified another published case of late-onset kidney disease in PA in the literature (Lam et al. 2011). The kidney failure in this patient was discovered in her fourth decade of life based on a rising serum creatinine. No additional laboratory characterization of the CKD was described and a kidney biopsy was not performed. Serum creatinine has often been deemed an unreliable marker of kidney function in some organic acidemias due to the low muscle mass of these patients and the restricted protein intake. Nevertheless, these two cases demonstrate the importance of monitoring eGFR in patients with PA. In our patient, creatinine-based estimation of GFR correlated acceptably well with measured GFR by 99mTc-DTPA scan. As no reversible lesion was identified on kidney biopsy, therapy has focused on surveillance for complications of CKD, avoidance of potentially nephrotoxic agents, and ongoing provision of optimal nutrition. The lisinopril dose was reduced to 2.5 mg daily and continued given the mild cardiomyopathy.
With advances in treatment and care of many inborn errors of metabolism, later-onset complications potentially related to the underlying disorder are now starting to emerge. While the etiology of this patient’s CKD cannot be established with certainty, the histopathological findings do demonstrate tubulointerstitial disease for which PA must be entertained as a cause. We expect that as more cases emerge, the clinical course and spectrum of renal pathology in this disorder will be better defined.
Synopsis
This is the second reported case of kidney disease in an individual with propionic acidemia, and the first report with pathologic information, indicating that chronic kidney disease may be a late-stage complication of propionic acidemia.
Footnotes
Competing interests: None declared
Contributor Information
H. J. Vernon, Email: hvernon1@jhmi.edu
Collaborators: Johannes Zschocke and K Michael Gibson
References
- Baumgartner D, Scholl-Bürgi S, Sass JO, et al. Prolonged QTc intervals and decreased left ventricular contractility in patients with propionic acidemia. J Pediatr. 2007;150:192–197. doi: 10.1016/j.jpeds.2006.11.043. [DOI] [PubMed] [Google Scholar]
- Carrillo-Carrasco N, Venditti C (2012) Propionic Acidemia. In: R.A. Pagon, T.D. Bird, C.R. Dolan, K. Stephens, M.P. Adam (eds) GeneReviews™ [Internet]. University of Washington, Seattle; 1993–2012. Available from: http://www.ncbi.nlm.nih.gov/books/NBK92946/
- Chandler RJ, Zerfas PM, Shanske S, et al. Mitochondrial dysfunction in mut methylmalonic acidemia. FASEB J. 2009;23:1252–1261. doi: 10.1096/fj.08-121848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Angio CT, Dillon MJ, Leonard JV. Renal tubular dysfunction in methylmalonic acidaemia. Eur J Pediatr. 1991;150:259–263. doi: 10.1007/BF01955526. [DOI] [PubMed] [Google Scholar]
- de Baulny HO, Dionisi-Vici C, Wendel U. Branched chain organic acidurias/acidaemias. In: Saudubray JM, van den Berghe G, Walter JH, editors. Inborn metabolic diseases- diagnosis and treatment. New York: Springer; 2012. pp. 277–296. [Google Scholar]
- Desviat LR, Pérez B, Pérez-Cerdá C, et al. Propionic acidemia: mutation update and functional and structural effects of the variant alleles. Mol Genet Metab. 2004;83:28–37. doi: 10.1016/j.ymgme.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Fenton WA, Grave RA, Rosenblatt DA (2001) Disorders of propionate and methylmalonate metabolism. In: Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A (eds) The online metabolic & molecular bases of inherited disease [Internet]. Available from: http://www.ommbid.com/OMMBID/a/c.html/organic_acids/disorders_propionate_methylmalonate_metabolism
- Grünert SC, Müllerleile S, de Silva L, et al. Propionic acidemia: neonatal versus selective metabolic screening. J Inherit Metab Dis. 2012;35:41–49. doi: 10.1007/s10545-011-9419-0. [DOI] [PubMed] [Google Scholar]
- Haas M. Histologic subclassification of IgA nephropathy: a clinicopathologic study of 244 cases. Am J Kidney Dis. 1997;29:829–842. doi: 10.1016/S0272-6386(97)90456-X. [DOI] [PubMed] [Google Scholar]
- Hörster F, Hoffmann GF. Pathophysiology, diagnosis, and treatment of methylmalonic aciduria-recent advances and new challenges. Pediatr Nephrol. 2004;19:1071–1074. doi: 10.1007/s00467-004-1572-3. [DOI] [PubMed] [Google Scholar]
- Ianchulev T, Kolin T, Moseley K, et al. Optic nerve atrophy in propionic acidemia. Ophthalmology. 2003;110:1850–1854. doi: 10.1016/S0161-6420(03)00573-6. [DOI] [PubMed] [Google Scholar]
- Lam C, Desviat LR, Perez-Cerdá C, et al. 45-Year-old female with propionic acidemia, renal failure, and premature ovarian failure: late complications of propionic acidemia? Mol Genet Metab. 2011;103:338–340. doi: 10.1016/j.ymgme.2011.04.007. [DOI] [PubMed] [Google Scholar]
- Lee TM, Addonizio LJ, Barshop BA, et al. Unusual presentation of propionic acidaemia as isolated cardiomyopathy. J Inherit Metab Dis. 2009;32:97–101. doi: 10.1007/s10545-009-1084-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lücke T, Pérez-Cerdá C, Baumgartner M, et al. Propionic acidemia: unusual course with late onset and fatal outcome. Metabolism. 2004;53:809–810. doi: 10.1016/j.metabol.2003.12.025. [DOI] [PubMed] [Google Scholar]
- Miyazaki T, Ohura T, Kobayashi M, et al. Fatal propionic acidemia in mice lacking propionyl-CoA carboxylase and its rescue by postnatal, liver-specific supplementation via a transgene. J Biol Chem. 2001;21:35995–35999. doi: 10.1074/jbc.M105467200. [DOI] [PubMed] [Google Scholar]
- Molteni KH, Oberley TD, Wolff JA, et al. Progressive renal insufficiency in methylmalonic acidemia. Pediatr Nephrol. 1991;5:323–326. doi: 10.1007/BF00867492. [DOI] [PubMed] [Google Scholar]
- Pouria S, Barratt J. Secondary IgA nephropathy. Semin Nephrol. 2008;28:27–37. doi: 10.1016/j.semnephrol.2007.10.004. [DOI] [PubMed] [Google Scholar]
- Rutledge SL, Geraghty M, Mroczek E, et al. Tubulointerstitial nephritis in methylmalonic acidemia. Pediatr Nephrol. 1993;7:81–82. doi: 10.1007/BF00861581. [DOI] [PubMed] [Google Scholar]
- Surtees RA, Matthews EE, Leonard JV. Neurologic outcome of propionic acidemia. Pediatr Neurol. 1992;8:333–337. doi: 10.1016/0887-8994(92)90085-D. [DOI] [PubMed] [Google Scholar]
- Sutton VR, Chapman KA, Gropman AL, et al. Chronic management and health supervision of individuals with propionic acidemia. Mol Genet Metab. 2012;105:26–33. doi: 10.1016/j.ymgme.2011.08.034. [DOI] [PubMed] [Google Scholar]
- Tumlin JA, Madaio MP, Hennigar R. Idiopathic IgA nephropathy: pathogenesis, histopathology, and therapeutic options. Clin J Am Soc Nephrol. 2007;2:1054–1061. doi: 10.2215/CJN.04351206. [DOI] [PubMed] [Google Scholar]
- Ugarte M, Pérez-Cerdá C, Rodríguez-Pombo P, et al. An overview of mutations in the PCCA and PCCB genes causing propionic acidemia. Hum Mutat. 1999;14:275–282. doi: 10.1002/(SICI)1098-1004(199910)14:4<275::AID-HUMU1>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Walter JH, Michalski A, Wilson WM, et al. Chronic renal failure in methylmalonic acidemia. Eur J Pediatr. 1989;148:344–348. doi: 10.1007/BF00444131. [DOI] [PubMed] [Google Scholar]
- Williams SR, Hurley PE, Altiparmak UE, et al. Late onset optic neuropathy in methylmalonic and propionic acidemia. Am J Ophthalmol. 2009;147:929–933. doi: 10.1016/j.ajo.2008.12.024. [DOI] [PubMed] [Google Scholar]