

Abstract
Objective The characterization of a novel large deletion in the galactose-1-phosphate uridyltransferase (GALT) gene accounting for the majority of disease alleles in Cypriot patients with classic galactosemia.
Methods DNA sequencing was used to identify the mutations followed by multiplex ligation-dependent probe amplification (MLPA) analysis in the cases suspected of harboring a deletion. In order to map the breakpoints of the novel deletion, a PCR walking approach was employed. A simple PCR assay was validated for diagnostic testing for the new deletion. Haplotype analysis was performed using microsatellite markers in the chromosomal region 9p. RT-PCR was used to study RNA expression in lymphoblastoid cell lines.
Results The new deletion spans a region of 8489 bp and eliminates all GALT exons as well as the non-translated sequences of the adjacent interleukin 11 receptor alpha (IL11RA) gene. In addition, the deletion is flanked by a 6 bp block of homologous sequence on either side suggesting that a single deletion event has occurred, probably mediated by a recombination mechanism. Microsatellite marker analysis revealed the existence of a common haplotype. The RNA expression studies showed a lack of IL11RA transcripts in patients homozygous for the deletion.
Conclusions We have identified and characterized a novel contiguous deletion which affects both the GALT enzyme and the IL11RA protein resulting in classic galactosemia with additional phenotypic abnormalities such as craniosynostosis, a feature that has been associated with defects in the IL11RA gene.
Electronic supplementary material: The online version of this chapter (doi:10.1007/8904_2013_249) contains supplementary material, which is available to authorized users.
Introduction
Classic galactosemia (OMIM# 230400) is the most common inherited disorder of carbohydrate metabolism resulting in the inability to metabolize galactose by the normal “Leloir” biochemical pathway (Holton et al. 2001). The incidence of classic galactosemia varies in different populations with an average incidence worldwide of 1:62,000 births (Levy and Hammersen 1978; Tyfield et al. 1999). An increased incidence has been reported in Ireland, 1:21,000 (Murphy et al. 1999), and Greece, 1 in 22,000 (Schulpis et al. 1997), and a lower incidence in Japan, 1:1,000,000 (Aoki and Wada 1988).
Classic galactosemia is caused by mutations in the GALT gene which encodes the enzyme galactose-1-phosphate uridyltransferase (GALT, EC 2.7.712) and is inherited in an autosomal recessive fashion (Leslie et al. 1992). The GALT gene is located on chromosome 9p13, is arranged into 11 exons, and spans about 4.3 kb of genomic DNA (Shih et al. 1984; Reichardt and Berg 1988; Flach et al. 1990). To date, 264 variants within the GALT gene have been identified according to the entries in the database at the ARUP Institute of Experimental Pathology (http://www.arup.utah.edu/database/GALT/GALT_welcome.php) with about 85 % being pathogenic. Most of the mutations are missense mutations (Calderon et al. 2007) with p.Gln188Arg (c.563A>G) being the most common mutation (64 %) in European populations (Tyfield et al. 1999). The p.Lys285Asn (c.855G>T) mutation is another frequent mutation in European populations with an overall frequency of about 8 %, but, as for the p.Gln188Arg mutation, there are large differences in its relative frequency in different ethnic groups. In many populations it is the second most frequent disease-causing GALT mutation, with a frequency in some countries of east and central Europe of 25–40 % (Greber-Platzer et al. 1997; Tyfield et al. 1999; Zekanowski et al. 1999). Together the p.Gln188Arg and p.Lys285Asn mutations account for approximately 70–80 % of classic galactosemia alleles in Caucasian populations (Tyfield et al. 1999). Although classic galactosemia occurs in numerous different countries and ethnic groups worldwide, only a few ethnicity-genotype associations have been established. For example, p.Ser135Leu, a relatively mild mutation, is almost exclusively found in individuals of black African origin (Lai et al. 1996), whereas a complex deletion of 5.5 kb in the GALT gene has been strongly linked to patients of Ashkenazi Jewish descent (Berry et al. 2001; Barbouth et al. 2006; Coffee et al. 2006). This large deletion in the GALT gene is characterized by the preservation of a segment of 117 bp that contains portions of exon 8 and intron 8 and an additional 12 bp insertion immediately downstream of the exon 8/intron 8 segment (Coffee et al. 2006).
In this study, we identified the mutations responsible for classic galactosemia in the Greek Cypriot population and characterized a novel large deletion that is present in the majority of disease alleles.
Methods
Subjects
Eight Greek Cypriot patients with classic galactosemia (Table 1) and 13 of their parents were studied (two patients were siblings). In addition, ten subjects unrelated to any of the galactosemia patients and found to be carriers were examined. Blood samples were collected after obtaining the approval of the National Bio-ethics Committee for the project and with signed consent.
Table 1.
Clinical, biochemical, and molecular data of Greek Cypriot patients with classic galactosemia
| Patient | Sex | Age at diagnosis | Age today | GALT activitya (μmol/h/g Hb) NR: 18–40 | Clinical features at presentation | Clinical picture today | Genotype |
|---|---|---|---|---|---|---|---|
| 1 | M | 6 months | 19 years | 1.1 | Jaundice, diarrhea, and hypoglycemia | Severe learning difficulties and dyspraxia, behavioral problems | 8.5Kb deletion/ p.Lys285Asn |
| 2 | M | 5 weeks | 14 years | 0 | Jaundice, cataracts, liver disease, ascites, FTT | Severe learning difficulties, poor vision (optic atrophy), microcephaly | Homozygous 8.5Kb deletion |
| 3b | M | 7 days | 13 years | 0 | Jaundice, feeding problems, high LFT | Normal neurological and motor development, very good at school | Homozygous p.Lys285Asn |
| 4 | F | 2 weeks | 9 years | 4.8 | Jaundice, liver disease | Normal neurological and motor development | 8.5Kb deletion/ p.Lys285Asn |
| 5 | F | 3 weeks | 7 years | 1.9 | Jaundice, FTT, high LFT, diarrhea, referred from neonatal screening due to mildly elevated phenylalanine | Normal neurological and motor development | 8.5Kb deletion/ p.Lys285Asn |
| 6 | F | 3 days | 7 years | 0.05 | Jaundice, FTT, high LFT | Craniosynostosis | Homozygous 8.5Kb deletion |
| 7b | M | 1 day | 3 years | <1.0 | NIL | Normal | Homozygous p.Lys285Asn |
| 8 | F | 13 days | 2 years | 5.9 | Jaundice, FTT, vomiting, palpable liver, ascites, coagulopathy, high LFT | Craniosynostosis | Homozygous 8.5Kb deletion |
M male, F female, FTT failure to thrive, LFT liver function test, NR normal range
aSpectrophotometric method of Kalckar et al.
bSiblings
GALT Enzyme Activity Measurement
Measurement of GALT activity in washed red blood cells was performed using the spectrophotometric method of Kalckar et al. (1956) which is based on UDP-glucose consumption.
PCR Amplification and DNA Sequencing
Patient DNA was isolated from whole blood using the Gentra Puregene Blood Kit (Qiagen), according to the manufacturer instructions. For bidirectional automated sequencing, GALT exons were amplified in eight fragments. Primers used for PCR amplification carried an M13 derived tag used for the subsequent cycle sequencing reaction. Sequences of all primers used in this study are listed in MOESM_1. Following PCR amplification, products were treated with ExoSAP enzyme and subjected to a cycle sequencing reaction in 96 well plates using forward and reverse M13 primers (Sigma-Proligo) (M13F: 5’-CACGACGTTGTAAAACGAC-3’, M13R: 5’-GGATAACAATTTCACACAGG-3’) and BigDye Terminator reagent mix (Applied Biosystems). Sequencing products were subsequently subjected to ethanol (85 %) cleanup using the Beckman Coulter Biomek NX liquid-handling robot. Final elution was in water. All samples were run on an ABI 3730 DNA analyzer (Applied Biosystems). Sequence analysis was performed using Soft Genetics “Mutation Surveyor®” DNA variant software.
Junction Fragment PCR Assay to Exclude a Previously Described Deletion
Patient samples in which none of the 11 exons could be amplified were examined by means of a junction fragment PCR assay for an already described complex deletion of 5.5 kb (Coffee et al. 2006). It is a PCR-based assay using three primers. The additional forward primer anneals to the deleted part; thus, a non-deleted allele results in a 680 bp product in contrast to the allele carrying the 5.5 kb deletion which gives a fragment of 500 bp.
MLPA Analysis
GALT exon copy number was tested using the P156 and P156-B1 SALSA MLPA (multiplex ligation-dependent probe amplification) kits from MRC Holland according to manufacturer instructions (version 11; 02-01-2007). Fragments were analyzed on a Beckman CEQ 8000 capillary analyzer and the raw data were processed using the Coffalyser software (MRC Holland).
Identification of Deletion Breakpoints
In order to map the breakpoints of the novel deletion, a PCR walking approach was employed using DNA samples from the three individuals identified as homozygous by MLPA. At first, the identification of a downstream breakpoint of the deletion was pursued. PCR primers targeting the adjacent IL11RA and the genes CCL21 and VCP, located further downstream, were designed. A PCR product was obtained in all samples suggesting that the downstream border is located within the IL11RA gene (data not shown). A subsequent series of primer design/PCR steps was performed to narrow down the deleted region (MOESM_2 and MOESM_3). Finally, using the primers DEL-10 F and DEL-9R that anneal 689 bp upstream of the GALT ATG start codon and 186 bp downstream of the IL11RA ATG, respectively (flanking a region of 10,047 bp), a 1.6 kb PCR product was obtained in homozygous and heterozygous deletion carriers but not in controls. PCR bidirectional sequencing of this PCR product using primers flanking the deletion (MOESM_4) was performed.
Screening for the New Deletion
A simple PCR assay was validated for diagnostic testing for the new deletion. PCR was performed using 0.5 μl AmpliTaq Gold DNA Polymerase (Applied Biosystems), with 1 μl DNA to 50 μl final volume. Three primers were added in the PCR mix: 1.2 μM forward primer DEL-10 F (5’-CCACCTAGATGGTGGCTGGAGCTT), 0.28 μM reverse primer DEL-9R (5’-ACTTACCCGGCAGTCACTCCAGG), and 0.04 μM forward primer DEL-Internal F (5’-GCGCACGCACATGCAAAGCA). The third primer was added to score the presence of the wild-type allele. PCR conditions were 94 °C for 10 min, then 40 cycles (94 °C, 20 sec; 68 °C, 30 sec; 72 °C, 1.5 min), and final extension at 72 °C for 5 min. The expected lengths of PCR amplicons for the deleted and the normal allele are 1.6 kb and 651 bp, respectively.
Haplotype Analysis
We performed haplotype analysis using the following microsatellite markers in the chromosomal region 9p: D9S1788, D9S1845, D9S165, D9S1878, D9S1817, D9S1805, D9S1804, D9S1791, D9S1859, D9S50, D9S1874, and D9S148. The forward primer for each was labeled with either Cy3 or Cy5 at the 5’ end. Primer sequences and amplification conditions for each PCR are shown in MOESM_5. PCR products were analyzed on a Beckman CEQ 8000 capillary analyzer. Haplotypes of individuals were constructed based on the map distance between markers estimated by means of MAP-O-MAT (Kong and Matise 2005), located at http://compgen.rutgers.edu/mapomat.
RT-PCR Assay
We studied RNA expression in Epstein-Barr virus (EBV)-transformed lymphoblastoid cell lines derived from two of the patients homozygous for the deletion. RNA extraction was performed using the RNeasy® Midi Kit (Qiagen, Cat. No. 75144) as per manufacturer instructions (Second edition, pages 26–31) and cDNA synthesis was achieved using the ProtoScript® M-MuLV First Strand Synthesis Kit (New England Biolabs, NEB#E6300S) following the manufacturer instructions (Version 1.0). We used primers specific for a region of the IL11RA cDNA downstream of the deletion (MOESM_6). A control PCR (of the β-actin housekeeping gene) was used to evaluate sample integrity.
Results
The GALT enzyme level of the eight patients was within the expected range for patients with classic galactosemia, using the spectrophotometric method, <6 μmol/h/gHb (Table 1). Interestingly, the most frequent mutation in Caucasian populations, p.Gln188Arg, was not found in any of the Cypriot patients with classic galactosemia. Two patients (siblings) were found to be homozygous for the p.Lys285Asn mutation and their parents were confirmed to carry the same mutation, while another three patients appeared to be homozygous for the p.Lys285Asn mutation but only one of their parents carried this mutation. For three patients, none of the 11 exons could be amplified. All the above findings could be explained by the presence of a large deletion covering the whole gene. Patient samples in which GALT exons could not be amplified were analyzed for the presence of the previously reported 5.5 kb GALT deletion characterized by an intervening complex junction fragment (Coffee et al. 2006). All patients were apparently negative for the 5.5 kb deletion (Fig. 1) suggesting the existence of a novel large GALT deletion.
Fig. 1.
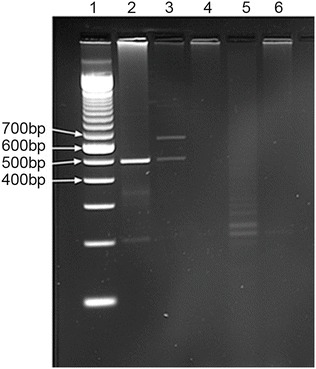
Agarose gel image of the result of the junction fragment PCR assay for the 5.5Kb deletion described by Coffee et al. Lane 1: 100 bp ladder. Lane 2: homozygote control for the 5.5 kb deletion. Lane 3: heterozygote control for the 5.5 kb deletion. Lanes 4–6: Cypriot patients
The MLPA results obtained for the three patient samples in which no exon could be amplified confirmed the presence of a large deletion encompassing all exons of the GALT gene, as no signal was produced for any of the GALT exons (Fig. 2). The parents of these patients were confirmed as deletion carriers. Samples from the three patients apparently homozygous for p.Lys285Asn and three of their parents in whom no mutation was detected by exon sequencing were also analyzed by MLPA and found to be heterozygous carriers of the same deletion. MLPA analysis further indicated lack of amplification of the probe targeted to the 5’ non-translated region of the adjacent IL11RA gene located 3.5 kb downstream of the GALT stop codon (Fig. 2). We repeated the MLPA analysis using the newer version of the MLPA kit (P156-B1), which contains a probe for exon 17 of the upstream gene DNAI1, and found that this probe was amplified in individuals carrying the deletion at comparable levels to normal individuals, indicating that the upstream border of the deletion lies within the 132.5 kb region between GALT and exon 17 of DNAI1 (MOESM_7).
Fig. 2.

Traces generated using the SALSA MLPA kit P156 GALT, for a patient homozygous for the new deletion, one of the parents and a normal control. Arrows indicate absent or reduced signal (red arrows for GALT, green arrows for IL11RA). Exon numbers are shown at the top of the peaks (E1–E11). Reference probes are marked by an asterisk (*)
Using a series of primer design/PCR steps, we narrowed down the deleted region until a fragment of 1.6 kb was obtained (Fig. 3b). Bidirectional sequencing of this PCR product using primers flanking the deletion (MOESM_4) revealed that the deletion spans a region of 8,489 bp (Fig. 3c, d). Identical results were obtained for all three patients found to be homozygous for the deletion. Interestingly, the deletion borders were found to be located within a homologous sequence block of six nucleotides (GTCAGT), present on either side, and could not therefore be precisely defined (Fig. 3d). Using the simple PCR screening assay that we validated, the new deletion was found in ten more galactosemia carriers (previously diagnosed in our laboratory). Haplotype analysis of these ten carriers plus the three homozygous patients revealed a common haplotype (MOESM_8).
Fig. 3.

(a) Schematic representation of the structure of the wild-type GALT gene (exons 1–11 are shown in blue) and of the first two exons of the adjacent IL11RA gene (red). The black arrows indicate the location of the forward and the reverse primers used for the PCR amplification, and the green arrow indicates the location of a third primer added to score the presence of the wild-type allele. The yellow boxed area includes the deleted part. (b) PCR screening test for the new deletion. 1 % agarose gel image of the PCR assay using three primers: DEL-10 F, DEL-internal F, and DEL-9R. A product of 1.6 kb was obtained for patients homozygous for the new deletion. In the normal control, a 651 bp product was obtained. In heterozygous carriers both products were obtained. Lane 1: 1 kb ladder. Lane 2: 100 bp ladder. Lane 3: normal control. Lane 4: homozygous patient for the novel 8.5 kb deletion. Lanes 5–6: Heterozygous carriers for the 8.5 kb deletion. Lane 7: blank. (c) Bidirectional sequence analysis of the junction fragment revealed that the deletion spans a region of 8489 bp. (d) Schematic representation of the GALT locus indicating the deleted part and showing the presence of a homologous sequence block of six nucleotides GTCAGT (red box) flanking the deletion
We studied RNA expression in EBV-transformed lymphoblastoid cell lines derived from two of the patients homozygous for the deletion. A PCR product was amplified in the control but not the patient samples suggesting the lack of IL11RA transcripts in the patients (Fig. 4). A control PCR (of the β-actin housekeeping gene) to evaluate sample integrity produced comparable results in the patient samples and the controls (Fig. 4). These findings indicate that the novel deletion simultaneously abrogates the expression of both the GALT gene and the adjacent IL11RA1 gene.
Fig. 4.
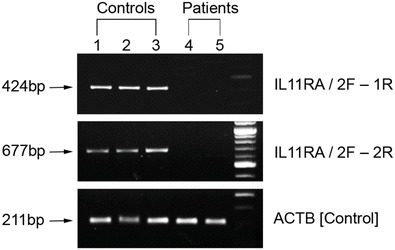
1.5 % agarose gel image of the RT-PCR assay using two primer sets specific for a region of the IL11RA cDNA downstream of the deletion. A product of 677 bp is obtained in the controls but not in the patients. Amplification of the housekeeping gene for β-actin using primers ACTB F and ACTB R was used as a check on sample integrity. Lanes 1–3: cDNAs from normal controls. Lanes 4–5: cDNAs from two Cypriot galactosemic patients homozygous for the 8.5 kb deletion. Lane 6: 100 bp ladder
Discussion
In this study, eight Greek Cypriot patients with classic galactosemia were characterized at the molecular level. These patients are, to our knowledge, the only patients diagnosed with galactosemia in Cyprus in the last 20 years (approximate number of births in this period 200,000). The most frequent mutation in Caucasian populations, p.Gln188Arg, was not found in any of our patients. Two siblings were found to be homozygous for the p.Lys285Asn mutation, three of the patients were found to be homozygous for a novel large deletion and three of the patients were found to be compound heterozygous for the p.Lys285Asn mutation and the new deletion. The presence of a large deletion on one allele can make the patient appear homozygous for the mutation on the other allele, as was the case in our three patients that were compound heterozygous, and as pointed out by Barbouth et al. as a possible cause of molecular misdiagnosis in galactosemia (Barbouth et al. 2006).
The MLPA results indicated that the new deletion encompasses all GALT exons and extends into the adjacent IL11RA gene while the upstream limit lies in the 132.5 kb region between the GALT gene and exon 17 of the DNAI1 gene. We proceeded to identify the deletion breakpoints by a PCR walking approach which revealed that the new deletion spans a region of 8,489 bp and the borders lie approximately 120 bp upstream of the GALT ATG and 140 bp upstream of the IL11RA ATG start codon. The presence of a homologous sequence of 6 bp flanking the deletion borders (Fig. 3d) may imply that a recombination mediated mutational event is responsible for the occurrence of the deletion. The process of microhomology-mediated end joining (MMEJ) is a repair mechanism for DNA double-strand breaks occurring during cell division and differentiation. Repeated sequences of 5–25 bp flanking the break are recombined following annealing of the complementary strands from each repeated sequence and resulting in deletion of intervening sequences (McVey and Lee 2008).
We performed haplotype analysis on 13 individuals bearing the new deletion, three homozygous and ten heterozygous, all unrelated. A common haplotype was found in all carriers of the deletion (MOESM_8) suggesting a single mutational event for the occurrence of this deletion in Cyprus. Although most individuals bearing the deletion originate from the western part of the island, a more extensive study is required in order to establish whether a cluster exists.
The level of GALT activity in patients homozygous for the new deletion would be expected to be zero since the deletion abolishes the whole of the gene. This was found to be so in two of our patients (0 and 0.05 μmol/h/g Hb), but the third patient had a residual activity of 5.9 μmol/h/g Hb (Table 1). This is probably due to the limitations of the spectrophotometric method we used to measure GALT. Use of a more specific and sensitive method like LC-MS/MS (Li et al. 2010) would probably reveal undetectable activity.
A comparison of the clinical phenotype of the galactosemic patients described in this study revealed the presence of additional clinical features in the three patients homozygous for the deletion that have so far not been described in patients with classic galactosemia (Table 1). In particular, one of the patients had microcephaly and optic atrophy and the other two displayed craniosynostosis. One of the patients with craniosynostosis was extensively studied at Great Ormond Street Hospital, London, for mutations in two genes associated with craniosynostosis (FGFR2 and FGFR3), but no mutations in these genes were found. Previous reports implicate mutations in the IL11RA gene as being responsible for craniosynostosis (Coussens et al. 2008; Nieminen et al. 2011). Nieminen et al. have shown that IL11 signaling is essential for the normal development of craniofacial bones and teeth and that its function is to restrict suture fusion and tooth number. The same authors identified five causative mutations in the IL11RA gene in patients with craniosynostosis of Pakistani and European origin. Given that the downstream border of the deletion found in the Cypriot patients extends up to the non-translated region of IL11RA, we addressed the possibility that the identified deletion eliminates the expression of IL11RA. The RNA expression studies showed lack of IL11RA transcripts in the patients (Fig. 4). These findings strongly suggest that the new deletion simultaneously eliminates GALT and IL11RA expression (contiguous deletion). Consequently, we attribute the craniosynostosis of our galactosemia patients to a defect in the IL11RA gene caused by the new large deletion we have described.
In conclusion, we have described a novel large deletion responsible for classic galactosemia in Greek Cypriot patients which covers the whole of the GALT gene and extends into the adjacent interleukin 11 receptor alpha (IL11RA) gene. Patients homozygous for this deletion show additional clinical features such as microcephaly and craniosynostosis, which can be attributed to the defect in the IL11RA gene.
Electronic Supplementary Material
Online Supporting Information_REVISED (DOC 426 KB)
Acknowledgments
We are grateful to:
Dr. Violetta Anastasiadou, Dr. Marianna Kousparou, and Dr. Andreas Hadjidemetriou, from Archbishop Makarios III Children’s Hospital in Nicosia, for offering their patients to be included in this study.
Dr. Kyproula Christodoulou, Head of the Department of Neurogenetics of the Cyprus Institute of Neurology & Genetics, for her help regarding haplotype analysis.
Mr. Mark Greenslade and Ms. Sarah Burton-Jones from Bristol Genetics Laboratory, Southmead Hospital, Bristol, for excellent technical support.
We would also like to thank all the children and their parents for participating in this study and for their cooperation.
This work was funded by Telethon Cyprus and by the Cyprus Research Promotion Foundation (Project PENEK/0609/64 was cofinanced by the European Regional Development Fund and the Republic of Cyprus through the Research Promotion Foundation).
Disclosure Statement: The authors declare no conflict of interest.
Synopsis
A novel contiguous deletion eliminates all GALT exons as well as the non-translated sequences of the adjacent interleukin 11 receptor alpha (IL11RA) gene causing classic galactosemia with additional phenotypic abnormalities such as craniosynostosis.
Compliance with Ethics Guidelines
Conflict of Interest
Rena Papachristoforou, Petros Petrou, Hilary Sawyer, Maggie Williams, and Anthi Drousiotou declare that they have no conflict of interest.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000 (5). Informed consent was obtained from all patients for being included in this study.
Footnotes
Competing interests: None declared
Contributor Information
Anthi Drousiotou, Email: anthidr@cing.ac.cy.
Collaborators: Johannes Zschocke and K Michael Gibson
References
- Aoki K, Wada Y. Outcome of the patients detected by newborn screening in Japan. Acta Paediatr Jpn. 1988;30(4):429–434. doi: 10.1111/j.1442-200X.1988.tb02533.x. [DOI] [PubMed] [Google Scholar]
- Barbouth D, Slepak T, Klapper H, Lai K, Elsas LJ. Prevention of a molecular misdiagnosis in galactosemia. Genet Med. 2006;8(3):178–182. doi: 10.1097/01.gim.0000204019.54509.40. [DOI] [PubMed] [Google Scholar]
- Berry GT, Leslie N, Reynolds R, Yager CT, Segal S. Evidence for alternate galactose oxidation in a patient with deletion of the galactose-1-phosphate uridyltransferase gene. Mol Genet Metab. 2001;72(4):316–321. doi: 10.1006/mgme.2001.3151. [DOI] [PubMed] [Google Scholar]
- Calderon FR, Phansalkar AR, Crockett DK, Miller M, Mao R. Mutation database for the galactose-1-phosphate uridyltransferase (GALT) gene. Hum Mutat. 2007;28(10):939–943. doi: 10.1002/humu.20544. [DOI] [PubMed] [Google Scholar]
- Coffee B, Hjelm LN, DeLorenzo A, Courtney EM, Yu C, Muralidharan K. Characterization of an unusual deletion of the galactose-1-phosphate uridyl transferase (GALT) gene. Genet Med. 2006;8(10):635–640. doi: 10.1097/01.gim.0000237720.78475.fb. [DOI] [PubMed] [Google Scholar]
- Coussens AK, Hughes IP, Wilkinson CR, et al. Identification of genes differentially expressed by prematurely fused human sutures using a novel in vivo – in vitro approach. Differentiation. 2008;76(5):531–545. doi: 10.1111/j.1432-0436.2007.00244.x. [DOI] [PubMed] [Google Scholar]
- Flach JE, Reichardt JK, Elsas LJ., 2nd Sequence of a cDNA encoding human galactose-1-phosphate uridyl transferase. Mol Biol Med. 1990;7(4):365–369. [PubMed] [Google Scholar]
- Greber-Platzer S, Guldberg P, Scheibenreiter S, et al. Molecular heterogeneity of classical and Duarte galactosemia: mutation analysis by denaturing gradient gel electrophoresis. Hum Mutat. 1997;10(1):49–57. doi: 10.1002/(SICI)1098-1004(1997)10:1<49::AID-HUMU7>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Holton JB, Walter JH, Tyfield LA. Galactosemia. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited diseases. New York: McGraw-Hill; 2001. pp. 1553–1587. [Google Scholar]
- Kalckar HM, Anderson EP, Isselbacher KJ. Galactosemia, a congenital defect in a nucleotide transferase. Biochim Biophys Acta. 1956;20(1):262–268. doi: 10.1016/0006-3002(56)90285-2. [DOI] [PubMed] [Google Scholar]
- Kong X, Matise TC. MAP-O-MAT: internet-based linkage mapping. Bioinformatics. 2005;21(4):557–559. doi: 10.1093/bioinformatics/bti024. [DOI] [PubMed] [Google Scholar]
- Lai K, Langley SD, Singh RH, Dembure PP, Hjelm LN, Elsas LJ., 2nd A prevalent mutation for galactosemia among black Americans. J Pediatr. 1996;128(1):89–95. doi: 10.1016/S0022-3476(96)70432-8. [DOI] [PubMed] [Google Scholar]
- Leslie ND, Immerman EB, Flach JE, Florez M, Fridovich-Keil JL, Elsas LJ. The human galactose-1-phosphate uridyltransferase gene. Genomics. 1992;14(2):474–480. doi: 10.1016/S0888-7543(05)80244-7. [DOI] [PubMed] [Google Scholar]
- Levy HL, Hammersen G. Newborn screening for galactosemia and other galactose metabolic defects. J Pediatr. 1978;92(6):871–877. doi: 10.1016/S0022-3476(78)80351-5. [DOI] [PubMed] [Google Scholar]
- Li Y, Ptolemy AS, Harmonay L, Kellogg M, Berry GT. Quantification of galactose-1-phosphate uridyltransferase enzyme activity by liquid chromatography-tandem mass spectrometry. Clin Chem. 2010;56(5):772–780. doi: 10.1373/clinchem.2009.140459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVey M, Lee SE. MMEJ repair of double-strand breaks (director’s cut): deleted sequences and alternative endings. Trends Genet. 2008;24(11):529–538. doi: 10.1016/j.tig.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy M, McHugh B, Tighe O, et al. Genetic basis of transferase-deficient galactosaemia in Ireland and the population history of the Irish Travellers. Eur J Hum Genet. 1999;7(5):549–554. doi: 10.1038/sj.ejhg.5200327. [DOI] [PubMed] [Google Scholar]
- Nieminen P, Morgan NV, Fenwick AL, et al. Inactivation of IL11 signaling causes craniosynostosis, delayed tooth eruption, and supernumerary teeth. Am J Hum Genet. 2011;89(1):67–81. doi: 10.1016/j.ajhg.2011.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichardt JK, Berg P. Cloning and characterization of a cDNA encoding human galactose-1-phosphate uridyl transferase. Mol Biol Med. 1988;5(2):107–122. [PubMed] [Google Scholar]
- Schulpis K, Papakonstantinou ED, Michelakakis H, Podskarbi T, Patsouras A, Shin Y. Screening for galactosaemia in Greece. Paediatr Perinat Epidemiol. 1997;11(4):436–440. doi: 10.1046/j.1365-3016.1997.d01-31.x. [DOI] [PubMed] [Google Scholar]
- Shih LY, Suslak L, Rosin I, Searle BM, Desposito F. Gene dosage studies supporting localization of the structural gene for galactose-1-phosphate uridyl transferase (GALT) to band p13 of chromosome 9. Am J Med Genet. 1984;19(3):539–543. doi: 10.1002/ajmg.1320190316. [DOI] [PubMed] [Google Scholar]
- Tyfield L, Reichardt J, Fridovich-Keil J, et al. Classical galactosemia and mutations at the galactose-1-phosphate uridyl transferase (GALT) gene. Hum Mutat. 1999;13(6):417–430. doi: 10.1002/(SICI)1098-1004(1999)13:6<417::AID-HUMU1>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Zekanowski C, Radomyska B, Bal J. Molecular characterization of Polish patients with classical galactosaemia. J Inherit Metab Dis. 1999;22(5):679–682. doi: 10.1023/A:1005511020607. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Online Supporting Information_REVISED (DOC 426 KB)