

Abstract
Background
The Streptococcus Anginosus Group (SAG) represents three closely related species of the viridans group streptococci recognized as commensal bacteria of the oral, gastrointestinal and urogenital tracts. The SAG also cause severe invasive infections, and are pathogens during cystic fibrosis (CF) pulmonary exacerbation. Little genomic information or description of virulence mechanisms is currently available for SAG. We conducted intra and inter species whole-genome comparative analyses with 59 publically available Streptococcus genomes and seven in-house closed high quality finished SAG genomes; S. constellatus (3), S. intermedius (2), and S. anginosus (2). For each SAG species, we sequenced at least one numerically dominant strain from CF airways recovered during acute exacerbation and an invasive, non-lung isolate. We also evaluated microevolution that occurred within two isolates that were cultured from one individual one year apart.
Results
The SAG genomes were most closely related to S. gordonii and S. sanguinis, based on shared orthologs and harbor a similar number of proteins within each COG category as other Streptococcus species. Numerous characterized streptococcus virulence factor homologs were identified within the SAG genomes including; adherence, invasion, spreading factors, LPxTG cell wall proteins, and two component histidine kinases known to be involved in virulence gene regulation. Mobile elements, primarily integrative conjugative elements and bacteriophage, account for greater than 10% of the SAG genomes. S. anginosus was the most variable species sequenced in this study, yielding both the smallest and the largest SAG genomes containing multiple genomic rearrangements, insertions and deletions. In contrast, within the S. constellatus and S. intermedius species, there was extensive continuous synteny, with only slight differences in genome size between strains. Within S. constellatus we were able to determine important SNPs and changes in VNTR numbers that occurred over the course of one year.
Conclusions
The comparative genomic analysis of the SAG clarifies the phylogenetics of these bacteria and supports the distinct species classification. Numerous potential virulence determinants were identified and provide a foundation for further studies into SAG pathogenesis. Furthermore, the data may be used to enable the development of rapid diagnostic assays and therapeutics for these pathogens.
Keywords: Streptococcus Milleri group, Streptococcus Anginosus group, Streptococcus anginosus, Streptococcus constellatus, Streptococcus intermedius, Phylogenetics, Virulence, Comparative genomics, Whole-genome sequencing
Background
The genus Streptococcus consists of Gram-positive cocci that are divided into sub-groups via numerous biochemical and molecular methods. The majority of Streptococcus species can be divided into either β-hemolytic causing complete zones of lysis on blood agar plates or α-hemolytic, formation of green zones due to oxidation of hemoglobin by hydrogen peroxide to form a green methemoglobin. Lancefield typing (based on specific carbohydrates within the bacterial cell wall) provides groupings that do not necessarily follow recognized species. The most clinically important are S. pyogenes known as Lancefield Group A Streptococcus (GAS), S. agalactiae Lancefield Group B Streptococcus (GBS), Lancefield group O (S. pneumoniae, S. mitis and S. pseudopneumoniae) and the variable Lancefield group species belonging to the Streptococcus Anginosus group (Which has also been referred to as the Streptococcus Milleri Group primarily by clinicians; SAG) that include non-typeable (using Lancefield typing) as well as strains that are Lancefield group C, F and G. The majority of α-hemolytic streptococci are non-pyogenic including the viridans group Streptococcus (VGS = Anginosus, Mitis and Salivarius groups), Mutans, and S. suis, which is a species that has not been assigned to a group [1]. VGS are considered to be part of the normal microbiota in the human oropharyngeal, urogenital and gastrointestinal tracts [2]. Many of the VGS are classified as α-hemolytic based on their activity on standard sheep’s blood agar. However, some strains can be β-hemolytic, including S. anginosus and S. constellatus that show beta-hemolytic activity and have been shown to produce a streptolysin S-like protein [3]. S. intermedius produces beta-hemolysis on human blood due to a human specific hemolysin called intermedilysin [4] and may also be beta-hemolytic on sheep blood agar. Many other VGS behave similarly and also show β-hemolytic activity under anaerobic conditions, but not aerobically under which these assays are usually conducted (Surette and Teal, unpublished data).
The taxonomic grouping of the SAG has historically been debated and the definitions have ranged from that of one to three species (with or without subspecies) [5]. The validity of S. anginosus (SA), S. intermedius (SI) and S. constellatus (SC) as individual species has been addressed through phenotypic analysis, DNA: DNA hybridization studies and genetic characterization, and currently there is little debate that there are at least three distinct species with additional subspecies [6,7]. A recent study has elucidated that SAG consist of 3 species with S. constellatus divided into 3 subspecies (subsp constellatus (SCC), subsp pharyngis (SCP) and subsp viborgensis) and S. anginosus divided into 2 subspecies [subsp anginosus (SAA) and subsp whileyi (SAW)], based on the use of seven core housekeeping genes [7]. The SAG are phenotypically diverse but most strains share common characteristics such as slow growth rate, distinctive ‘caramel smell’, ability to hydrolyze arginine, acetoin production from glucose, and an inability to ferment sorbitol [8]. Lancefield sero-grouping is variable with SA Lancefield types of A, C, F or G, while SC is typically Lancefield C, F or no antigen, and SI is generally not typeable using the Lancefield method. Almost half of all human SAG clinical isolates are Lancefield F type [9]. Due to this phenotypic variability, molecular methods must be used for proper classification of SAG.
The SAG are part of the microbiota of the respiratory, gastrointestinal, and genitourinary tract with variable carriage levels [8]. The SAG are also medically important for their ability to cause suppurative infections and have been isolated from numerous body sites [10,11]. Of particular interest, SAG has been identified as the most common organism isolated from brain abscess [12,13], liver abscess [14] and empyema [13,15]. Their capacity to elicit pulmonary exacerbation and contribute to disease pathology in CF has also been demonstrated [14,16,17]. However, the exact mechanism for virulence within SAG has yet to be determined.
Although many Streptococcus species have the ability to cause disease, virulence studies within the Streptococcus genus often focus on GAS, GBS and S. pneumoniae[18-20]. SAG virulence and pathogenesis mechanisms have not yet been well studied; however, virulence mechanisms have been identified within SAG that allow for the invasion of host cells, evasion of host immune activity, spreading, and allow for the colonization of host tissues to occur. Intermedilysin is a cholesterol-dependent cytolysin produced by all SI strains that demonstrates specificity for human erythrocytes [21], and is essential for invasion of human cells by SI [4]. S. constellatus and SA also exhibit the β-hemolytic phenotype on sheep’s blood agar [22], this hemolytic activity has been attributed to the cytolytic factor Streptolysin S-like peptides encoded by the sagA gene within a sag operon [23]. Mutation to the luxS gene has also been shown to decrease hemolytic activity in SI [23]. Capsules allow evasion of the host immune system and encapsulated SAG strains have been isolated having a greater virulence potential than non-encapsulated strains [24]. They are more likely to cause larger abscesses, earlier spontaneous drainage and death in mice compared to non-encapsulated strains [25]. Hyaluronan (HA) is a major component of the extra cellular matrix of human connective tissue and is expressed by many cell types [26]. HA up regulates the hyl gene increasing SAG spreading and colonization within the host [27]. Most SAG isolates have both hyaluronidase and chondroitin sulfatase activity [9]. A detailed analysis of SAG virulence targets are required to achieve a firm understanding for the overall virulence potential within SAG.
The number of sequenced bacterial genomes has exploded with the advent of new sequencing technologies, which has allowed for comparative genomic analysis. Using Roche GS20 and Illumina technologies, we have sequenced to closure and, sequence polished and fully annotated seven SAG genomes including representatives of each species. With the abundance of sequenced strains within the Streptococcus genus, we conducted a detailed comparative analysis utilizing 66 streptococcal genomes including representatives from SAG, Mitis, Pyogenic, Salivarius, Bovis, Mutans groups and S. suis strains. Such genomic comparisons allowed for detailed characterization with insights gained into SAG phylogenomics, core genome, virulence potential, horizontally transferred genetic material, and microevolution within the host.
Results and discussion
An introduction to the SAG genome structure
The SAG strains were chosen for study based on being the numerically dominant bacterial isolate cultured during CF pulmonary exacerbation or primary culture from an invasive infection (Table 1). All strains were sequenced using Roche GS20 and Illumina sequencing technologies. The sequence coverage for each genome can be observed in Additional file 1. All seven genomes were sequenced to full closure with additional Sanger and Illumina sequence polishing to correct sequence errors and false pseudogenes. Polishing was used to correct homopolymer base-calling errors known to occur with the GS20 pyrosequencing reactions. Illumina sequencing was not done for SCP C232 as Sanger sequencing was done previously to evaluate homopolymer errors and achieve genome closure.
Table 1.
Background information for SAG strains used in this study
| Species | Strain | Lancefield type | Casea | Extent of SAG disease | Clinical outcome | Genotype | Age/Sex |
|---|---|---|---|---|---|---|---|
| SA |
C238 |
C |
3 |
Recurrent broncho-pulmonary |
Transplant |
∆F508/∆F508 |
18/F |
| SA |
C1051 |
C |
NA |
Blood sepsis |
NA |
Non-CF |
NA |
| SCP |
C232 |
C |
4 |
Recurrent broncho-pulmonary |
Stabilized |
∆F508/∆F508 |
30/M |
| SCP |
C818* |
C |
4 |
Recurrent broncho-pulmonary |
Stabilized |
∆F508/∆F508 |
31/M |
| SCP |
C1050 |
C |
NA |
Blood sepsis |
NA |
Non-CF |
NA |
| SI |
C270 |
NT |
5 |
Broncho-pulmonary |
Stabilized |
∆F508/∆F508 |
32/F |
| SI | B196 | NT | 1 | Broncho-pulmonary, septic arthritis, Osteomyelitis, pyomyositis | Total hip arthroplasy | ∆F508/∆F508 | 18/M |
aCase history as described in Parkins et al. [17]; *SCP C818 was isolated from the same individual 13 months after SCP C232; NT, Non-typable; NA, Not available for these isolates.
Each finished genome of the seven in-house sequenced SAG was comprised of a single circular chromosome ranging in size from 1.91 Mbp to 2.23 Mbp and is within the range of genome sizes observed for all available sequenced SAG strains (Table 2). A schematic view of all sequenced SAG genomes, including the draft genome sequences available from NCBI are provided in Figure 1, with an overall SAG comparison showing the pan-genome in Figure 1A and a comparison of each species in Figure 1B-D. The average genome size for SAG is very similar to the 2.02 Mbp average among the streptococci used for this study (n = 66). An overall comparison, using MAUVE, of the different species within SAG revealed that SI and SC had long stretches of synteny with regions of rearrangement. SA had a greater amount of variation, although 11 of the genomes were draft genomes (contigs) meaning that synteny is difficult to infer (Additional file 2). The BLAST atlas comparison for Figure 1B-D allows for a comparison of all strains to a reference strain for each species of SAG, and reveals regions of divergence within the genomes relative to the reference strain. The general features of the SAG genomes are summarized in Table 2, and an overall summary of all genomes used in this study with publicly available genomic sequence data (Additional file 3). As previously identified SI F0395 was incorrectly submitted as an SI and has subsequently been shown to be a SCC [7].
Table 2.
Summary of genome characteristics for sequenced SAG
| Species | Sub-species | Strain | Genome size | G + C% | CDSs | tRNA | Avg length CDS (nt) | Coding% | Pseudogenes | GenBank accession |
|---|---|---|---|---|---|---|---|---|---|---|
|
SA |
Anginosus |
C1051 |
1,911,706 |
38.97 |
1728 |
58 |
941 |
85.04 |
40 |
CP003860 |
|
SA |
Whileyi |
C238 |
2,233,640 |
38.23 |
1976 |
58 |
945 |
83.55 |
80 |
CP003861 |
| SA |
Whileyi |
CCUG39159T |
2,294,730 |
38.50 |
2146 |
40 |
930 |
86.58 |
0* |
GCA_000257765 |
| SA |
Anginosus |
SK1138 |
1,958,191 |
38.61 |
1852 |
33 |
943 |
88.36 |
0 |
GCA_000287595 |
| SA |
Anginosus |
SK52T |
1,892,386 |
38.65 |
1805 |
31 |
921 |
88.36 |
0 |
GCA_000214555 |
| SA |
Anginosus |
F0211 |
1,993,790 |
38.44 |
1844 |
31 |
956 |
87.63 |
0 |
GCA_000184365 |
| SA |
Anginosus |
62CV |
1,816,149 |
38.80 |
1751 |
61 |
912 |
87.95 |
0 |
GCA_000186545 |
|
SC |
Pharyngis |
C232 |
1,935,414 |
38.18 |
1756 |
59 |
916 |
83.12 |
69 |
CP003800 |
|
SC |
Pharyngis |
C818 |
1,935,662 |
38.18 |
1754 |
59 |
916 |
83.02 |
71 |
CP003840 |
|
SC |
Pharyngis |
C1050 |
1,991,156 |
38.13 |
1797 |
59 |
921 |
83.27 |
73 |
CP003859 |
| SC |
Pharyngis |
SK1060T |
1,963,771 |
37.99 |
2186 |
50 |
754 |
83.45 |
0 |
GCA_000223295 |
| SC |
Constellatus |
SK53T |
1,840,061 |
37.92 |
1730 |
32 |
943 |
87.83 |
0 |
GCA_000257785 |
| SC |
Constellatus |
F0395 |
1,927,278 |
37.94 |
1861 |
61 |
916 |
87.63 |
0 |
GCA_000234015 |
|
SI |
|
C270 |
1,960,728 |
37.65 |
1778 |
60 |
949 |
86.09 |
24 |
CP003858 |
|
SI |
|
B196 |
1,996,214 |
37.56 |
1815 |
60 |
952 |
86.53 |
18 |
CP003857 |
| SI |
|
SK54T |
1,919,718 |
37.58 |
1778 |
28 |
949 |
87.49 |
0 |
GCA_000258445 |
| SI |
|
JTH08 |
1,933,610 |
37.71 |
1780 |
67 |
979 |
86.59 |
0 |
AP010969 |
| SI | F0413 | 1,921,346 | 37.63 | 1820 | 62 | 925 | 87.02 | 0 | GCA_000234035 |
*There are no genes annotated as pseudogenes, however, there are genes that are annotated with potential sequence errors or frameshift mutations. Strains that have been bolded are high quality finished genomes from this study. T: denotes reference strain, SA: Streptococcus anginosus, SC: Streptococcus constellatus, SI: Streptococcus intermedius.
Figure 1.

Schematic view of the Pan-genome and the circular SAG genomes based on nucleotide identity of >70%. An overall pan-genome Figure was constructed as well as figures for each species within SAG. The figures are arranged as follows; A) Pan-genome comparison of SAG; the genomes were constructed by adding sequences unique to the pangenome at the end of the chromosome it is found in; B) Circular genome comparison of SA with SAW C238 as the reference genome; C) Circular genome comparison of SC with SCP C232 as reference genome; and D) Circular genome comparison of SI with B196 as the reference genome; For all diagrams the order of rings from innermost to outermost is: the ruler in kbp, the G + C skew (purple ring) with positive strand >0, and negative strands <0; G + C content positive strand >0 and negative strand <0 (black ring), followed by the genomes as listed in the legends. The numbers on the outer rings indicate regions of horizontal gene transfer including ICE; Integrative conjugative element and phi; bacteriophage regions.
The G + C content for SAG strains ranged from 37.56 to 38.97% (avg 38.14, n = 18), with the overall G + C content increasing from SI, SC to SA (Table 2). Overall the average of 38.14% is similar to the 38.57% average G + C content for all Streptococcus strains analyzed (Additional file 3). None of the SAG strains contained plasmid DNA similar to other Streptococcus[28-30]. All seven in-house sequenced SAG contained four rRNA operons and 58, 59 and 60 tRNA genes in SA, SC and SI respectively, with the majority of tRNA genes situated around rRNA operons, as seen with other sequenced Streptococcus strains [31]. With regard to the number of tRNA and rRNA found within the finished genomes, SAG were most similar to S. mitis and S. suis[30], having fewer numbers of both these RNA genes than streptococci in the pyogenic, mutans, bovis and salivarius groups [28,29,32-35].
The SAG strains had on average 1800 CDSs (coding DNA sequence: does not include pseudogenes) with an average length of 934 bp, while the streptococcal average was 1943 CDSs with an average size of 893 bp. This difference in number of CDSs may be due in part to the differing size of streptococci genomes, but it may also be partially due to the extra care taken to manually annotate the in-house SAG genomes resulting in a higher number of pseudogenes. This has been demonstrated using S. pyogenes MGAS315, MGAS8232, SF370 and SSI-1, which in the original annotation had zero pseudogenes [29,32-34], but when retrospectively examined for the presence of pseudogenes they were shown to have 42, 50, 60 and 51 respectively [36]. The seven in-house sequenced SAG genomes harbor 16 to 80 pseudogenes depending on the strain, with the most found in SAW C238 and the least in SI B196 (Table 2). When pseudogenes are included in the total CDSs, the average number of CDSs increased to 1855. An example of how sequence errors or pseudogenes can affect the average CDS size is seen in the whole-genome shotgun sequence for SCP SK1060. This genome had an average CDS size of 754 bp (Table 2), however, a closer look at this genome revealed a large number of essential genes that were truncated including, parC, gyrA, rpoA, uvrA, dnaG, dnaE, and ftsA. The lack of extra genetic material as shown in Figure 1A for SCP SK1060 compared to other SC strains also shows that the extra CDSs are created owing to sequence errors and not novel genetic materials. In this study, even after manual sequence verification, generating high quality finished genomes, all seven in-house sequenced SAG strains still contained predicted pseudogenes, defined as a full-length CDS present within another SAG strain from this study or GenBank or another genome in GenBank. This demonstrates the value of having single contig, high quality reference genomes for SAG to aid in the analysis of comparative genomic studies.
Accurate Phylogeny of SAG requires multiple genetic loci
With the increased genomic sequences available, the capacity for applying phylogenetic analysis methods has increased; however, there is urgent need to evaluate their equivalency. Phylogenetic analysis using the single locus 16S rRNA has historically been the primary molecular method determining species within the SAG and, indeed this is how the seven in-house sequenced SAG strains were originally speciated. To assess the discriminatory power of 16S rRNA sequencing we compared the results to those acquired from an in-house core-SNP pipeline, a multi-locus automated pipeline for phylogenomic analysis called AMPHORA [37], and two alternate single loci targets cpn60 and rpoB. For all analysis strategies, 66-sequenced streptococcal genomes were included in the SAG clustering analysis. In all iterations SI, SC, and SA strains clustered together by species with one exception-SA F0211 clustered with the SC strains when using cpn60 as a reference.
Compared to the dendrogram constructed for 16S rRNA (Figure 2A), dendrograms of similar overall topology were acquired for all methods (Figure 2B, core-SNP pipeline; 2C, rpoB and 2D, cpn60) [Amphora (not shown)]; in general the branch lengths were longer with the multi-locus approaches and a greater degree of statistical certainty was achieved. It has been suggested that rpoB is a sensitive single locus method for differentiating Streptococcus. Housekeeping genes, such as rpoB and cpn60 are believed to be either selectively neutral or subject to purifying selection. The rate of synonymous substitutions (dS) should be equal to or slightly greater than the rate of nonsynonymous substitutions (dN), giving a dN : dS ratio of less than 1. The ratio of mean dN : dS per site (dN:dS ratio) for rpoB was found to be 0.106 and the ratio for cpn60 was 0.056 indicating purifying selection and demonstrating their suitability for use in phylogenetic analysis. Our analysis revealed cpn60 to provide the longest branch lengths discriminating one strain from another (Figure 2D), thus outperforming both rpoB (Figure 2C) and 16S rRNA (Figure 2A), supporting the findings of Glazunova (2006) [38]. For all analytic approaches, except for 16S rRNA, the closest related species to the SAG were S. sanguinis and S. gordonii. These cluster in a tight group away from all other sequenced streptococcal strains as previously demonstrated [7]. Schouls (2003) have previously demonstrated that speciation based on 16S rRNA is problematic given there is demonstrated lateral transfer within the SAG of portions of the 16S rRNA gene [39]; our study supports this fundamental limitation of 16S rRNA as a phylogenetic marker and demonstrates that 16S rRNA has lower discriminatory power for speciation than any of the other approaches tested herein. Although cpn60 and rpoB each performed reasonably well for SAG phylogenetic discrimination, we advocate that single locus results will be less reliable than results achieved with multiple genetic loci.
Figure 2.

Phylogenetic relationship among Streptococcus strains. Four phylogenetic trees were constructed, each using a different genetic locus or multiple loci for analysis as follows: A) 16S rRNA, B) In-house core-SNP pipeline, C)rpoB, and D)cpn60. Bootstrap values of equal or greater than 75%,or approximate maximum likelihood ratio test values equal to or greater than 0.8 are shown. The blue boxes highlight SCC, purples boxes highlight SCP, the red boxes highlight SI, dark green boxes highlight SAA and light green highlights SAW.
Global ortholog analysis for SAG within the genus Streptococcus
A total genetic content comparison was conducted to determine the Streptococcus pan- and core genomes via ortholog comparison between 66 Streptococcus reference strains using OrthoMCL [40]. A total of 11587 orthologous groups were identified within the 66 Streptococcus strains, with 7669 ortholog groups absent within SAG (Additional file 4). In a similar study using 11 species and 45 strains of Streptococcus, 9053 orthologous groups were identified with 7442 absent within the S. dysgalactiae group [41]. The differences in numbers are due to an increased number of species (16) and strains (66) used in the present study, which is the largest study of this type done for Streptococcus. The Streptococcus pan-genome increases by an average of 45 genes for each of the 21 additional strains used [41]. Most genes are from the five new Streptococcus species generated in this study. This shows that the Streptococcus pan-genome should still be considered ‘open’ with new genes added with additional genomes analyzed.
The core genome of Streptococcus contains 626 genes, accounting for 32.2% (626/1943 (average genome #CDS) × 100) of the Streptococcus genome. Previous analysis suggests that the core genome should account for >25% of the average genome [42]. The core genome common to all seven in-house sequenced SAG was 1234 genes (Figure 3A), and decreased to 1145 when the 11 SAG draft genomes (WGS contigs) available in GenBank are added to the analysis (Figure 3B). The three comparator groups of pyogenic Streptococcus including Group A Streptococcus (GAS; S. pyogenes), Group B Streptococcus (GBS; S. agalactiae) and Group C Streptococcus (GCS; S. equi) have 1285, 1305 and 1325 core genes respectively [32]. The S. dysgalactiae group has 1471 genes common to both S. dysgalactiae subsp. dysgalactiae and S. dysgalactiae subsp. equisimilis[41]. The average core genome percentage for SAG is lower than other groups at only 62.2% (1145/1842 n = 18), however, it is more than the 60% threshold suggested as a limit for core genomes within streptococcal groups [42]. The majority of groups analyzed were for a single species rather than multiple species as done for SAG.
Figure 3.
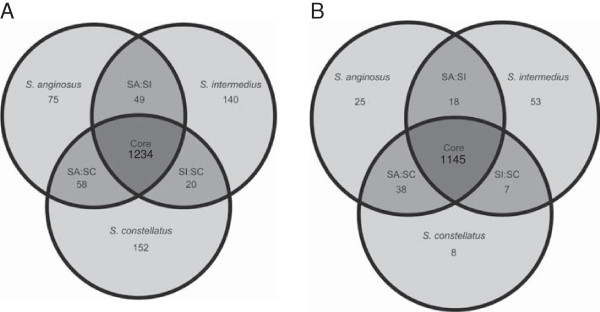
Genomic content analysis of the sequenced SAG strains. A) Comparison of seven SAG strains sequenced in this study showing the overall core genomes. B) Comparison of all available sequenced SAG strains. Numbers within circles correspond to number of CDSs that are conserved within the strains analyzed (intersection of strain circles) or that are unique to each strain analyzed.
Comparison of individual species within SAG revealed that SC had the most core genes (1617) followed by SA (1323) and SI (1316), this is consistent with other species of streptococcus; S. thermophilus (1271) [43]; S. pneumoniae (1619) [44]; and S. agalactiae (1806) [45]. Within SAG the average percentage of core genes is 75.23% for SC, 70.7% for SA and 72.9% for SI. The values for SAG are similar to those found for other Streptococcus species including S. pyogenes (80.3%), S. agalactiae (79.1%) and S. thermophilus (81.4%) [42].
Extended analysis demonstrates that SAG shares the most core genes with S. gordonii and S. sanguinis (Table 3; Additional file 5). S. anginosus shares a slightly greater percentage of their core genome with S. sanguinis, whereas SI and SC share a very similar percentage of their core genome with both S. gordonii and S. sanguinis (Additional file 6). The finding that S. gordonii and S. sanguinis were most closely related to the SAG strains confirms previous studies [38]. Genome comparisons also confirmed that SAG shared a decreased portion of their core genome with less taxonomically related Streptococcus species including S. pneumoniae, S. pyogenes, and S. agalactiae (Table 3). The greatest numbers of orthologs from the core genome were found in S. gordonii (1073) and S. sanguinis SK36 (1067), while the fewest were found in S. pyogenes with 887 (Table 3, Additional file 5). This is partly due to the closer taxonomic relationship between SAG, S. sanguinis and S. gordonii, however, it may also be attributed to the number of genomes used in the analysis, with groups that have a greater number of sequenced genomes having a fewer number of core gene orthologs. Interestingly, S. mutans (which is less related to the SAG based on taxonomic analysis) shared more genes than some of the more closely related strains (Table 3, Additional file 5). This was also seen in a previous study that showed S. sanguinis shared more genes with S. mutans than the more closely related S. pneumoniae[31]. The most plausible explanation is the shared ecological niche that they all inhabit, however, S. pneumoniae is known to have increased virulence relative to S. sanguinis and S. mutans.
Table 3.
Comparison of gene content of SAG to clinically important Streptococcus species
| Species | Average Core genes | # of SAG core genes not found | Total SAG genes analyzed | % of SAG core genes found |
|---|---|---|---|---|
|
S. gordoniia |
1073* |
72 |
1145 |
93.71 |
|
S. sanguinisb |
1069 |
76 |
1145 |
93.36 |
|
S. mitisc |
992 |
153 |
1145 |
86.64 |
|
S. pneumoniaed |
954 |
117 |
1071 |
89.08 |
|
S. suise |
969 |
148 |
1117 |
86.75 |
|
S. agalactiaef |
956 |
163 |
1121 |
85.43 |
|
S. pyogenesg |
887 |
209 |
1096 |
80.93 |
| S. mutansh | 958 | 180 | 1138 | 84.18 |
*This table was constructed using results from OrthoMCL; aS. gordonii analysis included 1 strain; bS. sanguinis analysis included 1 strain; cS. mitis analysis included 1 strain; dS. pneumoniae analysis included 12 strains; eS. suis analysis included 6 strains; fS. agalactiae analysis included 3 strains; gS. pyogenes analysis included 13 strains; hS. mutans analysis included 2 strains.
Differences in protein functional classifications within SAG
Protein functions for the ortholog groups (discussed above) were analyzed observing COG superfamily protein for all 66 Streptococcus reference strains this revealed that all strains had a similar distribution of proteins within the COG categories (Figure 4). Indeed previous studies have revealed the majority of Streptococcus strains have a similar percentage of their genome dedicated to each COG category [29-31]. Analysis of the total predicted coding ORFs shows that approximately 68% (66.5 to 70.6%) of all proteins in SAG code for a characterized protein from the COG superfamily of proteins, except for in SCP SK1060 (for reasons discussed above), which has only 51% of its proteins hitting to characterized proteins. Focusing on the most closely related strains to SAG reveals that S. gordonii and S. sanguinis, both have approximately 68% of their genomes coding for characterized COG superfamily proteins (Figure 4B).
Figure 4.
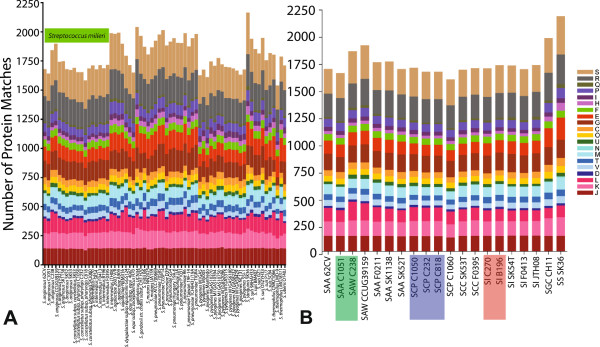
Comparison SAG to the genus Streptococcus based on COG categories. A) eggNOG comparison of available Streptococcus genomes from NCBI, SAG are highlighted with a green bar. B) Comparison of all SAG genomes (SAW; S. anginosus subsp. whileyi; SAA; S. anginosus subsp. anginosus; SCP, S. constellatus subsp. pharyngis; SCC, S. constellatus subsp. constellatus; SI, S. intermedius) with the most closely related Streptococcus strains (SS; S. sanguinis, and SGC; S. gordonii), strains sequenced in this study are highlighted with: red for SI, blue for SC, and green for SA. Only functional categories with at least one protein present are represented in the graphs. Significant BlastP hits were filtered using the following criteria: 80% PID over a minimum of 80% of the protein length or proteins over 100 amino acids in length with a minimum of 50% length and 30% PID to eggNOG categories. Category abbreviations as follows: J, Translation, ribosomal structure and biogenesis; K, Transcription; L Replication, recombination and repair; D, cell cycle control, cell division, chromosomal partitioning; V, Defense mechanisms; T, Signal transduction mechanisms; M, Cell wall/membrane/envelope biogenesis; N, Cell motility, U, Intracellular trafficking, secretion, and vesicular transport; O, Posttranslational modification, protein turnover, chaperones; C, Energy production and conversion, G, Carbohydrate transport and metabolism; E, Amino acid transport and metabolism, F Nucleotide transport and metabolism; H, Coenzyme transport and metabolism; I, Lipid transport and metabolism; P, Inorganic ion transport and metabolism; Q, Secondary metabolites biosynthesis, transport and catabolism; R, General function predicted only; S, Function unknown.
Comparing SAG to S. gordonii and S. sanguinis there are only a few noticeable differences in the number of proteins within different COG categories (Figure 4B). Within SA there is an increase of replication, recombination, and repair proteins for SAW C238 (172) and SAW CCUG39159 (159), compared to an average of 117 for the other strains in Figure 4B. This difference is likely due to the increase in phage-related proteins within a recently identified SAW subspecies, which may have increased propensity for the uptake of foreign DNA (largest SAG genomes in this study) as compared to SAA (Figure 1A). There is a decrease in amino acid transport and metabolism proteins (Group E), and poorly characterized protein (Groups R and S) in SAG (112 and 472) compared to S. gordonii (173 and 570) and S. sanguinis (202 and 620). Some of these differences are due to the size of the genomes, as the genomes for both S. gordonii and S. sanguinis are larger than the SAG genomes, accounting for some of the increase in proteins found. In S. mutans, COG groups L, E, R and S are also known to be variable within the genus, with many proteins from these groups present within the accessory genome for S. mutans[46]. The analysis of these differences will provide insight into the species-specific characteristics for SAG.
Orthologs identified as SAG-unique signatures
The delineation of SAG as a unique group of Streptococcus species relies on the presence of genomic material not present within other Streptococcus species. SAG-unique signatures were identified as regions conserved within SAG and absent in all other Streptococcus strains. A total of 688 ortholog groups were present in at least one strain of SAG and not in any of the other Streptococcus strains included in this study. Small conserved regions were identified from the overall large COG figure and a drill down figure focusing on the potential unique regions within SAG was created (Additional file 5). Of the 114-ortholog groups that were identified within SAG, only eight unique proteins were conserved within most of the sequenced SAG strains (Table 4). These eight proteins had <50 percent identity (PID) to anything in the NCBI protein database and <40 PID to any streptococcal protein (Table 4). This analysis was conducted for each individual species with SA, SI and SC having 1, 14 and 42 ortholog groups respectively that were unique to each species. A number of orthologs were unique to each strain of SAG, however, almost all of these genes encoded for hypothetical or conserved hypothetical proteins (Additional file 7). Very few unique proteins were observed for SA and SI. Interestingly, the unique genes for SC had a G + C content of 33.4%, which is lower than the average G + C content for sequenced SC strains (38.1%), suggesting these genes were likely acquired via horizontal transfer. A small number of unique genes found in SAG is not unexpected since a large number of Streptococcus strains have been sequenced to date; further analysis of these proteins may provide specific markers for detection or typing assays.
Table 4.
Unique genes found in SAG strains, as determined by OrthoMCL analysis
| Locus namea | Length (AA)b | Gene product | % G + C | COG #c | % HSPd | PIDe | Organismf | Accession # |
|---|---|---|---|---|---|---|---|---|
| SCRE_0567 |
128 |
Conserved hypothetical protein |
22.22 |
3514 |
108.5 |
37 |
S. macacae |
ZP_09135394 |
| SCRE_0686* |
202 |
Putative phosphoglycerate mutase |
36.95 |
4151 |
100.0 |
50 |
Paenibacillus sp |
YP_003241041 |
| SCRE_1286 |
126 |
Conserved hypothetical protein |
32.55 |
4048 |
100.0 |
41 |
Leuconostoc fallax |
ZP_08312902 |
| SCRE_1287 |
278 |
Conserved hypothetical protein |
38.47 |
4049 |
100.0 |
57 |
Lactobacillus casei |
YP_805494 |
| SCRE_1374* |
316 |
Tagatose 1,6-diphosphate aldolase |
39.43 |
|
100.0 |
65 |
Pasterurella mutocida |
ZP_18466110 |
| SCRE_1851* |
333 |
Conserved hypothetical protein |
38.82 |
3609 |
100.6 |
28 |
S. gordonii |
YP_001449372 |
| SCRE_1887* |
229 |
Triose-phosphate isomerase |
28.41 |
4057 |
100.0 |
40 |
Leuconostoc citreum |
YP_001728668 |
| SCRE_1888 | 179 | Conserved hypothetical protein | 27.96 | 3880 | 100.0 | 48 | Bacillus sp | ZP_01723913 |
*Genes found in all sequenced SAG from GenBank and this study. aS. constellatus subsp. pharyngis C232 was used as reference for all SAG strains; bLength of protein within SAG strains in amino acids; cCOG number generated during orthoMCL analysis; dPercent coverage of protein from SAG compared to best match in NCBI database using BlastP; ePID is equal to the percent aa identity for the best match using BlastP from NCBI ; fOrganisms with best protein match to a non-SAG strain from NCBI database using BlastP.
SAG virulence factor repertoire identified for future pathogenomics investigations
Owing to a paucity of genome and experimental data, limited virulence traits have been previously identified for the SAG, and studies to date have focused on specific virulence traits and not their global virulence potential [3,22-25,27,47]. The curated genomic sequence data sets herein provide the opportunity for in silico ortholog gene searches to identify potential virulence traits within SAG. A custom virulence database (VirDB) constructed for this study contains 189 known or putative virulence factors from the genus Streptococcus (Additional file 8). VirDB was used to query all sequenced Streptococcus strains (Additional file 3) to identify virulence genes within the Streptococcus pan-genome (Figure 1). A more detailed analysis of the putative virulence genes within S. gordonii and S. sanguinis is also presented (Figure 5). There are 55 virulence proteins identified within SAG that had greater than 35 PID over at least 50% of the protein when compared to this virulence database (Additional files 9, 10 and 11). The importance of using these respective cut-offs was to limit the over-inference of short domains or incomplete proteins in our analysis. Overall, there were 16 known Streptococcus virulence proteins conserved within the sequenced SAG strains and each individual SAG strain contained 27 to 36 virulence proteins. The breakdown of virulence gene by SAG species was 30–34 (SA), 30–35 (SC) and 27–36 (SI). The overall division of virulence traits showed that SI and SC share greater than half of their virulence traits, although the overall repertoire of virulence traits differed between the two species. There was less conservation of virulence traits among the seven strains of SA, with some strains showing greater similarity to either SC or SI for their overall virulence gene composition (Figure 5). Although the phylogenetic groupings in this study are from our core-SNP method, the examination of the genes reveals the same similarities mentioned above.
Figure 5.
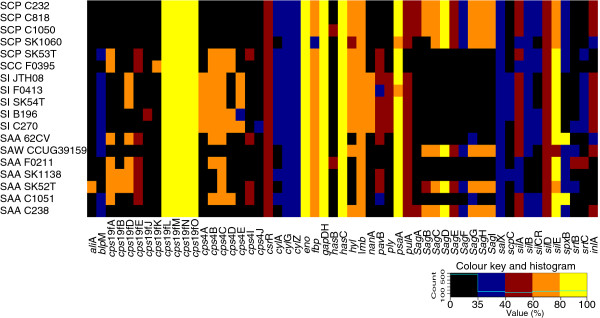
Streptococcus virulence protein detection within SAG, S. gordonii and S. sanguinis strains. A total of 66 Streptococcus genomes (Additional file 2) were queried using a custom virulence database (VirDB) of 189 known Streptococcus virulence determinants (Additional file 6) and a heatmap was constructed to demonstrate relatedness of virulence determinants within each queried strain. The legend on the lower right hand corner depicts the PID of the ortholog from the specific strain to the reference protein within the VirDB and the sum of hits (histogram). The heatmap uses 5 colors to represent percent protein identity as follows: SCP, S. constellatus subsp. pharyngis; SCC, S. constellatus subsp. constellatus; SI, S. intermedius; SAA, S. anginosus subsp anginosus; SAW, S. anginosus subsp. whileyi; Black loci not present (0-35%), Blue (36-40%), Red (40-60%), Orange (60-80%) and Yellow (80–100).
Five of the 55 virulence proteins newly identified within SAG are inferred to be adhesion proteins including: a fibronectin binding protein (Fbp54), important in adhesion for S. gordonii[48], a S. pneumoniae and S. mitis surface adhesion protein (PsaA) [49], a laminin-binding protein important for adherence in GBS [50], pullulanase protein important in Streptococcus adhesion [51] and Streptococcus enolase a strong plasminogen-binding protein [52].
Twelve loci involved in invasion or evasion from host proteins were also identified in SAG. All five proteins encoded for by the Streptococcus invasion locus silA to silE [53], four capsule proteins (Cps19FL to O) from S. pneumoniae[54], hemolysin proteins from the S. agalactiae hemolysin loci cylZ and cylG [55], and UDP-glucose pyrophosphorylase protein (HasC) [56], were present in the conserved SAG virulence proteins. Finally, three regulator function proteins were present, including a two component response regulator CsrR (CovR), known to regulate expression of extracellular carbohydrates in Streptococcus[57]; a protein with homology to salivaricin-A (SalX), a bacteriocin known to inhibit growth of streptococci [58]; and GAPDH an immunomodulatory protein important in streptococcal colonization [59].
Some virulence proteins were present in more than one species of SAG revealing that the SAG virulence repertoire may vary with strain. Homologs for S. pneumoniae polysaccharide capsule operon (Cps4) [60]; four proteins from the hemolysin complex from S. agalactiae (Cyl) [55] and a S. pneumoniae surface-expressed adhesion protein (PavB) [61] were found in SI, SCC and some SA. Interestingly only some SA (C238, CCUG39159 and SK52T) and SCP contained homologs to sagA through sagI that form the streptolysin S cytolytic toxin complex of GAS. Streptolysin S is a strong cytolytic toxin that provides the ability for transepithelial migration [62] and sagA homologs have recently been shown to confer β-hemolytic activity in SA [3]. The cytolytic function can be detected via β-hemolysis on sheep’s blood agar plates. Strains predicted to be β-hemolytic based on genomic analysis (SAW C238 and SCP C818, C232 and C1050) were congruent with β-hemolytic phenotype observed on sheep blood (results not shown).
There was also a homolog present in SAG to a hyaluronidase precursor protein (HylA) that provides the ability to survive on host hyaluronic acid as a sole carbon source, as well as aiding in bacterial spreading by allowing for detachment from biofilms [27]. This gene was found in all SC and SI strains and SAW strains, and the presence of the HylA protein was confirmed through traditional phenotypic growth testing for in house sequenced SAG strains [9].
Internalin A (InlA) is a major invasion protein from Listeria monocytogenes that mediates the attachment and invasion of hepatocytes by L. monocytogenes and is encoded by the inlA gene [63]. A homolog to this gene has been found in Streptococcus spp. and was termed the streptococcal leucine-rich (Slr) protein [64]. A homolog to this protein was identified in all SCP, SI and some SA (C238, 62CV and CCUG39159), and is highly conserved within SAG having 97.4% nucleotide identity and 97.2 PID. Internalin A orthologs have also been identified in many other sequenced streptococcal species including S. sanguinis VMC66 [GenBank: EFX94353.1], S. pyogenes MGAS8232 [GenBank; AAL97968.1] and S. agalactiae 2603 V/R [GenBank; NC_004116]. Within sequenced S. sanguinis, inlA is found inserted between pyrR and a hypothetical protein, while in S. pyogenes this gene is located between metK and birA and in S. agalactiae it is located between lepA and a histidine diad domain protein encoding gene. The region around the inlA gene is conserved in all SAG, with inlA inserted between pyrR and a conserved hypothetical protein as previously seen in S. sanguinis. In SAG without inlA, the pyrR and the gene encoding the conserved hypothetical protein are present, thus it appears that the inlA was lost or never integrated into some SA and SCC. In S. gordonii [GenBank: NC_009009], SA 62CV [GenBank: EFW07950.1] and SC SK1060 [GenBank; EGV09572.1], there are remnants of a leucine-rich protein that has been truncated located next to pyrR, which shows that inlA has been gained and lost in some Streptococcus strains. It has been shown in L. monocytogenes that loss or truncation of the inlA gene causes decreased invasive ability [65]. Similar results were shown for slr, where an isogenic GAS strain lacking slr was significantly less virulent in a mouse model and more susceptible to phagocytosis by human polymorphonuclear leukocytes [64].
Two virulence genes were identified in SI that were not found in any of the other SAG. Both genes have been previously identified including; nanA, a sialidase A precursor that may influence host bacterial interactions [47], a pneumolysin-like protein (Ply), identified as intermedilysin, which is a human erythrocyte specific cytotoxin [4,21]. There were no virulence traits found to be specific to either SC or SA. Indeed, the use of VirDB has identified numerous virulence genes within SAG that may allow SAG to adhere, invade and spread within the host.
Evaluation of colonization potential: a genetic look at SAG LPXTG proteins
Bacterial attachment to host cells is essential in host colonization. Colonization occurs through interactions between bacterial surface-exposed proteins and host cell receptors [66]. One of the most common motifs found in bacterial cell surface-exposed proteins is the LPxTG motif. The motif anchors the C-terminus of these externally facing surface fibrillar proteins [67]. These LPxTG motif proteins are covalently attached to the cell wall by sortases, with sortase A (SrtA) being the most common within streptococci [68]. Many LPxTG motif proteins have been associated with virulence in Gram-positive microorganisms, however many also have no known associated function [20,68]. Each SAG strain had at least one srtA sortase ortholog, with all SI having two srtC orthologs. SA F0211 is the only SA strain to have multiple sortases, with a SrtA and two SrtC orthologs. Increased sortase function may increase virulence potential through assembly of surface structures [69].
A total of 58 LPxTG motif proteins were identified within SAG, however, only 50 of these proteins had a signal peptidase in addition to the LPxTG motif to allow expression on the cell surface. The number of LPxTG motif proteins in SAG strains was in the mid range for Streptococcus spp. ranging from 14 to 22 [28,29,32,70]. SA had the most LPxTG motif proteins with 21 and 22 for SAW C238 and C1051 respectively, while SC and SI ranged from 14 to 17 (Additional file 12). One LPxTG motif protein, hyaluronate lyase precursor protein (HylA) was found in all SI, SC and SAW strains. This protein is known to be an important virulence factor as discussed above. Twelve of the LPxTG motif proteins had collagen-binding domains, the same number found in S. equi subsp. zooepidemicus[32], and thus could be involved in SAG virulence. Also identified were eight proteins important as either pili or in fibrinogen-binding. The presence of numerous potential adherence proteins is common for oral microbiota, and thus, high numbers of these proteins within SAG is not unexpected.
Genetic analysis of two component (histidine kinases/response regulator) systems (TCS)
Bacterial survival is dependent on a pathogen’s capacity to sense and respond to their environment. Two-component histidine kinase systems (TCS) have been shown to play vital roles in virulence as well as the ability to respond to specific environmental signals [71,72]. In total there were 14 TCS found in SAG, 9 of which were conserved in all SAG strains, although the response regulator (RR) for SAGTCS6 is truncated in SAW C238 and the SAGTCS6 histidine kinase (HK) is truncated in SCP strains. S. intermedius and SAW had the most TCS with 13, while SCP had the least with 10 (Table 5). These numbers are similar to other sequenced Streptococcus strains including: 14 in S. sanguinis[31], 13 in S. pneumoniae[73-75], 14 in S. mutans[28,76], 13 in S. pyogenes[29] and 17 in S. agalactiae[70].
Table 5.
Comparative summary of two component system (TCS) histidine kinases identified in SAG with other Streptococcus species
| SAGTCS1 | Equivalent TCS HK in S. pneumoniae2 | Gene name | Linked to virulence | SAG strain found in | Loci3 | Best non-SAG BlastP results | PID4 | Accession # |
|---|---|---|---|---|---|---|---|---|
| 1 |
9 |
zmpSR |
Yes |
All SAG |
SIR_0800 |
S. sanguinis |
69 |
YP_001035081.1 |
| 2 |
5 |
ciaH |
Yes |
All SAG |
SIR_0858 |
S. oralis |
70 |
EGL88510.1 |
| 3 |
2 |
vicK |
Yes |
All SAG |
SIR_0929 |
S. cristatus |
84 |
ZP_08058686.1 |
| 4 |
None |
|
NDa |
All SAG |
SIR_1092 |
S. sanguinis |
67 |
EGG40097.1 |
| 5 |
13 |
blpH |
Yes |
All SAG except SAb SK1138, SA F0211 and SA SK52T |
SIR_1152 |
S. equi |
50 |
YP_002746951.1 |
| 6 |
1 |
|
Yes |
All SAG |
SIR_1196 |
S. cristatus |
63 |
ZP_08059673.1 |
| 7 |
3 |
|
No |
All SAG |
SIR_1292 |
S. cristatus |
67 |
ZP_08060583.1 |
| 8 |
None |
|
ND |
All SAG |
SIR_1635 |
S. cristatus |
89 |
ZP_07864009.1 |
| 9 |
8 |
|
Yes |
All SAG |
SIR_1771 |
S. mitis |
64 |
ZP_07642266.1 |
| 10 |
12 |
comD |
Yes |
All SAG |
SIR_1900 |
S. cristatus |
59 |
ZP_08060066.1 |
| 11 |
6 |
|
Yes |
SIc and SA |
SIR_0289 |
S. sanguinis |
65 |
EGJ40992.1 |
| 12 |
7 |
|
Yes |
SI and SA C1051 |
SIR_1062 |
S. cristatus |
71 |
ZP_08060320.1 |
| 13 |
11 |
|
No |
SI and SCCd |
SIR_1373 |
S. sanguinis |
96 |
EGF13754.1 |
| 14 | None | ND | SA C1051, SA 62CV and SA SK52T | SAIN_0922 | S. oralis | 95 | ZP_13522336.1 |
1Two component system histidine kinases systems found in SAG; 2The equivalent TCS found in S. pneumoniae as reviewed by Paterson et al. [74] , SAG orthologs found using blast hits from NCBI; 3All SAGTCS present in more than one SAG are listed using the loci from histidine kinase from SI B196 since this strain had the most TCS orthologs.; 4PID is equal to the percent aa identity for the best match using BlastP from NCBI ; aND; No data for this HK, bSA = S. anginosus, cSI = S. intermedius, dSCC = S. constellatus subsp. constellatus.
Thirteen of the 14 TCS found in SAG were found to have orthologs in other species of Streptococcus including nine orthologs to TCS that have been linked to virulence in S. pneumoniae[75]. Of these nine, four are well characterized, including VicRK, CiaRH, ComDE, and BlpR (Table 5). These TCS are important to S. pneumoniae virulence, quorum sensing, competence, bacteriocin production, and stress response [71,77]. Four additional SAG TCS (designated herein as SAGTCS1, SAGTCS6, SAGTCS9, and SAGTCS11) show homology to less characterized TCS that are also important for virulence in S. pneumoniae [78,79]. Another TCS sequence (SAGTCS13) is poorly characterized showing the most similarity to a characterized TCS from S. mutans[76]. SAGTCS13 has been disrupted in SCP and SA except SK52T, CCUG39159 and C1051, to leave only a truncated portion of the HK with an absent cognate RR. This truncation appears to be due to the insertion of the SAG operon within these strains of SAG, as a complete HK/RR system is observed in all strains that lack the SAG operon; the importance of this TCS has not been well characterized [74]. Finally SAGTCS14 was only found in SA C1051, 62CV and SK52T, with homology to uncharacterized TCS from S. oralis, S. mutans, S. macacae and Lactobacillus salivarius. The importance of TCS in many aspects of virulence is well known and shows the virulence potential for SAG, however, more work is required to fully characterize TCS within SAG.
Potential natural immunity conveyed by clusters of regularly interspaced short palindromic repeats (CRISPRs) within SAG
Clusters of regularly interspaced short palindromic repeats (CRISPRs) contribute to bacterial immunity to invasion from foreign DNA such as bacteriophage and plasmid DNA [80]. CRISPR analysis in SAG strains showed that seven of the 18 strains analyzed contained CRISPRs (Additional file 13). Three of the seven strains had more than one CRISPR for a total of 12 CRISPRs identified within SAG (Additional file 13). Based on the Cas1 protein all CRISPRs except for one that had a Cas1 protein were of the CRISPR subtype II-A also known as the NMENI subtype [80,81].
The CRISPRs SintB196-1, Sang62-1 and Sang1051-2 were closely related to each other based on Cas1 AA identity and are all inserted between a transcriptional regulator (SI B196 SIR_0753, SAA C1051 SAIN_0897 and 62CV_1302) and a conserved hypothetical protein (LPXTG motif protein: SI B196 SIR_0758, C1051 SAIN_0902 and 62CV_1297). CRISPRs SconSK53-1, SangSK52-1, SangSK1138-1 and F0211-1 and F0211-3 were also similar to each other based on Cas1 AA identity. F0211-1 and F0211-3 may be part of the same CRISPR as inferred from their location within the genome; however, as this strain is only publically available as a draft WGS project data set, this inference cannot be confirmed. Gene composition was the same for all three CRISPR elements including known CRISPR associated proteins cas9 (csn1), cas1, cas2 and csn2 followed by the CRISPR direct repeat (DR) and spacer regions (Figure 6A). This genetic arrangement and Blast results suggests that this CRISPR is most closely related to the CRISPR subtype II-A family. CRISPRs with similar genetic content have been identified within S. thermophilus, S. gordonii, S. suis and S. infantarius[82]. The DR length was 28 to 36 bp for all three strains (it appears the smaller direct repeats are truncated versions of the larger direct repeats), while there was variation in the number of spacers present within each CRISPR (Additional file 13). The DR regions were similar for all subtype II-A CRISPR elements within SAG with the last repeat showing variation as has been shown for other CRISPR elements [80].
Figure 6.

Comparative analysis of the Type II-A (Nmeni) SAG CRISPR. A) Schematic view of the Type II-A CRISPR regions identified within SAG. B) Schematic view of the Type I-C CRISPR region identified within SA. Similar colored arrows indicate gene loci conserved between SAG. The number of DR and spacer regions are indicated by black bars, with the number of bars correlating to number of regions present within each CRISPR.
The CRISPR SconSK53-2 was the only non Type II-A CRISPR (identified as Type 1-C), which is located next to an integrase and is composed of cas3, cas5, cas8, cas7, cas4, cas1 and cas2 genes from 5′ to 3′ and followed by a CRISPR region with a DR of 32 bp and 19 spacers (Figure 6B). This type of CRISPR has been previously identified within Streptococcus[83], and showed the most similarity to CRISPR-associated loci from S. sanguinis, S. mutans, S. parasanguinis, and S. pyogenes. The localization next to an integrase gene and high similarity (85 to 93% AA identity) to a similar region within S. mutans LJ23 (GenBank NC_017768), while the surrounding area shows less similarity (<78% AA identity) suggests that this region was horizontally transferred into SCC SK53T.
S. anginosus C1051 had a total of three CRISPR regions, with Sang1051-1 and Sang1051-3 both appearing to be degenerate. Sang1051-1 has cas1 and cas2 genes and the CRISPR region with 21 DR of 37 bases in length (Figure 6B), similar to Sthe2c from S. thermophilus LMD-9 [82], however, compared to other Cas1 proteins there is less than 65% AA identity as compared to all other Cas1 proteins that have matches greater than 90% based on AA identity. The third CRISPR region, Sang1051-3, had three spacers of 28 bases (Additional file 13). Although neither CRISPR-1 nor CRISPR-3 had a full complement of CRISPR-associated proteins, the fact the CRISPR-1 had 21 spacers suggests that this CRISPR may still be functional [84]. S. anginosus C1051 and SI B196 did not have any detectable prophage integrated into their genomes. This may be due to the presence of multiple CRISPR elements within these genomes.
For the 12 CRISPRs described above, there were a total of 201 spacers (Additional file 14). Of these 201 a total of 50 had greater than 80% identity over 50% of their sequence to a sequence in GenBank. However, none of the spacers from SAG showed 100% DNA identity to anything in the NCBI nucleotide database, which is significant as it has been shown that 100% match is required for immunity to foreign DNA [85]. A total of 11 spacers showed >90% nucleotide similarity to previously identified Streptococcal-specific phages including five phages from S. pneumoniae; 11865, 8140, V22 [86], Cp-1 [87] and EJ-1 [88]; two phages from S. pyogenes, phi370.2 [34], and phiNIH1 [89]; streptococcus phage C1 isolated from Group C Streptococcus[90]; S. oralis phage PH10 [91]; S. gordonii phage PH15 [92]; and a phage from S. gallolyticus subsp. gallolyticus UCN34 [93]; (Additional file 14). All spacers were unique except for 16 spacers from SA F0211, this one spacer was located within each of the three CRISPRs identified for SA F0211, these spacers were similar to an integrative conjugative element (ICE) from Vibrio fluvialis Ind1 (ICEVflInd1) and showed similarity to genes encoding for pili and conjugative machinery. It appears that F0211 had multiple encounters with one or multiple conjugative elements similar to ICEVflInd1 and has acquired a means to prevent integration of this type of ICE. Perhaps CRISPRs have played a role in the evolution of SAG and may be responsible for the lack of foreign genetic material seen in some in-house sequenced SAG strains. We are currently investigating whether SAG CRISPRs might be variable enough to be useful in a subtyping scheme.
Extensive horizontal gene transfer and accessory gene content in SAG
None of the SAG strains sequenced in this study possessed extracellular plasmids, which would indicate that this is not a common method of HGT among SAG strains; although loss of plasmid on in vitro culture is not uncommon and cannot be completely ruled out. This was shown in a previous study where only two plasmids were identified within 27 strains of SAG [5]. However, all strains from this study showed evidence of HGT via presence of prophage, insertion sequences, transposons and ICEs and those account for 12 to 28.5% of the total predicted coding sequences within the seven genomes sequenced for this study (Table 6; Figure 1; Additional file 2). The prophage and ICEs identified within this study are highlighted in Figure 1B-D and can be seen in the MAUVE analysis also (Additional file 2). These SAG mobile elements will be described in detail elsewhere (Olson et al., manuscript in preparation).
Table 6.
SAG accessory regions
| Genome | Genome size (nt) | # AR1 | Combined size ARs (nt) | AR% of Genome | AR CDS | HGT ARs2 | HGT CDS3 | Combined Size HGT ARs (nt) | % of Genome |
|---|---|---|---|---|---|---|---|---|---|
| SA C1051 |
1911706 |
47 |
437231 |
22.87 |
353 |
18 |
218 |
306042 |
16.01 |
| SAW C238 |
2233640 |
53 |
745460 |
33.37 |
608 |
26 |
481 |
636082 |
28.48 |
| SCP C232 |
1935414 |
34 |
476022 |
24.60 |
412 |
12 |
287 |
330294 |
17.07 |
| SCP C818 |
1935662 |
34 |
476022 |
24.59 |
412 |
12 |
287 |
330294 |
17.07 |
| SCP C1050 |
1991156 |
35 |
531952 |
26.72 |
461 |
14 |
344 |
402267 |
20.21 |
| SI C270 |
1960728 |
47 |
524216 |
26.74 |
437 |
7 |
202 |
243715 |
12.43 |
| SI B196 | 1996214 | 52 | 554383 | 27.77 | 470 | 11 | 234 | 279114 | 13.98 |
1AR; Accessory region, 2HGT AR, Accessory region with evidence of horizontal gene transfer; 3HGT CDS, total number of predicted coding regions within the HGT ARs. SA, S. anginosus subspecies anginosus; SAW, S. anginosus subspecies whileyi; SCP S. constellatus subspecies pharyngies, SI; S. intermedius.
Investigation of SAG natural competence
Uptake of naked DNA from the environment, known as natural competence, allows bacteria to survive and thrive in variable environmental conditions. Natural competence has been studied in-depth for the streptococci with S. pneumoniae serving as the model organism [94]. The complexity of natural competence systems and diversity of streptococcal competence genes has made in-silico prediction of natural competence difficult [95]. Genome transcriptomics and tiling microarrays have shown that there are 22 essential genes required for natural transformation in S. pneumoniae[96]. Orthologs to 21 of the essential competence genes were identified in all seven in-house sequenced SAG (Additional file 15). For all remaining WGS strains, there were truncated or absent genes likely owing to sequence errors; however, this would have to be experimentally confirmed. The only homolog absent in all strains was comW, which is also missing in the naturally competent S. zooepidemicus[97], S. sanguinis[31] and S. mutans[98] suggesting that it may not be absolutely required for natural competence.
All SCP examined to date (n = 34) are not naturally competent, but competence has been shown in SI (Lacroix, Grinwis and Surette, unpublished data). Sequence analysis has revealed that the lack of natural competence in SC may be due to an insertion within comEA gene. This truncation results in a gene product approximately half that of the original comEA and when this gene is inactivated in S. pneumoniae, competence is abrogated [96]. The detected insertion has been experimentally validated using Sanger sequencing. All SCP strains had a nine nt in-frame deletion in the comD gene. This has been found to be a feature of all SCP (n = 34), but does produce a functional ComD (Lacroix, Grinwis and Surette, unpublished data). The only other difference seen in the complement of competence genes within SAG was the number of comX orthologs. For most Streptococcus strains there are two comX orthologs (comX1 and comX2), for the SAG there were three comX loci in most strains. However, one copy of comX was truncated in SCP C1050, and in some of the publicaly available WGS strains included in our analysis (Additional file 16). Further functional studies will have to be completed in SAG to identify and characterize which competence proteins are essential.
Microevolution within SAG
Two strains (SCP C232 and C818) that were sequenced within this study were cultured from the same individual almost one year apart. These isolates were both the numerically dominant strain present during a pulmonary exacerbation. Genomic comparisons revealed that these strains were almost identical, differing by only 18 SNPs (Additional file 17). The SNPs were found within intergenic regions (6), and causing both synonymous (3) and non-synonymous mutations (9). Two of the non-synonymous mutations introduced stop codons, resulting in two truncated genes within SCP C818; one is a putative ABC transporter (ATPase portion) while the other is a 3-dehydroquinate dehydratase (aroD). Mutations in aroD have been extensively characterized in Salmonella typhi[99] and have been shown to render bacteria auxotrophic for aromatic amino acids p-aminobenzoate (pABA) and 2, 3-dihydroxybenzoate. This results in the inability to produce ubiqinone and menaquinone causing cellular respiration defects [100], as well as defects in the cell envelope [101]. Indeed, it has been shown that aromatic amino acids are abundant within the sputum produced within the lungs of CF lung patients and thus may provide an adequate source of these essential nutrients [102]. Further studies will be required to determine the importance of this aroD mutation within SCP C818. Two SNPs were located within a region of the rpoB gene known to cause rifampicin resistance [103]. Finally, a second type of divergence between SCP C232 and C818 were tandem repeat and microsatellite regions, which are potential targets for multilocus variable number tandem repeat analysis (MLVA). There were a total of five regions of increased/decreased copies of tandem repeats ranging in size from four to 41 nt (Additional file 18). Of these five regions, three are located in truncated genes, whereas the other two were within non-coding regions. Further analysis into potential MLVA targets within SAG is warranted. Although both these strains are highly similar, appreciating the evolution that occurred during a year in a CF lung is critical to understanding the fitness advantage required for chronic bacterial pathogens.
Conclusions
This study presented the analysis and comparison of the whole-genome sequences for the three species within SAG, important pathogens with the capacity to cause serious infections throughout the body. Sequencing strains from both respiratory and invasive infections, we identified no clear differences in gene content between these types of isolates, suggesting that it may be host factors that promote certain types of infection as opposed to bacteria-specific virulence factors. There were only eight genes detected that were uniquely common to all seven in-house sequenced SAG; these were also found in most SAG draft genomes that are currently publically available. The comparison of SC, SI and SA strains revealed significant differences with respect to virulence factors, surface proteins and carriage of horizontal genetic elements, with SA showing the most intra-species variability. Horizontal gene transfer between SAG and other pathogens within their environment has clearly played a significant role in the evolution of species within SAG, which will need to be studied in far greater detail. The detailed comparison of microevolution within SAG has identified potential targets for molecular typing methods as well as potential research questions regarding survival of a bacterial pathogen within the lung of a CF patient. The generation of our high quality finished reference genomes for the seven in-house sequenced SAG strains will provide a valuable resource for the analysis of future SAG draft genomes. This comparative genomic analysis provides a key genetic framework for assessing and understanding the molecular events contributing to SAG pathogenesis.
Methods
Bacterial isolates
Four respiratory SAG isolates were cultivated on McKay agar from adult patients at the time of a pulmonary exacerbation. The remaining three isolates were from infections at other body sites to provide a comparison between respiratory and non-respiratory (invasive) SAG strains. A total of seven SAG strains were sequenced with at least one respiratory and invasive strain from each of the three species within the SAG. Three SCP strains were sequenced: C232 and C818, both respiratory strains isolated from one individual almost a year apart, and C1050, an invasive isolate from a non-related individual. Two SI strains were sequenced: C270, a respiratory isolate, and B196, an invasive isolate obtained from a CF patient coinciding with a pulmonary exacerbation. Finally, two SA strains were sequenced: SAW C238, a respiratory isolate, and SAA C1051, an invasive isolate (Table 1). All isolates were obtained in accordance with the University of Calgary ethics approval and written approval was obtained for participation in the study from all human subjects providing bacterial isolates.
Chromosomal DNA isolation
Strains were cultured on Columbia blood agar plates (CBAB) or Brain heart infusion (BHI) agar and incubated for 24 hr at 37°C plus 5% CO2. A single colony was inoculated into 20 mL of BHI broth and incubated as above. These cultures were then centrifuged at 5000 × g for 30 min and washed three times with sterile PBS. Cells were re-suspended in 1 mL of sterile PBS and lysed by physical disruption using the MiniBeadbeater-8™ (BioSpec Products, Inc). DNA was purified from lysates using standard phenol: chloroform and ethanol precipitation. All genomic preps were run on an agarose gel to ensure chromosomal integrity. Finally, the DNA was quantified using the Qubit® (Invitrogen, Burlington, ON).
Genome sequencing, assembly, and Gap closure
Genomic DNA was sequenced using the Roche GS20 standard platform as per manufacturer’s protocols (454 Life-sciences, Brandford, CT). For gap closure, fosmid libraries were created as per manufacturer’s protocols using the CopyControl™ Fosmid Library production kit (Epicentre Technologies, Madison, WI) and Sanger sequencing was done on selected clones. Traditional PCR was done using proof-reading (HiFi platinum Taq; Invitrogen) Taq polymerase as per manufacturer’s protocols. PCR products were purified using the QIAquick PCR purification kit (Qiagen, Mississauga, ON) and sequenced with an ABI3730XL capillary electrophoresis instrument (Applied Biosystems, Foster City, CA). After sequencing on the Roche GS20 genome sequencer the raw reads were assembled using the Newbler assembler software package v1.0.53.17. After closure of the genome to a single contigous sequence, the ori was located by performing a simple Blastp using dnaA as a reference.
Sequence errors and SNP confirmation
Potential sequence errors attributed to homonucleotide runs were manually tested in SCP C232 using primers designed to flank the region of interest. PCR amplification was conducted using Invitrogen HiFi Platinum proof-reading Taq polymerase (Invitrogen), following manufacturer’s instructions, with 1 μM of each oligonucleotide and the following thermocycling conditions: 94°C for 5 min, 40 cycles of 94°C for 30 s, 55°C for 30 s and extension at 68°C for at least 30 s (time varied for larger size fragments) followed by 68°C for 5 min. All SNPs found between SCP C232 and C818 were confirmed by Sanger sequencing as described above. For all other SAG strains (except SCP C232), a combination of standard sequencing and Illumina sequence technology were applied to correct base calling errors caused by homonucleotide runs. Illumina sequencing runs were completed at the Iowa State University DNA facility using an Illumina GAII, (Illumina Inc, San Diego, CA). For Illumina sequencing, 5 μg of genomic DNA was submitted. The DNA facility created libraries and bar-coded five strains (SCP C1050 and C818, SAA C1051 and SI C270 and B196) which were then pooled in a single lane using 36 bp reads, while SAW C238 was run in a single lane using 75 bp reads. To ensure that Illumina data was reliable, 100 regions showing variation between the GS20 data and Illumina data were analyzed by Sanger sequenceing. In all cases, the Illumina data was observed to be correct. Illumina data and GS20 data were aligned and co-analyzed using CLC Genomics Workbench version 4.0 (CLC genomics, Cambridge, MA).
Genome auto-annotation
Draft and finished genomes were automatically annotated using an in-house version of the GenDB 2.2 genome annotation system. In the annotation pipeline, genes are predicted using a combination of CRITICA [104] and Glimmer 3.0 [105]de novo gene predictors. Locations of Ribosomal Binding Sites (RBS) were also predicted via CRITICA. RNAmmer [106] and tRNAScan-SE [107] were used to predict all rRNA (5S, 16S, 23S) subunits and tRNA, respectively. Functional observations were collected from BLASTN [108] alignments to the NCBI nucleotide (nt) database [109], and from BLASTP [108] alignments to the Kyoto Encyclopedia of Genes and Genomes (KEGG) [110] and the NCBI non-redundant protein (nr) database [109]. Observations related to protein families were collected using PSI-BLAST [108] alignments to SWISS-PROT (sp) [111] and Clusters of Orthologous Groups (COG) databases [112]. Conserved protein domain observations were collected as RPS-Blast [108] alignments to the Conserved Domain Database (CDD) [113]. Additional observations collected for each CDS included Hidden Markov Model protein sequence classification searches against the TIGRFAM [114] and PFAM [115] databases, membrane spanning helices searches via EMBOSS helix-turn-helix [116] TMHMM [117] and presence of signal peptide sequences [118]. All functional observations were analyzed within the annotation system using a set of pre-defined heuristics to automatically assign a gene name and biological role for each CDS, when possible. Bi-directional BLASTP [108] for all predicted CDS was run between each pair of genomes and within each genome to identify potential orthologs and paralogs, respectively.
All intergenic regions (with a 25 base pair (bp) elongation on either end) were analyzed through a separate customized pipeline for identification of potential reading frame shifts, short genes overlooked by the automatic pipeline, or genes in regions of localized atypical nucleotide composition. Regions with BLASTX [108] alignments and EMBOSS getORF [116] open reading frames (ORF) were identified as potential CDS regions, run through the function prediction pipeline and automatically marked for manual curation.
Manual curation of SAG genomes
All predicted CDSs were manually inspected with their GenDB observations to validate inference of the auto annotation. Predicted genes were inspected for potential frameshift errors and alternative start sites. All potential frameshift errors were experimentally validated with Sanger sequencing (described above). Orthologs between genomes were multi-annotated manually with inference from a chosen reference genome using the GenDB ortholog finding tool based on ClustalW multiple alignments of sequences flagged from the Bidirectional Blastp observations. To facilitate annotating the genomes in a timely manner, sequencing corrections and annotation were finished concurrently. The final version of each corrected sequence was re-mapped to the annotations and observations using custom perl scripts built to work within the GenDB system.
Accession numbers
The manually curated, high quality finished genomes sequenced generated within this study have been deposited at GenBank with the accessions, SAA C1051 [GenBank: CP003860], SAW C238 [GenBank: CP003861], SCP C232 [GenBank: CP003800], SCP C818 [GenBank: CP003840], SCP C1050 [GenBank: CP003859], SI B196 [GenBank: CP003857], SI C270 [GenBank: CP003858].
Genome visualization and analysis
The pan-genome analysis was done using Gview version 1.6 [119] with SAW C238 as the seed genome and adding all SAG genomes as listed in Figure 2 to create a pangenome reference, this pangenome was then used to compare all individual SAG genomes to the pangenome using a BLAST atlas with the default settings on Gview version 1.6 [119]. Blast atlas for each of the species within SAG were constructed by using the basic setting on Gview version 1.6 and creating a circular comparison with SAW C238, SCP C232 and SIB196 as the reference for SA, SC, and SI respectively. MAUVE version 3.2.1 [120] was used with default settings to construct linear views of the SAG chromosomes.
Phylogenetic analysis
Orthologs were identified by OrthoMCL v2.0.2 [40]; for the in-house core-SNP pipeline those orthologs present as single copies and common to all data sets were included for analysis. Each orthologous group was aligned using ClustalW (v1.8.2) and manually edited to correct for incorrectly predicted start sites. A subset of SNP loci present among all data sets (core) were identified and used to generate a meta-alignment using an in-house Perl script for downstream analysis. Alternatively a set of core gene alignments was generated using AMPHORA [37]. Of the 31 gene sets generated, 3 core genes (rplB, rplD, rplL) were discarded from further analysis owing to the fact they were not all present in all WGS genomes undergoing analysis. Phylogenetic trees were generated for comparison of each analysis method; in-house core-SNP pipeline, AMPHORA, 16S rRNA, groEL, and rpoB using Phylogenetic estimation using Maximum Likelihood (PhyML 3.0) [121] with nucleotide substitution models LG. To assess the stability of the tree branching patterns in rpoB, cpn60 and 16S rRNA gene trees bootstrap analysis with 100 pseudoreplicates was performed using evolutionary models and tree building as described above. The in-house core-SNP tree was analyzed using the approximate likelihood ratio test, using a selection threshold of 0.8, which is comparable to bootstrap supports of 75% [121]. The ratio of mean non-synonymous (dN) to synonymous substitutions (dS) per site (dN : dS ratio) within the two selected genes (rpoB,cpn60) was calculated using MEGA software using the Nei-Gojobori Method with Jukes Cantor correction [122,123].
Ortholog analysis
OrthoMCL [40] was used to identify orthologous gene groups from the proteomes of 66 sequenced Streptococcus strains (Additional file 2). Sequences and annotations for the WGS projects were obtained from the Broad Institute download site (http://broadinstitute.org/annotation/genome/Streptococcus_group/GenomesIndex.html) and all other fully annotated Streptococcus spp. were obtained from NCBI [109], excluding the seven in-house SAG. A BLASTP [108] e-value of 1e 10-5 and percent match length of 50% were used as cut-offs in orthoMCL [40] analysis. Signature genes for each strain were appended to the orthoMCL output, since it does not report signature genes, defined as genes that are only present in one strain. VENN diagrams for ortholog groups were generated using an in-house Perl script. VENN diagrams do not represent all genes from all genomes due to the fact that accessory genes are not included.
Clustered COG analysis
A comparative COG analysis was performed using a custom set of Perl scripts. One representative from each accessory gene group from the orthoMCL [40] output, as well as any non-orthologous predicted genes from each of the 66 Streptococcus genomes above, were blasted against the non-supervised Orthologous Groups (eggNOG) database [124]. Significant BlastP [108] matches were filtered using the following criteria: 80% PID over a minimum of 80% of the protein length or proteins over 100 amino acids in length, with a minimum of 75% length and 30% PID. Matches were scored on a binary scale, wherein a significant match score was assigned a value of one; non-significant matches were assigned a zero. Accessory gene matches to each COG group were hierarchically column-clustered with distance correlation. The resulting dendrogram was represented as a heatmap generated in R (http://www.r-project.org) using heatmap.2 from gplots package (http://cran.r-project.org/web/packages/gplots/index.html) with each tile shade representing a functional COG category associated with the gene group. Gene shading of black indicates exclusion from the gene cluster. The row dendrogram of the heatmap was not hierarchically clustered and simply represents the order of the phylogenetic tree resulting from the in-house core-SNP analysis (described above). Gene groups with no significant hits or hits to proteins with no assigned functional category from the eggNOG database [124] were excluded from the heatmap representation depicted.
COG table
A list of the number of protein matches to the eggNOG database [124] matching a COG functional category for each of the 66 Streptococcus reference strains was generated. Significant BlastP hits were filtered using the following criteria: 80% PID over a minimum of 80% of the protein length, or proteins over 100 amino acids in length with a minimum of 50% length and 30% PID.
CRISPR identification
Genomes were scanned for presence of CRISPRs using the online program CRISPRFinder and CRISPRcompar [125,126]. CRISPRs were classified based on their composition of CRISPR associated proteins [81]. Spacers were identified within the CRISPR regions and BlastN was used to determine if there were matches to known mobile elements within the GenBank database.
Virulence analysis (Heatmaps)
A virulence database (VirDB) was constructed in-house using a combination of a literature search from public NCBI protein database [109] and Streptococcus specific virulence genes from the Virulence Factors of the Pathogenic Bacteria Database (VFDB), http://www.mgc.ac.cn/VFs/main.htm[127]. The VirDB was made non-redundant using CD-HIT [128] with default values, and then curated manually to ensure that genes from the same operon were not collapsed into a single entry. The initial virulence database was comprised of 234 genes, but later reduced to 189 virulence-associated genes. A protein BLAST [108] was performed using cut-off scores of 35% percent identity (PID) and highest scoring pair (hsp) length of 50% for the longest genes. A similar database was constructed to specifically identify TCS and LPxTG proteins within SAG. Heatmaps were generated in R (http://www.r-project.org) using a modified version of heatmap.2 as described above. Genome order for presentation of data was predefined by the in-house core-SNP pipeline (described above). Gene order was organized alphabetically, although genes within an operon were clustered together. Virulence genes that did not have hits to any genomes were eliminated from heatmaps in Figure 5 showing SAG, S. sanguinis and S. gordonii.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
ABO performed manual annotation, genomic data analysis and wrote the manuscript. HK provided bioinformatics analysis, manual annotation, and contributed to writing the manuscript. CDS participated in study design, coordination, manual annotations of genomes, and provided critical analysis of manuscript. MEG participated in manual annotation, provided critical insights to data analysis and contributed to drafting of the manuscript. PM provided bioinformatics analysis for virulence analysis, VENN diagram analysis, overall support of bioinformatics tools, and aided in drafting the manuscript. CO and ST were instrumental in genome closure. MG provided input on design of project and contributed to analysis of data. SDT provided support for acquisition of genomic sequence data, genome closure, and overall project design. GV provided overall bioinformatics support. CRC and MGS conceived of the study, participated in design and coordination of the study. All authors read and approved the final manuscript.
Supplementary Material
Genome coverage by sequence method for seven in-house sequenced SAG strains.
MAUVE analysis of SAG genomes. A multiple alignment of SA, SI and SC to highlight accessory regions and potential synteny within the genomes. MAUVE 2.3.1 was used with default settings with and a reference genome for each species; SAW C238 (SA), SCC C232 (SC) and SI B196 (SI).
Genome summary of 66 Streptococcus strains used for comparative genomic analysis studies.
Genomic content analysis comparison of SAG to other clinically important species within the genus Streptococcus. A) S. suis, B) S. pyogenes, C) S. agalactiae, D) S. pneumoniae, E) S. mutans F) S. mitis, G) S. sanguinis, H) S. gordonii. Numbers within circles correspond to number of CDSs that are conserved within the strains analyzed or that are unique to each strain analyzed.
Clusters of orthologous group (COG) analysis of 66 Streptococcus genomes. The phylogenetic tree was constructed using an in-house core-SNP pipeline as described previously. This Figure is a small portion of the overall comparison of all orthologs representing COGs present or absent in SAG while absent or present within the majority of other analysed Streptococcus. The COG map was created using OrthoMCL, COG functional categories are shown on the far left portion of the Figure. The Streptococcus species used for analysis are listed in Additional file 2, Table S2.
Comparison of gene content of SAG species to S. gordonii str. Challis substr. CH1 and S. sanguinis SK36.
Genes unique to sequenced SC, SI or SA.
Virulence genes used in virulence database to query SAG for potential virulence genes.
Streptococcus constellatus genes with a match to the virulence gene database.
S. intermedius genes with a match to the virulence gene database.
S. anginosus genes with a match to the virulence gene database.
SAG LPxTG proteins.
Summary of CRISPRs found in SAG.
Spacer regions within SAG CRISPRs.
Comparative analysis of essential competence genes from S. pneumoniae TIGR4.
Comparison of competence protein carriage within SAG.
SNPs found within chromosomal sequence for SCP C232 as compared to SCP C818.
Tandem-repeats and microsatellite differences identified between SCP C232 and SCP C818.
Contributor Information
Adam B Olson, Email: adam.olson@phac-aspc.gc.ca.
Heather Kent, Email: heatkent@gmail.com.
Christopher D Sibley, Email: Christopher.sibley@gmail.com.
Margot E Grinwis, Email: margot.chilcott@gmail.com.
Philip Mabon, Email: philip.mabon@phac-aspc.gc.ca.
Claude Ouellette, Email: claude.ouellette@phac-aspc.gc.ca.
Shari Tyson, Email: shari.tyson@phac-aspc.gc.ca.
Morag Graham, Email: morag.graham@phac-aspc.gc.ca.
Shaun D Tyler, Email: shaun.tyler@phac-aspc.gc.ca.
Gary Van Domselaar, Email: gary.vandomselaar@phac-aspc.gc.ca.
Michael G Surette, Email: surette@mcmaster.ca.
Cindi R Corbett, Email: cindi.corbett@phac-aspc.gc.ca.
Acknowledgments
This work was supported by a Genomics Research and Development Initiative project awarded to CRC. Operating grants from the Canadian Institute of Health Research (CIHR) and Cystic Fibrosis Canada awarded to MGS. MGS is supported as a Canada Research Chair in Interdisciplinary Microbiome Research. CDS was supported by an Alberta foundation for medical research studentship and a Canada Graduate Scholarship from CIHR. The authors would like to thank all members of PHAC Genomics Core facility for generation of sequence data and primer synthesis. The authors would also like to thank all members of PHAC Bioinformatics Core for input on bioinformatics questions. The views and opinions expressed herein are those of the authors only, and do not necessarily represent the views and opinions of the PHAC or the Government of Canada.
References
- Facklam R. What happened to the streptococci: overview of taxonomic and nomenclature changes. Clin Microbiol Rev. 2002;14(4):613–630. doi: 10.1128/CMR.15.4.613-630.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell J. Streptococcus mitis: walking the line between commensalism and pathogenesis. Mol Oral Microbiol. 2011;14(2):89–98. doi: 10.1111/j.2041-1014.2010.00601.x. [DOI] [PubMed] [Google Scholar]
- Tabata A, Nakano K, Ohkura K, Tomoyasu T, Kikuchi K, Whiley RA, Nagamune H. Novel twin streptolysin S-like peptides encoded in the sag operon homologue of beta-hemolytic Streptococcus anginosus. J Bacteriol. 2013;14(5):1090–1099. doi: 10.1128/JB.01344-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukeno A, Nagamune H, Whiley RA, Jafar SI, Aduse-Opoku J, Ohkura K, Maeda T, Hirota K, Miyake Y, Kourai H. Intermedilysin is essential for the invasion of hepatoma HepG2 cells by Streptococcus intermedius. Microbiol Immunol. 2005;14(7):681–694. doi: 10.1111/j.1348-0421.2005.tb03647.x. [DOI] [PubMed] [Google Scholar]
- Coykendall AL, Wesbecher PM, Gustafson KB. “Streptococcus milleri,” Streptococcus constellatus, and Streptococcus intermedius are later synonyms of Streptococcus anginosus. Int J Syst Bacteriol. 1987;14(3):222–228. doi: 10.1099/00207713-37-3-222. [DOI] [Google Scholar]
- Whiley RA, Hall LM, Hardie JM, Beighton D. A study of small-colony, beta-haemolytic, Lancefield group C streptococci within the anginosus group: description of Streptococcus constellatus subsp. pharyngis subsp. nov., associated with the human throat and pharyngitis. Int J Syst Bacteriol. 1999;14(4):1443–1449. doi: 10.1099/00207713-49-4-1443. [DOI] [PubMed] [Google Scholar]
- Jensen A, Hoshino T, Kilian M. Taxonomy of the Anginosus group of the genus Streptococcus and description of Streptococcus anginosus subsp. whileyi subsp. nov. and Streptococcus constellatus subsp. viborgensis subsp. nov. Int J Syst Evol Microbiol. 2013;14(Pt 7):2506–2519. doi: 10.1099/ijs.0.043232-0. [DOI] [PubMed] [Google Scholar]
- Gossling J. Occurrence and pathogenicity of the Streptococcus milleri group. Rev Infect Dis. 1988;14(2):257–285. doi: 10.1093/clinids/10.2.257. [DOI] [PubMed] [Google Scholar]
- Grinwis ME, Sibley CD, Parkins MD, Eshaghurshan CS, Rabin HR, Surette MG. Characterization of Streptococcus milleri group isolates from expectorated sputum of adult patients with cystic fibrosis. J Clin Microbiol. 2010;14(2):395–401. doi: 10.1128/JCM.01807-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiley RA, Beighton D, Winstanley TG, Fraser HY, Hardie JM. Streptococcus intermedius, Streptococcus constellatus, and Streptococcus anginosus (the Streptococcus milleri group): association with different body sites and clinical infections. J Clin Microbiol. 1992;14(1):243–244. doi: 10.1128/jcm.30.1.243-244.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yassin M, Yadavalli GK, Alvarado N, Bonomo RA. Streptococcus anginosus (Streptococcus milleri group) pyomyositis in a 50-year-Old Man with acquired immunodeficiency syndrome: case report and review of literature. Infection. 2010;14(1):65–68. doi: 10.1007/s15010-009-6002-9. [DOI] [PubMed] [Google Scholar]
- Prasad KN, Mishra AM, Gupta D, Husain N, Husain M, Gupta RK. Analysis of microbial etiology and mortality in patients with brain abscess. J Infect. 2006;14(4):221–227. doi: 10.1016/j.jinf.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Sibley CD, Church DL, Surette MG, Dowd SE, Parkins MD. Pyrosequencing reveals the complex polymicrobial nature of invasive pyogenic infections: microbial constituents of empyema, liver abscess, and intracerebral abscess. Eur J Clin Microbiol Infect Dis. 2012;14(10):2679–2691. doi: 10.1007/s10096-012-1614-x. [DOI] [PubMed] [Google Scholar]
- Sibley CD, Grinwis ME, Field TR, Parkins MD, Norgaard JC, Gregson DB, Rabin HR, Surette MG. McKay agar enables routine quantification of the ’Streptococcus milleri’ group in cystic fibrosis patients. J Med Microbiol. 2010;14(Pt 5):534–540. doi: 10.1099/jmm.0.016592-0. [DOI] [PubMed] [Google Scholar]
- Ahmed RA, Marrie TJ, Huang JQ. Thoracic empyema in patients with community-acquired pneumonia. Am J Med. 2006;14(10):877–883. doi: 10.1016/j.amjmed.2006.03.042. [DOI] [PubMed] [Google Scholar]
- Sibley CD, Parkins MD, Rabin HR, Duan K, Norgaard JC, Surette MG. A polymicrobial perspective of pulmonary infections exposes an enigmatic pathogen in cystic fibrosis patients. Proc Natl Acad Sci USA. 2008;14(39):15070–15075. doi: 10.1073/pnas.0804326105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkins MD, Sibley CD, Surette MG, Rabin HR. The Streptococcus milleri group–an unrecognized cause of disease in cystic fibrosis: a case series and literature review. Pediatr Pulmonol. 2008;14(5):490–497. doi: 10.1002/ppul.20809. [DOI] [PubMed] [Google Scholar]
- Mitchell AM, Mitchell TJ. Streptococcus pneumoniae: virulence factors and variation. Clin Microbiol Infect. 2010;14(5):411–418. doi: 10.1111/j.1469-0691.2010.03183.x. [DOI] [PubMed] [Google Scholar]
- Maisey HC, Doran KS, Nizet V. Recent advances in understanding the molecular basis of group B Streptococcus virulence. Expert Rev Mol Med. 2008;14:e27. doi: 10.1017/S1462399408000811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl G, Stalhammar-Carlemalm M, Areschoug T. Surface proteins of Streptococcus agalactiae and related proteins in other bacterial pathogens. Clin Microbiol Rev. 2005;14(1):102–127. doi: 10.1128/CMR.18.1.102-127.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagamune H, Ohnishi C, Katsuura A, Taoka Y, Fushitani K, Whiley RA, Yamashita K, Tsuji A, Matsuda Y, Maeda T, Korai H, Kitamura S. Intermedilysin. A cytolytic toxin specific for human cells of a Streptococcus intermedius isolated from human liver abscess. Adv Exp Med Biol. 1997;14:773–775. doi: 10.1007/978-1-4899-1825-3_182. [DOI] [PubMed] [Google Scholar]
- Jacobs JA, Schot CS, Schouls LM. Haemolytic activity of the Streptococcus milleri group’ and relationship between haemolysis restricted to human red blood cells and pathogenicity in S. intermedius. J Med Microbiol. 2000;14(1):55–62. doi: 10.1099/0022-1317-49-1-55. [DOI] [PubMed] [Google Scholar]
- Pecharki D, Petersen FC, Scheie AA. LuxS and expression of virulence factors in Streptococcus intermedius. Oral Microbiol Immunol. 2008;14(1):79–83. doi: 10.1111/j.1399-302X.2007.00395.x. [DOI] [PubMed] [Google Scholar]
- Toyoda K, Kusano N, Saito A. Pathogenicity of the Streptococcus milleri group in pulmonary infections–effect on phagocytic killing by human polymorphonuclear neutrophils. Kansenshogaku Zasshi. 1995;14(3):308–315. doi: 10.11150/kansenshogakuzasshi1970.69.308. [DOI] [PubMed] [Google Scholar]
- Kanamori S, Kusano N, Shinzato T, Saito A. The role of the capsule of the Streptococcus milleri group in its pathogenicity. J Infect Chemother. 2004;14(2):105–109. doi: 10.1007/s10156-004-0305-7. [DOI] [PubMed] [Google Scholar]
- Agren UM, Tammi M, Ryynanen M, Tammi R. Developmentally programmed expression of hyaluronan in human skin and its appendages. J Invest Dermatol. 1997;14(2):219–224. doi: 10.1111/1523-1747.ep12319412. [DOI] [PubMed] [Google Scholar]
- Pecharki D, Petersen FC, Scheie AA. Role of hyaluronidase in Streptococcus intermedius biofilm. Microbiology. 2008;14(Pt 3):932–938. doi: 10.1099/mic.0.2007/012393-0. [DOI] [PubMed] [Google Scholar]
- Ajdic D, McShan WM, McLaughlin RE, Savic G, Chang J, Carson MB, Primeaux C, Tian R, Kenton S, Jia H, Lin S, Qian Y, Li S, Zhu H, Najar F, Lai H, White J, Roe BA, Ferretti JJ. Genome sequence of Streptococcus mutans UA159, a cariogenic dental pathogen. Proc Natl Acad Sci USA. 2002;14(22):14434–14439. doi: 10.1073/pnas.172501299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferretti JJ, McShan WM, Ajdic D, Savic DJ, Savic G, Lyon K, Primeaux C, Sezate S, Suvorov AN, Kenton S, Lai HS, Lin SP, Qian Y, Jia HG, Najar FZ, Ren Q, Zhu H, Song L, White J, Yuan X, Clifton SW, Roe BA, McLaughlin R. Complete genome sequence of an M1 strain of Streptococcus pyogenes. Proc Natl Acad Sci USA. 2001;14(8):4658–4663. doi: 10.1073/pnas.071559398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden MT, Hauser H, Sanders M, Ngo TH, Cherevach I, Cronin A, Goodhead I, Mungall K, Quail MA, Price C, Rabbinowitsch E, Sharp S, Croucher NJ, Chieu TB, Mai NT, Diep TS, Chinh NT, Kehoe M, Leigh JA, Ward PN, Dowson CG, Whatmore AM, Chanter N, Iversen P, Gottschalk M, Slater JD, Smith HE, Spratt BG, Xu J, Ye C, Bentley S, Barrell BG, Schultsz C, Maskell DJ, Parkhill J. Rapid evolution of virulence and drug resistance in the emerging zoonotic pathogen Streptococcus suis. PLoS One. 2009;14(7):e6072. doi: 10.1371/journal.pone.0006072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Alves JM, Kitten T, Brown A, Chen Z, Ozaki LS, Manque P, Ge X, Serrano MG, Puiu D, Hendricks S, Wang Y, Chaplin MD, Akan D, Paik S, Peterson DL, Macrina FL, Buck GA. Genome of the opportunistic pathogen Streptococcus sanguinis. J Bacteriol. 2007;14(8):3166–3175. doi: 10.1128/JB.01808-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beres SB, Sylva GL, Barbian KD, Lei B, Hoff JS, Mammarella ND, Liu MY, Smoot JC, Porcella SF, Parkins LD, Campbell DS, Smith TM, McCormick JK, Leung DY, Schlievert PM, Musser JM. Genome sequence of a serotype M3 strain of group A Streptococcus: phage-encoded toxins, the high-virulence phenotype, and clone emergence. Proc Natl Acad Sci USA. 2002;14(15):10078–10083. doi: 10.1073/pnas.152298499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smoot JC, Barbian KD, Van Gompel JJ, Smoot LM, Chaussee MS, Sylva GL, Sturdevant DE, Ricklefs SM, Porcella SF, Parkins LD, Beres SB, Campbell DS, Smith TM, Zhang Q, Kapur V, Daly JA, Veasy LG, Musser JM. Genome sequence and comparative microarray analysis of serotype M18 group A Streptococcus strains associated with acute rheumatic fever outbreaks. Proc Natl Acad Sci USA. 2002;14(7):4668–4673. doi: 10.1073/pnas.062526099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa I, Kurokawa K, Yamashita A, Nakata M, Tomiyasu Y, Okahashi N, Kawabata S, Yamazaki K, Shiba T, Yasunaga T, Hayashi H, Hattori M, Hamada S. Genome sequence of an M3 strain of Streptococcus pyogenes reveals a large-scale genomic rearrangement in invasive strains and new insights into phage evolution. Genome Res. 2003;14(6A):1042–1055. doi: 10.1101/gr.1096703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guedon E, Delorme C, Pons N, Cruaud C, Loux V, Couloux A, Gautier C, Sanchez N, Layec S, Galleron N, Almeida M, van de Guchte M, Kennedy SP, Ehrlich SD, Gibrat JF, Wincker P, Renault P. Complete genome sequence of the commensal Streptococcus salivarius strain JIM8777. J Bacteriol. 2011;14(18):5024–5025. doi: 10.1128/JB.05390-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerat E, Ochman H. Recognizing the pseudogenes in bacterial genomes. Nucleic Acids Res. 2005;14(10):3125–3132. doi: 10.1093/nar/gki631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Eisen JA. A simple, fast, and accurate method of phylogenomic inference. Genome Biol. 2008;14(10):R151. doi: 10.1186/gb-2008-9-10-r151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glazunova OO, Raoult D, Roux V. Streptococcus massiliensis sp. nov., isolated from a patient blood culture. Int J Syst Evol Microbiol. 2006;14(Pt 5):1127–1131. doi: 10.1099/ijs.0.64009-0. [DOI] [PubMed] [Google Scholar]
- Schouls LM, Schot CS, Jacobs JA. Horizontal transfer of segments of the 16S rRNA genes between species of the Streptococcus anginosus group. J Bacteriol. 2003;14(24):7241–7246. doi: 10.1128/JB.185.24.7241-7246.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Mackey AJ, Stoeckert CJ Jr, Roos DS. OrthoMCL-DB: querying a comprehensive multi-species collection of ortholog groups. Nucleic Acids Res. 2006;14(Database issue):D363–D368. doi: 10.1093/nar/gkj123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Lefebure T, Hubisz MJ, Pavinski Bitar P, Lang P, Siepel A, Stanhope MJ. Comparative genomic analysis of the Streptococcus dysgalactiae species group: gene content, molecular adaptation, and promoter evolution. Genome Biol Evol. 2011;14:168–185. doi: 10.1093/gbe/evr006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebure T, Stanhope MJ. Evolution of the core and pan-genome of Streptococcus: positive selection, recombination, and genome composition. Genome Biol. 2007;14(5):R71. doi: 10.1186/gb-2007-8-5-r71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen TB, Danielsen M, Valina O, Garrigues C, Johansen E, Pedersen MB. Streptococcus thermophilus core genome: comparative genome hybridization study of 47 strains. Appl Environ Microbiol. 2008;14(15):4703–4710. doi: 10.1128/AEM.00132-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding F, Tang P, Hsu MH, Cui P, Hu S, Yu J, Chiu CH. Genome evolution driven by host adaptations results in a more virulent and antimicrobial-resistant Streptococcus pneumoniae serotype 14. BMC Genomics. 2009;14:158. doi: 10.1186/1471-2164-10-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tettelin H, Masignani V, Cieslewicz MJ, Donati C, Medini D, Ward NL, Angiuoli SV, Crabtree J, Jones AL, Durkin AS, Deboy RT, Davidsen TM, Mora M, Scarselli M, Margarit y Ros I, Peterson JD, Hauser CR, Sundaram JP, Nelson WC, Madupu R, Brinkac LM, Dodson RJ, Rosovitz MJ, Sullivan SA, Daugherty SC, Haft DH, Selengut J, Gwinn ML, Zhou L, Zafar N. et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”. Proc Natl Acad Sci USA. 2005;14(39):13950–13955. doi: 10.1073/pnas.0506758102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Foxman B, Drake DR, Srinivasan U, Henderson J, Olson B, Marrs CF, Warren JJ, Marazita ML. Comparative whole-genome analysis of Streptococcus mutans isolates within and among individuals of different caries status. Oral Microbiol Immunol. 2009;14(3):197–203. doi: 10.1111/j.1399-302X.2008.00495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takao A, Nagamune H, Maeda N. Sialidase of Streptococcus intermedius: a putative virulence factor modifying sugar chains. Microbiol Immunol. 2010;14(10):584–595. doi: 10.1111/j.1348-0421.2010.00257.x. [DOI] [PubMed] [Google Scholar]
- Christie J, McNab R, Jenkinson HF. Expression of fibronectin-binding protein FbpA modulates adhesion in Streptococcus gordonii. Microbiology. 2002;14(Pt 6):1615–1625. doi: 10.1099/00221287-148-6-1615. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Ma Q, Su D, Li Q, Yao W, Wang C. Identification of horizontal gene transfer and recombination of PsaA gene in streptococcus mitis group. Microbiol Immunol. 2010;14(6):313–319. doi: 10.1111/j.1348-0421.2010.00216.x. [DOI] [PubMed] [Google Scholar]
- Spellerberg B, Rozdzinski E, Martin S, Weber-Heynemann J, Schnitzler N, Lutticken R, Podbielski A. Lmb, a protein with similarities to the LraI adhesin family, mediates attachment of Streptococcus agalactiae to human laminin. Infect Immun. 1999;14(2):871–878. doi: 10.1128/iai.67.2.871-878.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hytonen J, Haataja S, Finne J. Streptococcus pyogenes glycoprotein-binding strepadhesin activity is mediated by a surface-associated carbohydrate-degrading enzyme, pullulanase. Infect Immun. 2003;14(2):784–793. doi: 10.1128/IAI.71.2.784-793.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pancholi V, Fischetti VA. Alpha-Enolase, a Novel Strong Plasmin(ogen) Binding Protein on the Surface of Pathogenic Streptococci. J Biol Chem. 1998;14(23):14503–14515. doi: 10.1074/jbc.273.23.14503. [DOI] [PubMed] [Google Scholar]
- Hidalgo-Grass C, Ravins M, Dan-Goor M, Jaffe J, Moses AE, Hanski E. A locus of group A Streptococcus involved in invasive disease and DNA transfer. Mol Microbiol. 2002;14(1):87–99. doi: 10.1046/j.1365-2958.2002.03127.x. [DOI] [PubMed] [Google Scholar]
- Morona JK, Morona R, Paton JC. Comparative genetics of capsular polysaccharide biosynthesis in Streptococcus pneumoniae types belonging to serogroup 19. J Bacteriol. 1999;14(17):5355–5364. doi: 10.1128/jb.181.17.5355-5364.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spellerberg B, Pohl B, Haase G, Martin S, Weber-Heynemann J, Lutticken R. Identification of genetic determinants for the hemolytic activity of Streptococcus agalactiae by ISS1 transposition. J Bacteriol. 1999;14(10):3212–3219. doi: 10.1128/jb.181.10.3212-3219.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crater DL, Dougherty BA, van de Rijn I. Molecular characterization of hasC from an operon required for hyaluronic acid synthesis in group A streptococci. Demonstration of UDP-glucose pyrophosphorylase activity. J Biol Chem. 1995;14(48):28676–28680. doi: 10.1074/jbc.270.48.28676. [DOI] [PubMed] [Google Scholar]
- Jiang SM, Ishmael N, Dunning Hotopp J, Puliti M, Tissi L, Kumar N, Cieslewicz MJ, Tettelin H, Wessels MR. Variation in the group B Streptococcus CsrRS regulon and effects on pathogenicity. J Bacteriol. 2008;14(6):1956–1965. doi: 10.1128/JB.01677-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton M, Tagg JR, Wescombe P, Jenkinson HF. Intra- and interspecies signaling between Streptococcus salivarius and Streptococcus pyogenes mediated by SalA and SalA1 lantibiotic peptides. J Bacteriol. 2001;14(13):3931–3938. doi: 10.1128/JB.183.13.3931-3938.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madureira P, Baptista M, Vieira M, Magalhaes V, Camelo A, Oliveira L, Ribeiro A, Tavares D, Trieu-Cuot P, Vilanova M, Ferreira P. Streptococcus agalactiae GAPDH is a virulence-associated immunomodulatory protein. J Immunol. 2007;14(3):1379–1387. doi: 10.4049/jimmunol.178.3.1379. [DOI] [PubMed] [Google Scholar]
- Smith HE, de Vries R, van’t Slot R, Smits MA. The cps locus of Streptococcus suis serotype 2: genetic determinant for the synthesis of sialic acid. Microb Pathog. 2000;14(2):127–134. doi: 10.1006/mpat.2000.0372. [DOI] [PubMed] [Google Scholar]
- Jensch I, Gamez G, Rothe M, Ebert S, Fulde M, Somplatzki D, Bergmann S, Petruschka L, Rohde M, Nau R, Hammerschmidt S. PavB is a surface-exposed adhesin of Streptococcus pneumoniae contributing to nasopharyngeal colonization and airways infections. Mol Microbiol. 2010;14(1):22–43. doi: 10.1111/j.1365-2958.2010.07189.x. [DOI] [PubMed] [Google Scholar]
- Sumitomo T, Nakata M, Higashino M, Jin Y, Terao Y, Fujinaga Y, Kawabata S. Streptolysin S contributes to group A streptococcal translocation across an epithelial barrier. J Biol Chem. 2011;14(4):2750–2761. doi: 10.1074/jbc.M110.171504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer RT, Drevets DA, Campbell PA, Potter TA. Internalin A can mediate phagocytosis of Listeria monocytogenes by mouse macrophage cell lines. J Leukoc Biol. 1996;14(5):603–610. doi: 10.1002/jlb.60.5.603. [DOI] [PubMed] [Google Scholar]
- Reid SD, Montgomery AG, Voyich JM, DeLeo FR, Lei B, Ireland RM, Green NM, Liu M, Lukomski S, Musser JM. Characterization of an extracellular virulence factor made by group A Streptococcus with homology to the Listeria monocytogenes internalin family of proteins. Infect Immun. 2003;14(12):7043–7052. doi: 10.1128/IAI.71.12.7043-7052.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nightingale KK, Windham K, Martin KE, Yeung M, Wiedmann M. Select Listeria monocytogenes subtypes commonly found in foods carry distinct nonsense mutations in inlA, leading to expression of truncated and secreted internalin A, and are associated with a reduced invasion phenotype for human intestinal epithelial cells. Appl Environ Microbiol. 2005;14(12):8764–8772. doi: 10.1128/AEM.71.12.8764-8772.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croucher NJ, Harris SR, Fraser C, Quail MA, Burton J, van der Linden M, McGee L, von Gottberg A, Song JH, Ko KS, Pichon B, Baker S, Parry CM, Lambertsen LM, Shahinas D, Pillai DR, Mitchell TJ, Dougan G, Tomasz A, Klugman KP, Parkhill J, Hanage WP, Bentley SD. Rapid pneumococcal evolution in response to clinical interventions. Science. 2011;14(6016):430–434. doi: 10.1126/science.1198545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarre WW, Schneewind O. Surface proteins of gram-positive bacteria and mechanisms of their targeting to the cell wall envelope. Microbiol Mol Biol Rev. 1999;14(1):174–229. doi: 10.1128/mmbr.63.1.174-229.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalioui L, Pellegrini E, Dramsi S, Baptista M, Bourgeois N, Doucet-Populaire F, Rusniok C, Zouine M, Glaser P, Kunst F, Poyart C, Trieu-Cuot P. The SrtA Sortase of Streptococcus agalactiae is required for cell wall anchoring of proteins containing the LPXTG motif, for adhesion to epithelial cells, and for colonization of the mouse intestine. Infect Immun. 2005;14(6):3342–3350. doi: 10.1128/IAI.73.6.3342-3350.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozzi R, Prigozhin D, Rosini R, Abate F, Bottomley MJ, Grandi G, Telford JL, Rinaudo CD, Maione D, Alber T. Structural basis for group B streptococcus pilus 1 sortases C regulation and specificity. PLoS One. 2012;14(11):e49048. doi: 10.1371/journal.pone.0049048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tettelin H, Masignani V, Cieslewicz MJ, Eisen JA, Peterson S, Wessels MR, Paulsen IT, Nelson KE, Margarit I, Read TD, Madoff LC, Wolf AM, Beanan MJ, Brinkac LM, Daugherty SC, DeBoy RT, Durkin AS, Kolonay JF, Madupu R, Lewis MR, Radune D, Fedorova NB, Scanlan D, Khouri H, Mulligan S, Carty HA, Cline RT, Van Aken SE, Gill J, Scarselli M, Mora M, Iacobini ET, Brettoni C, Galli G, Mariani M, Vegni F, Maione D, Rinaudo D, Rappuoli R, Telford JL, Kasper DL, Grandi G, Fraser CM. Complete genome sequence and comparative genomic analysis of an emerging human pathogen, serotype V Streptococcus agalactiae. Proc Natl Acad Sci USA. 2002;14(19):12391–12396. doi: 10.1073/pnas.182380799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halfmann A, Schnorpfeil A, Muller M, Marx P, Gunzler U, Hakenbeck R, Bruckner R. Activity of the two-component regulatory system CiaRH in Streptococcus pneumoniae R6. J Mol Microbiol Biotechnol. 2011;14(2):96–104. doi: 10.1159/000324893. [DOI] [PubMed] [Google Scholar]
- Hoch JA. Two-component and phosphorelay signal transduction. Curr Opin Microbiol. 2000;14(2):165–170. doi: 10.1016/S1369-5274(00)00070-9. [DOI] [PubMed] [Google Scholar]
- Lange R, Wagner C, de Saizieu A, Flint N, Molnos J, Stieger M, Caspers P, Kamber M, Keck W, Amrein KE. Domain organization and molecular characterization of 13 two-component systems identified by genome sequencing of Streptococcus pneumoniae. Gene. 1999;14(1):223–234. doi: 10.1016/S0378-1119(99)00266-8. [DOI] [PubMed] [Google Scholar]
- Paterson GK, Blue CE, Mitchell TJ. Role of two-component systems in the virulence of Streptococcus pneumoniae. J Med Microbiol. 2006;14(Pt 4):355–363. doi: 10.1099/jmm.0.46423-0. [DOI] [PubMed] [Google Scholar]
- Tettelin H, Nelson KE, Paulsen IT, Eisen JA, Read TD, Peterson S, Heidelberg J, DeBoy RT, Haft DH, Dodson RJ, Durkin AS, Gwinn M, Kolonay JF, Nelson WC, Peterson JD, Umayam LA, White O, Salzberg SL, Lewis MR, Radune D, Holtzapple E, Khouri H, Wolf AM, Utterback TR, Hansen CL, McDonald LA, Feldblyum TV, Angiuoli S, Dickinson T, Hickey EK, Holt IE, Loftus BJ, Yang F, Smith HO, Venter JC, Dougherty BA, Morrison DA, Hollingshead SK, Fraser CM. Complete genome sequence of a virulent isolate of Streptococcus pneumoniae. Science. 2001;14(5529):498–506. doi: 10.1126/science.1061217. [DOI] [PubMed] [Google Scholar]
- Biswas I, Drake L, Erkina D, Biswas S. Involvement of sensor kinases in the stress tolerance response of Streptococcus mutans. J Bacteriol. 2008;14(1):68–77. doi: 10.1128/JB.00990-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay YD, Lo H, Li YH, Halperin SA, Lee SF. Expression of the Streptococcus mutans essential two-component regulatory system VicRK is pH and growth-phase dependent and controlled by the LiaFSR three-component regulatory system. Microbiology. 2009;14(Pt 9):2856–2865. doi: 10.1099/mic.0.028456-0. [DOI] [PubMed] [Google Scholar]
- Throup JP, Koretke KK, Bryant AP, Ingraham KA, Chalker AF, Ge Y, Marra A, Wallis NG, Brown JR, Holmes DJ, Rosenberg M, Burnham MK. A genomic analysis of two-component signal transduction in Streptococcus pneumoniae. Mol Microbiol. 2000;14(3):566–576. doi: 10.1046/j.1365-2958.2000.01725.x. [DOI] [PubMed] [Google Scholar]
- McKessar SJ, Hakenbeck R. The two-component regulatory system TCS08 is involved in cellobiose metabolism of Streptococcus pneumoniae R6. J Bacteriol. 2007;14(4):1342–1350. doi: 10.1128/JB.01170-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haft DH, Selengut J, Mongodin EF, Nelson KE. A guild of 45 CRISPR-associated (Cas) protein families and multiple CRISPR/Cas subtypes exist in prokaryotic genomes. PLoS Comput Biol. 2005;14(6):e60. doi: 10.1371/journal.pcbi.0010060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, Horvath P, Moineau S, Mojica FJ, Wolf YI, Yakunin AF, van der Oost J, Koonin EV. Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol. 2011;14(6):467–477. doi: 10.1038/nrmicro2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath P, Coute-Monvoisin AC, Romero DA, Boyaval P, Fremaux C, Barrangou R. Comparative analysis of CRISPR loci in lactic acid bacteria genomes. Int J Food Microbiol. 2009;14(1):62–70. doi: 10.1016/j.ijfoodmicro.2008.05.030. [DOI] [PubMed] [Google Scholar]
- Lopez-Sanchez MJ, Sauvage E, Da Cunha V, Clermont D, Ratsima Hariniaina E, Gonzalez-Zorn B, Poyart C, Rosinski-Chupin I, Glaser P. The highly dynamic CRISPR1 system of Streptococcus agalactiae controls the diversity of its mobilome. Mol Microbiol. 2012;14(6):1057–1071. doi: 10.1111/j.1365-2958.2012.08172.x. [DOI] [PubMed] [Google Scholar]
- van der Ploeg JR. Analysis of CRISPR in Streptococcus mutans suggests frequent occurrence of acquired immunity against infection by M102-like bacteriophages. Microbiology. 2009;14(Pt 6):1966–1976. doi: 10.1099/mic.0.027508-0. [DOI] [PubMed] [Google Scholar]
- Deveau H, Barrangou R, Garneau JE, Labonte J, Fremaux C, Boyaval P, Romero DA, Horvath P, Moineau S. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J Bacteriol. 2008;14(4):1390–1400. doi: 10.1128/JB.01412-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croucher NJ, Vernikos GS, Parkhill J, Bentley SD. Identification, variation and transcription of pneumococcal repeat sequences. BMC Genomics. 2011;14:120. doi: 10.1186/1471-2164-12-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser R, Sabri M, Moineau S, Uetz P. The proteome and interactome of Streptococcus pneumoniae phage Cp-1. J Bacteriol. 2011;14(12):3135–3138. doi: 10.1128/JB.01481-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz E, Lopez R, Garcia JL. EJ-1, a temperate bacteriophage of Streptococcus pneumoniae with a Myoviridae morphotype. J Bacteriol. 1992;14(17):5516–5525. doi: 10.1128/jb.174.17.5516-5525.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikebe T, Wada A, Inagaki Y, Sugama K, Suzuki R, Tanaka D, Tamaru A, Fujinaga Y, Abe Y, Shimizu Y, Watanabe H. Working Group for Group A Streptococcus in Japan. Dissemination of the phage-associated novel superantigen gene speL in recent invasive and noninvasive Streptococcus pyogenes M3/T3 isolates in Japan. Infect Immun. 2002;14(6):3227–3233. doi: 10.1128/IAI.70.6.3227-3233.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson D, Schuch R, Zhu S, Tscherne DM, Fischetti VA. Genomic sequence of C1, the first streptococcal phage. J Bacteriol. 2003;14(11):3325–3332. doi: 10.1128/JB.185.11.3325-3332.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Ploeg JR. Genome sequence of the temperate bacteriophage PH10 from Streptococcus oralis. Virus Genes. 2010;14(3):450–458. doi: 10.1007/s11262-010-0525-0. [DOI] [PubMed] [Google Scholar]
- van der Ploeg JR. Characterization of Streptococcus gordonii prophage PH15: complete genome sequence and functional analysis of phage-encoded integrase and endolysin. Microbiology. 2008;14(Pt 10):2970–2978. doi: 10.1099/mic.0.2008/018739-0. [DOI] [PubMed] [Google Scholar]
- Rusniok C, Couve E, Da Cunha V, El Gana R, Zidane N, Bouchier C, Poyart C, Leclercq R, Trieu-Cuot P, Glaser P. Genome sequence of Streptococcus gallolyticus: insights into its adaptation to the bovine rumen and its ability to cause endocarditis. J Bacteriol. 2010;14(8):2266–2276. doi: 10.1128/JB.01659-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnsborg O, Havarstein LS. Pneumococcal LytR, a protein from the LytR-CpsA-Psr family, is essential for normal septum formation in Streptococcus pneumoniae. J Bacteriol. 2009;14(18):5859–5864. doi: 10.1128/JB.00724-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claverys JP, Martin B. Bacterial “competence” genes: signatures of active transformation, or only remnants? Trends Microbiol. 2003;14(4):161–165. doi: 10.1016/S0966-842X(03)00064-7. [DOI] [PubMed] [Google Scholar]
- Peterson SN, Sung CK, Cline R, Desai BV, Snesrud EC, Luo P, Walling J, Li H, Mintz M, Tsegaye G, Burr PC, Do Y, Ahn S, Gilbert J, Fleischmann RD, Morrison DA. Identification of competence pheromone responsive genes in Streptococcus pneumoniae by use of DNA microarrays. Mol Microbiol. 2004;14(4):1051–1070. doi: 10.1046/j.1365-2958.2003.03907.x. [DOI] [PubMed] [Google Scholar]
- Beres SB, Sesso R, Pinto SW, Hoe NP, Porcella SF, Deleo FR, Musser JM. Genome sequence of a Lancefield group C Streptococcus zooepidemicus strain causing epidemic nephritis: new information about an old disease. PLoS One. 2008;14(8):e3026. doi: 10.1371/journal.pone.0003026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein MI, Bang S, Florio FM, Hofling JF, Goncalves RB, Smith DJ, Mattos-Graner RO. Genetic diversity of competence gene loci in clinical genotypes of Streptococcus mutans. J Clin Microbiol. 2006;14(8):3015–3020. doi: 10.1128/JCM.02024-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canals R, Xia XQ, Fronick C, Clifton SW, Ahmer BM, Andrews-Polymenis HL, Porwollik S, McClelland M. High-throughput comparison of gene fitness among related bacteria. BMC Genomics. 2012;14:212–2164. doi: 10.1186/1471-2164-13-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hone DM, Harris AM, Chatfield S, Dougan G, Levine MM. Construction of genetically defined double aro mutants of Salmonella typhi. Vaccine. 1991;14(11):810–816. doi: 10.1016/0264-410X(91)90218-U. [DOI] [PubMed] [Google Scholar]
- Sebkova A, Karasova D, Crhanova M, Budinska E, Rychlik I. aro mutations in Salmonella enterica cause defects in cell wall and outer membrane integrity. J Bacteriol. 2008;14(9):3155–3160. doi: 10.1128/JB.00053-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer GC, Palmer KL, Jorth PA, Whiteley M. Characterization of the Pseudomonas aeruginosa transcriptional response to phenylalanine and tyrosine. J Bacteriol. 2010;14(11):2722–2728. doi: 10.1128/JB.00112-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JY, Fung CP, Chang FY, Huang LY, Chang JC, Siu LK. Mutations of the rpoB gene in rifampicin-resistant Streptococcus pneumoniae in Taiwan. J Antimicrob Chemother. 2004;14(2):375–378. doi: 10.1093/jac/dkh073. [DOI] [PubMed] [Google Scholar]
- Badger JH, Olsen GJ. CRITICA: coding region identification tool invoking comparative analysis. Mol Biol Evol. 1999;14(4):512–524. doi: 10.1093/oxfordjournals.molbev.a026133. [DOI] [PubMed] [Google Scholar]
- Delcher AL, Bratke KA, Powers EC, Salzberg SL. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics. 2007;14(6):673–679. doi: 10.1093/bioinformatics/btm009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagesen K, Hallin P, Rodland EA, Staerfeldt HH, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;14(9):3100–3108. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;14(5):955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. BLAST+: architecture and applications. BMC Bioinforma. 2009;14:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayers EW, Barrett T, Benson DA, Bolton E, Bryant SH, Canese K, Chetvernin V, Church DM, DiCuccio M, Federhen S, Feolo M, Fingerman IM, Geer LY, Helmberg W, Kapustin Y, Landsman D, Lipman DJ, Lu Z, Madden TL, Madej T, Maglott DR, Marchler-Bauer A, Miller V, Mizrachi I, Ostell J, Panchenko A, Phan L, Pruitt KD, Schuler GD. et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2011;14(Database issue):D38–D51. doi: 10.1093/nar/gkq1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999;14(1):29–34. doi: 10.1093/nar/27.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt Consortium. The Universal Protein Resource (UniProt) in 2010. Nucleic Acids Res. 2010;14(Database issue):D142–D148. doi: 10.1093/nar/gkp846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov DM, Mazumder R, Mekhedov SL, Nikolskaya AN, Rao BS, Smirnov S, Sverdlov AV, Vasudevan S, Wolf YI, Yin JJ, Natale DA. The COG database: an updated version includes eukaryotes. BMC Bioinforma. 2003;14:41. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Lu F, Marchler GH, Mullokandov M, Omelchenko MV, Robertson CL, Song JS, Thanki N, Yamashita RA, Zhang D, Zhang N, Zheng C, Bryant SH. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 2011;14(Database issue):D225–D229. doi: 10.1093/nar/gkq1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selengut JD, Haft DH, Davidsen T, Ganapathy A, Gwinn-Giglio M, Nelson WC, Richter AR, White O. TIGRFAMs and Genome Properties: tools for the assignment of molecular function and biological process in prokaryotic genomes. Nucleic Acids Res. 2007;14(Database issue):D260–D264. doi: 10.1093/nar/gkl1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, Gavin OL, Gunasekaran P, Ceric G, Forslund K, Holm L, Sonnhammer EL, Eddy SR, Bateman A. The Pfam protein families database. Nucleic Acids Res. 2010;14(Database issue):D211–D222. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000;14(6):276–277. doi: 10.1016/S0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- Moller S, Croning MD, Apweiler R. Evaluation of methods for the prediction of membrane spanning regions. Bioinformatics. 2001;14(7):646–653. doi: 10.1093/bioinformatics/17.7.646. [DOI] [PubMed] [Google Scholar]
- Bendtsen JD, Nielsen H, von Heijne G, Brunak S. Improved prediction of signal peptides: SignalP 3.0. J Mol Biol. 2004;14(4):783–795. doi: 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- Petkau A, Stuart-Edwards M, Stothard P, Van Domselaar G. Interactive microbial genome visualization with GView. Bioinformatics. 2010;14(24):3125–3126. doi: 10.1093/bioinformatics/btq588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling AE, Mau B, Perna NT. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One. 2010;14(6):e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criscuolo A. morePhyML: Improving the phylogenetic tree space exploration with PhyML 3. Mol Phylogenet Evol. 2011;14(3):944–948. doi: 10.1016/j.ympev.2011.08.029. [DOI] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;14(10):2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986;14(5):418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- Muller J, Szklarczyk D, Julien P, Letunic I, Roth A, Kuhn M, Powell S, von Mering C, Doerks T, Jensen LJ, Bork P. eggNOG v2.0: extending the evolutionary genealogy of genes with enhanced non-supervised orthologous groups, species and functional annotations. Nucleic Acids Res. 2010;14(Database issue):D190–D195. doi: 10.1093/nar/gkp951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grissa I, Vergnaud G, Pourcel C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007;14(Web Server issue):W52–W57. doi: 10.1093/nar/gkm360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grissa I, Vergnaud G, Pourcel C. CRISPRcompar: a website to compare clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2008;14(Web Server issue):W145–W148. doi: 10.1093/nar/gkn228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Chen L, Sun L, Yu J, Jin Q. VFDB 2008 release: an enhanced web-based resource for comparative pathogenomics. Nucleic Acids Res. 2008;14(Database issue):D539–D542. doi: 10.1093/nar/gkm951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;14(13):1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Genome coverage by sequence method for seven in-house sequenced SAG strains.
MAUVE analysis of SAG genomes. A multiple alignment of SA, SI and SC to highlight accessory regions and potential synteny within the genomes. MAUVE 2.3.1 was used with default settings with and a reference genome for each species; SAW C238 (SA), SCC C232 (SC) and SI B196 (SI).
Genome summary of 66 Streptococcus strains used for comparative genomic analysis studies.
Genomic content analysis comparison of SAG to other clinically important species within the genus Streptococcus. A) S. suis, B) S. pyogenes, C) S. agalactiae, D) S. pneumoniae, E) S. mutans F) S. mitis, G) S. sanguinis, H) S. gordonii. Numbers within circles correspond to number of CDSs that are conserved within the strains analyzed or that are unique to each strain analyzed.
Clusters of orthologous group (COG) analysis of 66 Streptococcus genomes. The phylogenetic tree was constructed using an in-house core-SNP pipeline as described previously. This Figure is a small portion of the overall comparison of all orthologs representing COGs present or absent in SAG while absent or present within the majority of other analysed Streptococcus. The COG map was created using OrthoMCL, COG functional categories are shown on the far left portion of the Figure. The Streptococcus species used for analysis are listed in Additional file 2, Table S2.
Comparison of gene content of SAG species to S. gordonii str. Challis substr. CH1 and S. sanguinis SK36.
Genes unique to sequenced SC, SI or SA.
Virulence genes used in virulence database to query SAG for potential virulence genes.
Streptococcus constellatus genes with a match to the virulence gene database.
S. intermedius genes with a match to the virulence gene database.
S. anginosus genes with a match to the virulence gene database.
SAG LPxTG proteins.
Summary of CRISPRs found in SAG.
Spacer regions within SAG CRISPRs.
Comparative analysis of essential competence genes from S. pneumoniae TIGR4.
Comparison of competence protein carriage within SAG.
SNPs found within chromosomal sequence for SCP C232 as compared to SCP C818.
Tandem-repeats and microsatellite differences identified between SCP C232 and SCP C818.