

Abstract
Background
Modern breeding and artificial selection play critical roles in pig domestication and shape the genetic variation of different breeds. China has many indigenous pig breeds with various characteristics in morphology and production performance that differ from those of foreign commercial pig breeds. However, the signatures of selection on genes implying for economic traits between Chinese indigenous and commercial pigs have been poorly understood.
Results
We identified footprints of positive selection at the whole genome level, comprising 44,652 SNPs genotyped in six Chinese indigenous pig breeds, one developed breed and two commercial breeds. An empirical genome-wide distribution of Fst (F-statistics) was constructed based on estimations of Fst for each SNP across these nine breeds. We detected selection at the genome level using the High-Fst outlier method and found that 81 candidate genes show high evidence of positive selection. Furthermore, the results of network analyses showed that the genes that displayed evidence of positive selection were mainly involved in the development of tissues and organs, and the immune response. In addition, we calculated the pairwise Fst between Chinese indigenous and commercial breeds (CHN VS EURO) and between Northern and Southern Chinese indigenous breeds (Northern VS Southern). The IGF1R and ESR1 genes showed evidence of positive selection in the CHN VS EURO and Northern VS Southern groups, respectively.
Conclusions
In this study, we first identified the genomic regions that showed evidences of selection between Chinese indigenous and commercial pig breeds using the High-Fst outlier method. These regions were found to be involved in the development of tissues and organs, the immune response, growth and litter size. The results of this study provide new insights into understanding the genetic variation and domestication in pigs.
Keywords: Pig, F-statistics, Breed, Positive selection, Network analysis
Background
Pigs and humans have interacted for approximately 10,000 years, and as a major protein source for humans, the pig is one of the most important domestic animals [1]. Domestic pigs originated from the Eurasian wild boar (Sus scrofa) approximately 9000 years ago. European and Asian pigs were domesticated independently and introgression of the Asian domestic pig into the European pig occurred after domestication [2,3]. Most of these breeds (especially commercial breeds) have been subjected to strong artificial selection to improve pork productivity. However, different breeds show large differences in morphology and production performance due to various breeding objectives, selection systems and rearing environments; nevertheless, very little is known on the molecular mechanisms of artificial selection on pigs.
The development of high-throughput sequencing and genotyping technologies makes it possible to investigate the selective pressures of various domestic animal species at the genomic level and to identify candidate genes associated with economic traits in order to better understand the mechanisms of adaptive evolution. For example, several important genes relevant to reproduction and growth such as GHR and MC1R have been identified in cattle [4-8], and Flori et al. [6] implemented a network analysis for detected genes that have been putatively subjected to selection. Akey et al. [9] identified 155 regions in the canine genome that have likely been subjected to strong artificial selection, including the HAS2 gene, which is involved in skin wrinkling. The thyroid stimulating hormone receptor (TSHR) gene was identified as having undergone strong artificial selection in domestic chickens [10]. The above studies used several types of approaches that were based on either the allele frequency spectrum or the properties of haplotype segregation in populations to detect signals of recent positive selection on a genome-wide scale [8]. For example, Fst (a measure of population differentiation) provides an estimate of the genetic variability between populations: a locus that shows significantly high Fst statistics compared with other loci provides evidence for positive selection [11]. Akey et al. [12] suggested that the loci in the tails of the empirical distribution of Fst be used as candidate targets of selection. Another method of identifying loci under selection is the EHH (Extended Haplotype Homozygosity) test [13], which identifies the genome regions that have unusually high LD and allele frequency.
The advent of the Illumina Porcine SNP60 BeadChip [14] allows for the investigation of selective pressure at the genome-wide level in pigs. Melanocortin receptor 1 gene (MC1R) was identified as an artificial selection gene related to coat colour in Chinese domestic pigs [15]. A missense mutation in the PPARD gene had an effect on the ear size of the pigs [16]. China has a number of indigenous pig breeds, most of which are fat-type and low degree of nurturing breeds. Therefore, using Chinese indigenous breeds would be a better way to obtain meaningful signatures of selection on genes implying for economic traits in the pig at genomic level. Therefore, the objective of this study was to identify regions subjected to recent artificial selection using a genome scan for SNP differences. The findings will contribute to the construction of a positive selection map, which could help us to understand the recent breeding history of different pig breeds. Our results will also facilitate the identification of candidate genes that are important for economic traits for breeding practices.
Results
Population structure and genome-wide distribution of Fst
To examine the genetic structure of the studied populations, the principle component analysis (PCA) was conducted based on all available SNP information. As shown in Figure 1, the first two components accounted for 42.43% and 8.94% of the variation, respectively. The Luchuan, Bama and Wuzhishan pigs were clustered closely, as were the Ningxiang and Tongcheng pigs and the Large White and Landrace pigs, while the Yutai and Laiwu were more distant from the other pig breeds.
Figure 1.
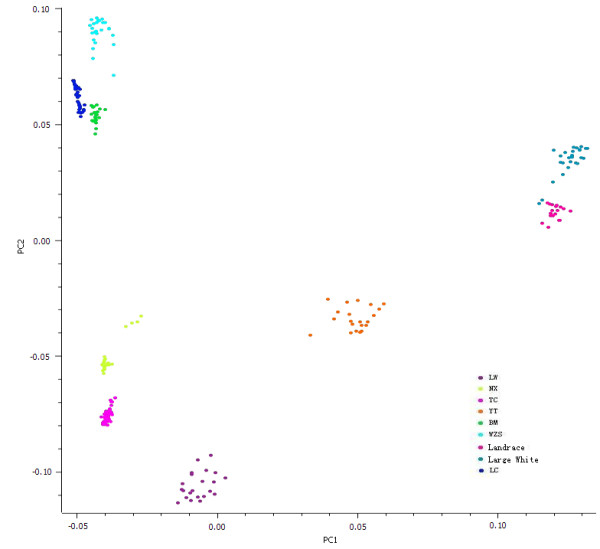
Principal component analysis results based on whole genome SNP data. (235 individuals, 44,652 SNPs). Individuals are plotted according to their coordinates on the biplot of PC1 versus PC2. Breed abbreviations are described in Materials and Methods.
We constructed the empirical genome-wide distribution of global Fst estimates based on 44,652 SNPs of the nine breeds (ALLPOP) in order to examine the inter-locus variation in allele frequencies (Figure 2). The average Fst of these loci was 0.3717 with standard deviation 0.16. Local environmental adaptation and artificial selection can change the allele frequencies of specific loci: the frequency of advantageous alleles at the selected loci will increase, leading to a higher than expected level of population differentiation (Fst) [12]. The genome-wide distribution of Fst revealed selection in the pig genome. To identify specific genomic regions containing signatures of selection, we constructed a chromosomal distribution of Fst as a function of chromosome position. As shown in Figure 3, the sex chromosomes have a smaller effective population size compared to the autosomes, which makes them more sensitive to demographic events and/or natural selection [12]. As a result, there was an unexpectedly high Fst level on the physical position 40-80 M of the X chromosome. Taking into account the PCA analysis results, the pairwise Fst between Chinese indigenous and European commercial breeds were calculated by merging Chinese indigenous breeds and commercial breeds into two groups (CHN VS EURO). In addition, the pairwise Fst between Northern (LW pigs) and Southern Chinese indigenous breeds was calculated by merging LC, WZS and BM into one group (Northern VS Southern).
Figure 2.
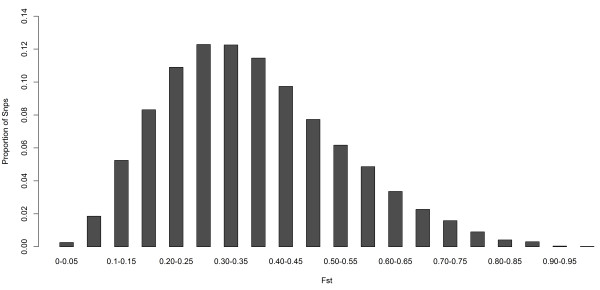
Genome-wide distribution of Fst in ALLPOP group.
Figure 3.
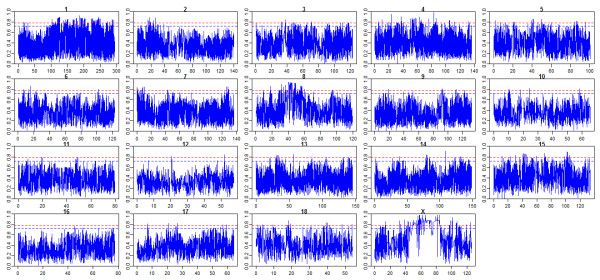
Fst for each SNP as a function of chromosome position in Mb in the ALLPOP group. The red dashed line corresponds to the 99% threshold on the corresponding empirical distributions, and the blue indicates the 97.5% threshold.
Candidate genes under selection
To identify loci subjected to selection, we focused on the high-Fst outlier method corresponding to the distribution of Fst. According to the empirical distribution of Fst estimates, we selected the high-Fst outlier SNPs that corresponded to the upper 1% of the distribution as the loci under selection. In the ALLPOP group, a total of 446 SNPs were determined to be subjected to natural or artificial selection following this criteria, and these SNPs were from a total of 81 candidate genes (Additional file 1: Table S1). In addition, a total of 84 and 79 candidate genes were identified in the CHN VS EURO group and the Northern VS Southern group, respectively (Additional file 2: Table S2 and Additional file 3: Table S3). Several candidate genes contain contiguous outlier-Fst SNPs; for example, the transient receptor potential three (TRPM3) gene contains five contiguous SNPs with Fst values that are consistently high, and the nuclear envelope spectrin repeat 2 (Nesprin-2) gene contained two outlier-Fst SNPs in the ALLPOP group.
Functional analysis of candidate genes under selection
Based on a system biology approach, we carried out network analysis using IPA software to identify the critical physiological pathways of the genes harbouring footprints of positive selection. The pig breeds selected have obvious differences in both morphology and performance. The Large White and Landrace pigs are well-known commercial breeds with high meat productivity, fast growth, and high adaptability; however, Chinese indigenous breeds vary in morphological and performance phenotypes and in local environmental suitability. For example, the Bama and Wuzhishan pigs from Southern China have a small body size, while the Laiwu pigs from Northern China are larger. First, 75 out of 81 genes in the ALLPOP group were mapped to the IPA database, and then three significance networks, namely N1, N2 and N3, were constructed. N2 and N3 were interconnected and further merged into a single network (N). Networks N and N1 are represented in Figures 4 and 5, respectively. The main hubs of the N network contained genes encoding protein kinases (Akt, Erk, Mapk, JAK2, PKC), transcription factors (NFκB, FOS), and several other signalling molecules (Insulin, CDKN1B, NR3C1, Vegf). The N network contained 32 candidate genes under selection (CD274, DHRS9, DIAPH2, ERO1L, GLP2R, GNAQ, HDAC8, HS6ST2, IGFBP7, IL17RD, JAK2, KLF13, MOV10, OTX2, PAX6, PPKCQ, PTPLAD1, SMG5, SORBS2, TFAP2A, ZBTB10, BBS9, CORO2A, DACH2, GFI1B, MMP16, POU3F4, PRIM1, RECQL, SERPINA7, TNMD, TRIM14), and the N1 network contained 29 candidate genes (AFF2, CEP78, COMMD8, EXT2, FAM184B, GCFC1, INO80D, KCNH5, KHDRBS3, LRP2BP, MED12, MLANA, MYO15A, NDST2, NDUFS1, NPAS2, OTC, PELI2, PHKA1, PSMB7, RGS22, SLC16A1, SLCO1A2, SYNE2, TIPRL, TRPM2, UBR2, UNC13C, WLS). These molecules were mainly involved in morphology, cellular function and maintenance, the cell cycle and signaling. The main hubs of the network contained IGF1R, JAK2 and calmodulin in the CHN VS EURO group (Additional file 4: Figure S1) and ESR1,PKC and insulin in Northern VS Southern group (Additional file 5: Figure S2).
Figure 4.

Representation of the gene network N. Symbols corresponding to genes under selection are colored in grey.
Figure 5.
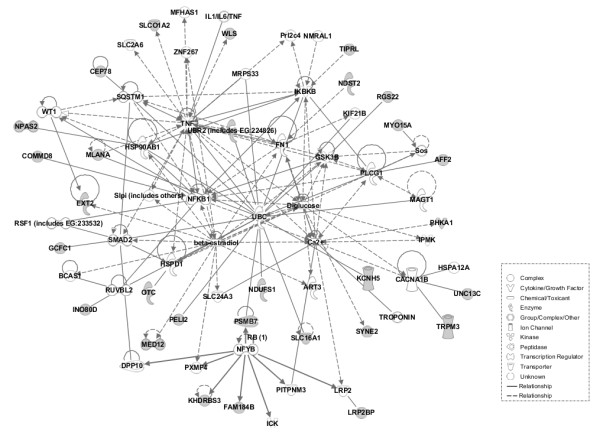
Representation of the gene network N1. Symbols corresponding to genes under selection are colored in grey.
Discussion
In this study, the population structure of the nine pig breeds was analyzed, and the PCA results showed that most of the individuals could be classified into their breeds using the first and second eigenvectors (Figure 1). As with other livestock species such as cattle and sheep [17,18], the combination of PC1 and PC2 separated individuals according to their geographic origin: of all the studied breeds, the indigenous breeds of Southern China (Wuzhishan, Bama, Luchuan) clustered together, as did the breeds of Central China (Nixiang, Tongcheng), the Northern Chinese breed (Laiwu) and a developed breed (Yutai) formed a separate single cluster, and two commercial breeds, the Large White and Landrace, formed a distinct cluster. There was almost no overlap between the nine different pig breeds. This opens the possibility that an informative SNP panel can be used to assign parentage, which has proven successful in cattle [19].
Pigs have been undergoing selection to enhance performance and productivity during domestication and breed formation. In the present study, global and pairwise Fst was utilized to detect genetic selection in Chinese indigenous and commercial pig breeds. First, the ALLPOP group showed evidence of selection on chromosomes 8 (Figure 3). We identified selection near KIT, which can affect coat colour in pigs when mutated [20] and also shows high evidence of selection in sheep [18]. In addition, as shown in Figures 4 and 5, the N network contained several hubs involved with physiological signaling molecules (NFκB, MAPK, ERK). These data indicate that these genes participate in the basic physiological processes, and the N network contained hubs (TNF and beta-estradiol) showing that the genes under selection are involved in the immune response and reproductive traits. The POU3F4 and OTX2 genes are important for the development of cochlea, and mutants of these two genes in mice cause developmental defects in the inner ear [21,22]. In mouse embryonic stem cells, the mutant zinc-finger proto-oncogene GFI1B gene decreases erythropoiesis of embryonic stem cells [23]. The PAX6 gene is necessary and sufficient to trigger the cascade of events required for eye formation [24]. The PAX6 and OTX2 genes also play important role in the development of the body axis [25,26]. In addition, several identified molecules are involved in the development of organs. The GNAQ gene can regulate cardiac growth and development, and mice lacking both GNAQ and GNA11 [Gaq(−/−); Ga11(−/−)] died at embryonic day 11 due to cardiomyocyte hypoplasia [27]. The TFAP2A gene is a critical transcription factor for epidermal differentiation and interacts with notch signaling molecules [28].
Several candidate genes are also involved in molecular transportation in the ALLPOP group. For example, the SLOC1A2 and SERPINA7 genes can increase the transport of thyroid hormone in the serum [29,30]. The SLC16A1 gene plays an important role in the transport of mevalonate and ketone bodies [31,32]. Among the candidate genes under selection, some are associated with genetic disorders and cancer in humans. GWAS results showed that a SNP substitution mutation of BBS9 was associated with amyotrophic lateral sclerosis [33]; furthermore, BANF2, SNX25, SAMD12 and GPR177 were associated with Crohn’s disease [34], and inflammatory bowel disease was associated with the upregulation of human CD274 at the cell surface from macrophage-derived dendritic cells of the inflamed colon [35]. In addition, three genes (DIAPH2, AFF2, POF1B) were involved in functions related to premature ovarian failure [36,37]. In the CHN VS EURO group, the network hubs are gathered at the centre with the IGF1R (Insulin-like growth factor 1 receptor) gene (Additional file 4: Figure S1), which is necessary for normal growth. IGF1R null mice die at birth of respiratory failure and exhibit only 45% of the body weight of their wild-type littermates [38]. European commercial pig breeds grow faster in contrast with Chinese indigenous breeds. In addition, the IGF1R gene also showed a strong signature of selection in European domestic pigs [39]. Interestingly, one of the most critical signalling molecules, JAK2, showed high evidence of positive selection both in the ALLPOP and the CHN VS EURO groups (Figure 4 and Additional file 4: Figure S1). JAK2 is an essential gene in mammals and participates in a variety of biological processes; the loss of JAK2 is lethal [40]. JAK2 is also involved in the immune response [41], and it has been suggested that these pig breeds may have different resistance to pathogens. In the Northern VS Southern group, the central hub of the network was the ESR1 (esotrogen receptor 1) gene (Additional file 5: Figure S2). The ESR1 gene was associated with litter size in pigs and was also a candidate gene for boar fertility and sperm quality [42-44]. Laiwu pigs have a higher reproductive capacity compared with the Bama and Wuzhishan pigs. Correspondingly, the ESR1 gene showed high evidence of positive selection in our study.
The high-Fst outlier method is a powerful tool for the detection of positive selection [45]; however, the high correlation between Fst estimates when loci are in strong disequilibrium makes it difficult to determine whether the Fst at particular SNP is markedly different from the expected values [46]. We also tested the correlation of Fst between pairs of SNPs as a function of marker distances; the correlation of Fst tended to drop quickly toward 0 when SNPs were more than 300 kb apart (data not shown). Modern pig breeds had much larger average linkage disequilibrium (LD) than humans and cattle [47], therefore, the results in pigs were greater than in humans and bovines [6,12].
Conclusions
Overall, a genome-wide scan was performed in Chinese indigenous pigs to help interpret artificial selection and adaptive evolution. We constructed population structures and genome-wide distributions of Fst. A number of genes were identified as displaying signatures of selection, and several critical physiological pathways of these genes were determined to have footprints of positive selection. Some of these genes play important roles in biological processes, which can be used to interpret the differences between these pig breeds.
Methods
DNA samples and SNP chip data quality control
DNA samples were obtained from 235 pigs from nine breeds from different areas of China, including six indigenous breeds: Tongcheng (TC, n = 35), Bama (BM, n = 22), Laiwu (LW, n = 23), Wuzhishan (WZS, n = 25), Ningxiang (NX, n = 24), Luchuan(LC, n = 40), two commercial breeds, Landrace (n = 18) and Large White (n = 26), and one developed breed, Yutai (YT, n = 22).
Genotyping was carried out using the Illumina Porcine SNP60 BeadChip [14], which contains a total of 62,123 SNPs. Quality control was determined using the PLINK programme [48]. A total of 8,383 unmapped markers (Based on Sus Scrofa Build 9.0) and 8,391 loci were filtered to exclude markers with a minor allele frequency (MAF) < 0.05. A total of 2,709 markers that were genotyped on less than 90% of all individuals were discarded from further analysis. The final data set consisted of 44,652 SNPs from nine breeds.
Population structure and Fst estimation
Principal component analysis (PCA) based on all available SNP information was performed using the SVS7 software (Golden Helix Inc., Bozeman, MT,USA). Fst statistics across populations were estimated using the Genepop 4.1 program [49]. Fst is a measure of population differentiation, which is defined as ,
where MSG, MSI and MSP represent the mean sums of squares for gametes, individuals and populations computed by an analysis of variance, respectively, and n c = (S1 − S2/S1)/(n − 1), where S 1 is the total sample size, S 2 is the sum of squared group sizes, and n is the number of non-empty groups.
Identification of candidate genes under selection
Genome regions containing the high-Fst outliers corresponding to the distribution of Fst were identified as follows: for all loci, a region was considered to be a high-Fst outlier if it corresponded to the upper 1% of the empirical genome-wide distribution of Fst. A gene was regarded as being under selection if it contained unexpectedly highly differentiated SNPs among the populations. All of the high-Fst outlier loci were mapped to gene-associated regions based on the pig genome annotation (Sus Scrofa Build 9.0 version). An SNP was considered to be from a particular gene if it mapped to either the 5′ upstream, 5′ UTR, coding, intronic, 3′UTR, or 3′ downstream region of the gene.
Network analysis of candidate genes
Network analysis was aimed at searching for the direct or indirect interactions between candidate molecules and the related property. The known interactions were annotated by experts according to the literature. Ingenuity Pathway Analysis (IPA) v7.0 (Ingenuity Systems Inc., USA, http://www.ingenuity.com/) was used to construct networks. We uploaded the genes being subjected to selection into this software and organized them into networks of interacting genes to identify several pathways containing important functionally related genes. This network analysis approach was similar to the described by Flori [6]. The genes that displayed evidence of selection were uploaded into IPA based on the eligible candidate genes, and IPA automatically constructed several networks that contained a limit of 70 molecules (including candidate genes).
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SY and XL carried out the statistical analysis and drafted the manuscript, BF participated in the study design and manuscript preparation, and ZT and KL contributed to the data collection and genotyped all of the samples. All authors contributed to editing the article and approved the final manuscript.
Supplementary Material
Candidate genes under selection with SNPs in high Fst in group ALLPOP.
Candidate genes under selection with SNPs in high Fst (CHN VS EURO).
Candidate genes under selection with SNPs in high Fst (Northern VS Southern).
Representation of the gene network group CHN VS EURO. Symbols corresponding to genes under selection are colored in grey.
Representation of the gene network group Northern VS Southern. Symbols corresponding to genes under selection are colored in grey.
Contributor Information
Songbai Yang, Email: a1688816@163.com.
Xiuling Li, Email: lixiuling_87@163.com.
Kui Li, Email: kuili@iascaas.net.cn.
Bin Fan, Email: fanbin@mail.hzau.edu.cn.
Zhonglin Tang, Email: zhonglinqy_99@sina.com.
Acknowledgements
The Work was supported by the National High Technology Research and Development Program of China (2011AA100302), the National Science Foundation of China (31172192), the National Basic Research Program of China (2012CB124706-6), the Program for New Century Excellent Talents (NCET-11-0646) and the Fundamental Research Funds for the Central Universities (2012PY009).
References
- Albarella U, Dobney K, Ervynk A, Rowley-Conwy P. Pigs and Humans: 10,000 Years of Interaction. Oxford: Oxford University Press; 2007. [Google Scholar]
- Giuffra E, Kijas JM, Amarger V, Carlborg O, Jeon JT, Andersson L. The origin of the domestic pig: independent domestication and subsequent introgression. Genetics. 2000;154(4):1785–1791. doi: 10.1093/genetics/154.4.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groenen MA, Archibald AL, Uenishi H, Tuggle CK, Takeuchi Y, Rothschild MF, Rogel-Gaillard C, Park C, Milan D, Megens HJ. et al. Analyses of pig genomes provide insight into porcine demography and evolution. Nature. 2012;491(7424):393–398. doi: 10.1038/nature11622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier M, Flori L, Riebler A, Jaffrezic F, Laloe D, Gut I, Moazami-Goudarzi K, Foulley JL. A whole genome Bayesian scan for adaptive genetic divergence in West African cattle. BMC Genomics. 2009;10:550. doi: 10.1186/1471-2164-10-550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier M, Naves M. Footprints of selection in the ancestral admixture of a New World Creole cattle breed. Mol Ecol. 2011;20(15):3128–3143. doi: 10.1111/j.1365-294X.2011.05163.x. [DOI] [PubMed] [Google Scholar]
- Flori L, Fritz S, Jaffrezic F, Boussaha M, Gut I, Heath S, Foulley JL, Gautier M. The genome response to artificial selection: a case study in dairy cattle. PLoS One. 2009;4(8):e6595. doi: 10.1371/journal.pone.0006595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qanbari S, Pimentel EC, Tetens J, Thaller G, Lichtner P, Sharifi AR, Simianer H. A genome-wide scan for signatures of recent selection in Holstein cattle. Anim Genet. 2010;41(4):377–389. doi: 10.1111/j.1365-2052.2009.02016.x. [DOI] [PubMed] [Google Scholar]
- Qanbari S, Gianola D, Hayes B, Schenkel F, Miller S, Moore S, Thaller G, Simianer H. Application of site and haplotype-frequency based approaches for detecting selection signatures in cattle. BMC Genomics. 2011;12(1):318. doi: 10.1186/1471-2164-12-318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akey JM, Ruhe AL, Akey DT, Wong AK, Connelly CF, Madeoy J, Nicholas TJ, Neff MW. Tracking footprints of artificial selection in the dog genome. Proc Natl Acad Sci U S A. 2010;107(3):1160–1165. doi: 10.1073/pnas.0909918107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin CJ, Zody MC, Eriksson J, Meadows JR, Sherwood E, Webster MT, Jiang L, Ingman M, Sharpe T, Ka S. et al. Whole-genome resequencing reveals loci under selection during chicken domestication. Nature. 2010;464(7288):587–591. doi: 10.1038/nature08832. [DOI] [PubMed] [Google Scholar]
- Nielsen R. Molecular signatures of natural selection. Annu Rev Genet 2005. 2005;39:197–218. doi: 10.1146/annurev.genet.39.073003.112420. [DOI] [PubMed] [Google Scholar]
- Akey JM, Zhang G, Zhang K, Jin L, Shriver MD. Interrogating a high-density SNP map for signatures of natural selection. Genome Res. 2002;12(12):1805–1814. doi: 10.1101/gr.631202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabeti PC, Reich DE, Higgins JM, Levine HZ, Richter DJ, Schaffner SF, Gabriel SB, Platko JV, Patterson NJ, McDonald GJ. et al. Detecting recent positive selection in the human genome from haplotype structure. Nature. 2002;419(6909):832–837. doi: 10.1038/nature01140. [DOI] [PubMed] [Google Scholar]
- Ramos AM, Crooijmans RP, Affara NA, Amaral AJ, Archibald AL, Beever JE, Bendixen C, Churcher C, Clark R, Dehais P. et al. Design of a high density SNP genotyping assay in the pig using SNPs identified and characterized by next generation sequencing technology. PLoS ONE. 2009;4(8):e6524. doi: 10.1371/journal.pone.0006524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Yang H, Li JR, Li HP, Ning T, Pan XR, Shi P, Zhang YP. Artificial selection of the melanocortin receptor 1 gene in Chinese domestic pigs during domestication. Heredity. 2010;105(3):274–281. doi: 10.1038/hdy.2009.191. [DOI] [PubMed] [Google Scholar]
- Ren J, Duan Y, Qiao R, Yao F, Zhang Z, Yang B, Guo Y, Xiao S, Wei R, Ouyang Z. et al. A missense mutation in PPARD causes a major QTL effect on ear size in pigs. PLoS Genet. 2011;7(5):e1002043. doi: 10.1371/journal.pgen.1002043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs RA, Taylor JF, Van Tassell CP, Barendse W, Eversole KA, Gill CA, Green RD, Hamernik DL, Kappes SM, Lien S. et al. Genome-wide survey of SNP variation uncovers the genetic structure of cattle breeds. Science. 2009;324(5926):528–532. doi: 10.1126/science.1167936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kijas JW, Lenstra JA, Hayes B, Boitard S, Porto Neto LR, San Cristobal M, Servin B, McCulloch R, Whan V, Gietzen K. et al. Genome-wide analysis of the world’s sheep breeds reveals high levels of historic mixture and strong recent selection. PLoS Biol. 2012;10(2):e1001258. doi: 10.1371/journal.pbio.1001258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaton MP, Harhay GP, Bennett GL, Stone RT, Grosse WM, Casas E, Keele JW, Smith TP, Chitko-McKown CG, Laegreid WW. Selection and use of SNP markers for animal identification and paternity analysis in U.S. beef cattle. Mamm Genome. 2002;13(5):272–281. doi: 10.1007/s00335-001-2146-3. [DOI] [PubMed] [Google Scholar]
- Fontanesi L, D’Alessandro E, Scotti E, Liotta L, Crovetti A, Chiofalo V, Russo V. Genetic heterogeneity and selection signature at the KIT gene in pigs showing different coat colours and patterns. Anim Genet. 2010;41(5):478–492. doi: 10.1111/j.1365-2052.2010.02054.x. [DOI] [PubMed] [Google Scholar]
- Morsli H, Tuorto F, Choo D, Postiglione MP, Simeone A, Wu DK. Otx1 and Otx2 activities are required for the normal development of the mouse inner ear. Development. 1999;126(11):2335–2343. doi: 10.1242/dev.126.11.2335. [DOI] [PubMed] [Google Scholar]
- Phippard D, Lu L, Lee D, Saunders JC, Crenshaw EB 3rd. Targeted mutagenesis of the POU-domain gene Brn4/Pou3f4 causes developmental defects in the inner ear. J Neurosci. 1999;19(14):5980–5989. doi: 10.1523/JNEUROSCI.19-14-05980.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleque S, Cameron S, Orkin SH. The zinc-finger proto-oncogene Gfi-1b is essential for development of the erythroid and megakaryocytic lineages. Genes Dev. 2002;16(3):301–306. doi: 10.1101/gad.959102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow RL, Altmann CR, Lang RA, Hemmati-Brivanlou A. Pax6 induces ectopic eyes in a vertebrate. Development. 1999;126(19):4213–4222. doi: 10.1242/dev.126.19.4213. [DOI] [PubMed] [Google Scholar]
- Kimura C, Shen MM, Takeda N, Aizawa S, Matsuo I. Complementary functions of Otx2 and Cripto in initial patterning of mouse epiblast. Dev Biol. 2001;235(1):12–32. doi: 10.1006/dbio.2001.0289. [DOI] [PubMed] [Google Scholar]
- Bishop KM, Rubenstein JL, O’Leary DD. Distinct actions of Emx1, Emx2, and Pax6 in regulating the specification of areas in the developing neocortex. J Neurosci. 2002;22(17):7627–7638. doi: 10.1523/JNEUROSCI.22-17-07627.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offermanns S, Zhao LP, Gohla A, Sarosi I, Simon MI, Wilkie TM. Embryonic cardiomyocyte hypoplasia and craniofacial defects in G alpha q/G alpha 11-mutant mice. EMBO J. 1998;17(15):4304–4312. doi: 10.1093/emboj/17.15.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Pasolli HA, Williams T, Fuchs E. AP-2 factors act in concert with Notch to orchestrate terminal differentiation in skin epidermis. J Cell Biol. 2008;183(1):37–48. doi: 10.1083/jcb.200804030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vranckx R, Rouaze-Romet M, Savu L, Mechighel P, Maya M, Nunez EA. Regulation of rat thyroxine-binding globulin and transthyretin: studies in thyroidectomized and hypophysectomized rats given tri-iodothyronine or/and growth hormone. J Endocrinol. 1994;142(1):77–84. doi: 10.1677/joe.0.1420077. [DOI] [PubMed] [Google Scholar]
- Xu C, Li CY, Kong AN. Induction of phase I, II and III drug metabolism/transport by xenobiotics. Arch Pharm Res. 2005;28(3):249–268. doi: 10.1007/BF02977789. [DOI] [PubMed] [Google Scholar]
- Pardridge WM. The blood–brain barrier: bottleneck in brain drug development. NeuroRx: J Am Soc Exp NeuroTherapeutics. 2005;2(1):3–14. doi: 10.1602/neurorx.2.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CM, Goldstein JL, Brown MS. cDNA cloning of MEV, a mutant protein that facilitates cellular uptake of mevalonate, and identification of the point mutation responsible for its gain of function. J Biol Chem. 1992;267(32):23113–23121. [PubMed] [Google Scholar]
- van Es MA, Van Vught PW, Blauw HM, Franke L, Saris CG, Andersen PM, Van Den Bosch L, de Jong SW, Van’t Slot R, Birve A. et al. ITPR2 as a susceptibility gene in sporadic amyotrophic lateral sclerosis: a genome-wide association study. Lancet Neurol. 2007;6(10):869–877. doi: 10.1016/S1474-4422(07)70222-3. [DOI] [PubMed] [Google Scholar]
- Consortium TWTCC. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447(7145):661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Chen L. Co-signaling molecules of the B7-CD28 family in positive and negative regulation of T lymphocyte responses. Microbes and Infection/Institut Pasteur. 2004;6(8):759–766. doi: 10.1016/j.micinf.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Goswami D, Conway GS. Premature ovarian failure. Hum Reprod Update. 2005;11(4):391–410. doi: 10.1093/humupd/dmi012. [DOI] [PubMed] [Google Scholar]
- Lacombe A, Lee H, Zahed L, Choucair M, Muller JM, Nelson SF, Salameh W, Vilain E. Disruption of POF1B binding to nonmuscle actin filaments is associated with premature ovarian failure. Am J Hum Genet. 2006;79(1):113–119. doi: 10.1086/505406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JP, Baker J, Perkins AS, Robertson EJ, Efstratiadis A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r) Cell. 1993;75(1):59–72. [PubMed] [Google Scholar]
- Rubin CJ, Megens HJ, Martinez Barrio A, Maqbool K, Sayyab S, Schwochow D, Wang C, Carlborg O, Jern P, Jorgensen CB. et al. Strong signatures of selection in the domestic pig genome. Proc Natl Acad Sci U S A. 2012;109(48):19529–19536. doi: 10.1073/pnas.1217149109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neubauer H, Cumano A, Muller M, Wu H, Huffstadt U, Pfeffer K. Jak2 deficiency defines an essential developmental checkpoint in definitive hematopoiesis. Cell. 1998;93(3):397–409. doi: 10.1016/S0092-8674(00)81168-X. [DOI] [PubMed] [Google Scholar]
- Zhong J, Yang P, Muta K, Dong R, Marrero M, Gong F, Wang CY. Loss of Jak2 selectively suppresses DC-mediated innate immune response and protects mice from lethal dose of LPS-induced septic shock. PLoS ONE. 2010;5(3):e9593. doi: 10.1371/journal.pone.0009593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz G, Ovilo C, Estelle J, Silio L, Fernandez A, Rodriguez C. Association with litter size of new polymorphisms on ESR1 and ESR2 genes in a Chinese-European pig line. Genet Sel Evol. 2007;39(2):195–206. doi: 10.1186/1297-9686-39-2-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunawan A, Kaewmala K, Uddin MJ, Cinar MU, Tesfaye D, Phatsara C, Tholen E, Looft C, Schellander K. Association study and expression analysis of porcine ESR1 as a candidate gene for boar fertility and sperm quality. Anim Reprod Sci. 2011;128(1–4):11–21. doi: 10.1016/j.anireprosci.2011.08.008. [DOI] [PubMed] [Google Scholar]
- van Rens BT, de Groot PN, van der Lende T. The effect of estrogen receptor genotype on litter size and placental traits at term in F2 crossbred gilts. Theriogenology. 2002;57(6):1635–1649. doi: 10.1016/S0093-691X(02)00671-4. [DOI] [PubMed] [Google Scholar]
- Karlsson S, Moen T. The power to detect artificial selection acting on single loci in recently domesticated species. BMC Res Notes. 2010;3:232. doi: 10.1186/1756-0500-3-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holsinger KE, Weir BS. Genetics in geographically structured populations: defining, estimating and interpreting FST. Nat Rev Genet. 2009;10(9):639–650. doi: 10.1038/nrg2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Plastow G. Genomic Diversity in Pig (Sus scrofa) and its Comparison with Human and other Livestock. Curr Genomics. 2011;12(2):138–146. doi: 10.2174/138920211795564386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond M, Rousset F. GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. J Hered. 1995;86:248–249. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Candidate genes under selection with SNPs in high Fst in group ALLPOP.
Candidate genes under selection with SNPs in high Fst (CHN VS EURO).
Candidate genes under selection with SNPs in high Fst (Northern VS Southern).
Representation of the gene network group CHN VS EURO. Symbols corresponding to genes under selection are colored in grey.
Representation of the gene network group Northern VS Southern. Symbols corresponding to genes under selection are colored in grey.